

Abstract
By studying primary isogenic murine embryonic fibroblasts (MEFs), we have shown that PLK3 null MEFs contain a reduced level of phosphatase and tensin homolog (PTEN) and increased Akt1 activation coupled with decreased GSK3β activation under normoxia and hypoxia. Purified recombinant Plk3, but not a kinase-defective mutant, efficiently phosphorylates PTEN in vitro. Mass spectrometry identifies threonine 366 and serine 370 as two putative residues that are phosphorylated by Plk3. Immunoblotting using a phosphospecific antibody confirms these sites as Plk3 phosphorylation sites. Moreover, treatment of MEFs with LiCl, an inhibitor of GSK3β and CK2, only partially suppresses the phosphorylation, suggesting Plk3 as an additional kinase that phosphorylates these sites in vivo. Plk3-targeting mutants of PTEN are expressed at a reduced level in comparison with the wild-type counterpart, which is associated with an enhanced activity of PDK1, an upstream activator of Akt1. Furthermore, the reduced level of PTEN in PLK3 null MEFs is stabilized by treatment with MG132, a proteosome inhibitor. Combined, our study identifies Plk3 as a new player in the regulation of the PI3K/PDK1/Akt signaling axis by phosphorylation and stabilization of PTEN.
Keywords: Hypoxia, Phosphatase, Protein Kinases, Protein Stability, Tumor Suppressor, Akt1, Cell Survival, PTEN, Polo-like Kinase
Introduction
Plk3, a member of the Polo-like kinase family, plays an important role in regulating cell cycle checkpoint control in response to genotoxic stresses (1, 2), as well as in cellular responses to hypoxia (3, 4). Plk3 is also strongly implicated in tumorigenesis. Aberrant expression of Plk3 is found in multiple tumors (5, 6). The human PLK3 gene is localized to the short arm of the chromosome 1 (1p32), a region that displays loss of heterozygosity or homozygous deletions in many types of cancer and has been proposed to harbor tumor susceptibility genes (7, 8). A recent mouse genetic study showed that PLK3−/− mice develop tumors in multiple organs at an enhanced rate (4). Many of the tumors developed in the PLK3 null mice are large in size and are highly vascularized (4), suggesting that this kinase may be involved in regulating the angiogenesis pathway.
The PTEN3 tumor suppressor is frequently mutated in cancer cells, and inherited PTEN mutations cause cancer-susceptibility conditions, including Cowden syndrome (9–13). The PTEN level, as well as its activity, profoundly influences tumor susceptibility because haplo-insufficiency of PTEN results in tumor development in many organs in animal models (14, 15). Biochemically, PTEN dephosphorylates the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate to generate phosphatidylinositol 3,4-bisphosphate and, by doing so, antagonizes the PI3K/Akt signaling pathway. Therefore, the PTEN tumor suppressor is a central negative regulator of the PI3K/PDK1/Akt signaling axis that controls multiple cellular functions, including cell growth, survival, proliferation, and angiogenesis (16). PTEN is also involved in regulating hypoxic responses and HIF-1α stability (17, 18). Loss of PTEN function and increased activities of PI3K/Akt are associated with enhanced expression of HIF-1α and its homologs during hypoxia (17–19). Increased levels of HIF-1α and VEGF play essential roles in the development and progression of human cancers (20, 21).
PTEN is also subjected to regulation by phosphorylation. Phosphorylation of several serine/threonine residues (e.g. Ser-370, Thr-382, Thr-383, and Ser-385) in the C-tail region of PTEN by casein kinase 2 (CK2) is essential for the tail-dependent regulation of stability (22, 23). PTEN phospho-defective mutant proteins exhibit decreased stability in comparison with the wild-type PTEN (22). GSK3β phosphorylates PTEN at Ser-362 and Thr-366. Interestingly, previous phosphorylation of PTEN at Ser-370 by CK2 promotes the phosphorylation at Thr-366 by GSK3β (24), suggesting that these enzymes may cooperate in the regulation of the phosphatase. PTEN can also be phosphorylated on tyrosine residues by Rak; this phosphorylation stabilizes PTEN (25, 26). Depletion of Rak via RNAi enhances the binding of PTEN to NEDD4-1, an E3 ligase, and promotes PTEN polyubiquitination and subsequent degradation (26).
To elucidate the molecular mechanism by which Plk3 regulates the cell survival pathway, we examined the expression and/or activation status of major components of the PI3K/Akt/GSK3β signaling axis in PLK3 null MEFs. PLK3 ablation resulted in a reduced level of PTEN, which was correlated with increased PDK1/Akt1 activation and decreased GSK3β activity. Protein kinase assays showed that recombinant Plk3, but not its kinase-defective mutant, phosphorylated PTEN on both Thr-366 and Ser-370 in vitro. Our further analysis revealed that phosphorylation of PTEN by Plk3 was important for regulating its stability and activity.
EXPERIMENTAL PROCEDURES
Cell Culture
A549 and HEK293T cell lines were obtained from the American Type Culture Collection. Cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 μg of penicillin and 50 μg of streptomycin sulfate per ml) under 5% CO2. Primary wild-type and PLK3−/− mouse embryonic fibroblasts (MEFs) were derived from embryonic day 14.5 embryos of the respective genotype, produced from the crossing of PLK3+/− mice. Primary MEF cells were cultured in DMEM supplemented with 15% FBS and antibiotics (as above) under 5% CO2. In all experiments that involved the use of MEFs, only primary MEFs of 3rd to 5th passages were used for various studies.
For experiments with LiCl or MG132 treatment, cells were cultured in the presence or absence of 10 mm LiCl or 10 μm MG132 for 4 h before harvesting for lysate preparation. The lysates were then subjected to Western blot analysis with the indicated antibodies.
Antibodies
Antibodies for Akt1, phospho-Akt1 (Ser-473), GSK3β, phospho-GSK3β (Ser-9), PTEN, phospho-PTEN (Ser-380/Ser-382/Ser-383), mTOR, phospho-mTOR (Ser-2448), PDK1, phospho-PDK1 (Ser-241), and β-actin were purchased from Cell Signaling Technology. Antibody for GFP was purchased from Santa Cruz Biotechnology. Antibody for phospho-PTEN (Thr(P)-366/Ser(P)-370) was purchased from Epitomics. Plk3 antibody was obtained from BD Biosciences.
Recombinant Proteins and Other Reagents
Recombinant His6-tagged Plk3 proteins were expressed using the baculoviral expression system and purified from Sf9 insect cells as described previously (27). Briefly, Sf9 cells (ATCC) cultured in Grace's insect cell culture medium were infected with Plk3 baculovirus. Three days after infection, cells were collected and lysed in a lysis buffer (50 mm NaH2PO4, 300 mm NaCl, 1% Nonidet P-40 20 mm imidazole, 1 mm PMSF, 2 μm pepstatin A, 10 units/ml aprotinin). Cell lysates were cleared by centrifugation and then incubated with Ni-NTA-agarose resins for 3 h at 4 °C. Plk3 protein was then eluted from Ni-NTA resins with lysis buffer containing 200 mm imidazole after extensive wash of the resins with the lysis buffer. The eluted protein was dialyzed into the storage buffer (25 mm Tris, pH 7.4, 5 mm EGTA, 2 mm DTT, 0.1% Triton X-100, and 50% glycerol) and stored at −80 °C for subsequent uses.
Recombinant proteins for full-length (catalog no. 7436) and partial GSK-3β (catalog no. 9237) were purchased from Cell Signaling Technology. Full-length inactive Akt1 protein was purchased from Novus Biologicals (catalog no. H00000207-P01). Full-length partially active Akt1 protein was purchased from GenWay (catalog no. 10-054-165007). Human PTEN protein was from Cayman Chemicals (catalog no. 10009746). Human PTEN with both His6 and HA tags was also expressed using the baculoviral system as above and purified to homogeneity for kinase and ubiquitination assays. Casein and wortmannin were purchased from Sigma.
Plk3 Kinase Assays
Plk3 kinase assays were carried out essentially using the protocol described (1). Briefly, indicated amounts of recombinant His6-Plk3 protein was added to kinase assay buffer (20 mm HEPES, pH 7.4, 2 mm EGTA, 10 mm MgCl2, 5 mm MnCl2, 1 mm DTT). Substrates (0.5 μg) were then added to achieve a total volume of 20 μl. Kinase reactions were initiated by the addition of 5 μl of either the radioactive ATP mix (50 mm MgCl2, 0.1 mm ATP, 5 μCi of [γ-32P]ATP) or the cold ATP mix (50 mm MgCl2, 5 mm ATP). After incubation for 30 min at 30 °C, SDS sample buffer was supplemented to each reaction. Proteins fractionated on SDS-denaturing gels were transferred to PVDF membranes followed by autoradiography or immunoblotting.
To detect phosphorylation of PTEN (p-PTENThr-366/Ser-370) by Plk3 in vitro using a phosphospecific antibody, His-tagged PTEN recombinant protein was first dephosphorylated using λ phosphatase (New England Biolab) according to the protocol provided by the supplier. Five hundred μl of IP buffer (20 mm HEPES, pH 7.4, 120 mm NaCl, 1% Triton X-100, 2 mm EGTA, 10% glycerol, 1 mm DTT, 500 μm PMSF, 2 μm pepstatin A, 1 IU/ml aprotinin) and 40 μl of Ni-NTA resin (50:50) was then added to the reaction. The mixture was rotated at 4 °C for 3 h. After incubation, the resin was washed three times with the IP buffer, three times with the kinase buffer (20 mm HEPES, pH 7.2, 2 mm EGTA, 10 mm MgCl2, 5 mm MnCl2, 1 mm DTT, 1 mm Na3VO4), and resuspended in the kinase buffer as a 50:50 (v/v) slurry. The suspension was then used as substrate for the cold kinase assay (without [32P]ATP in the kinase reaction) containing Plk3 or Plk3-K91R as above. After the reaction, the reaction mixtures were subjected to Western blot analysis with antibodies to phospho-Thr-366/Ser-370 and to total PTEN.
Transfection and Western Blot
All PTEN plasmids (wild-type and phosphorylation mutants) were obtained from Addgene. Plasmid transfection was performed using Lipofectamine reagents from Invitrogen according to the protocol provided by the manufacturer. Standard Western blot procedure was using throughout the study. SDS-PAGE was carried out using the mini gel system from Bio-Rad. Proteins were transferred to PVDF membranes that were blocked with PBST containing 5% nonfat dry milk. The membranes were then incubated overnight with primary antibodies using dilutions suggested by the manufacturers. The membranes were then incubated with secondary antibodies for 1 h at room temperature. After thorough washing with PBST buffer, signals on membranes were developed with an ECL system (Pierce).
Immunoprecipitation and Pulldown Assays
For immunoprecipitation, cells were lysed in the lysis buffer (20 mm HEPES, pH 7.6, 400 mm NaCl, 0.1% Nonidet P-40, 1 mm DTT, 1 mm EDTA, 1 mm DTT, 1 mm NaF, 1 mm sodium orthovanadate, 500 μm PMSF, 2 μm pepstatin A, 10 units/ml aprotinin) and cleared by centrifugation. One μg of antibody and 40 μl of protein G-agarose resin (50:50, Upstate Biotechnology) were then added to 1–3 mg of cell lysates and incubated at 4 °C for 3 h to overnight followed by extensively washing with the lysis buffer. Proteins on the resins were eluted with SDS sample buffer and then subjected to analysis by SDS-PAGE followed by Western blot with appropriate antibodies.
For pulldown assay, recombinant His6-Plk3 was affinity-purified and subsequently conjugated to Ni-NTA resin (Qiagen). Plk3-conjugated Ni-NTA resin or the resin alone was incubated for 3 h at 4 °C with A549 lysates. The resin was extensively washed, and proteins that were bound to either His6-Plk3 or the control resin were eluted in the SDS sample buffer and subjected to SDS-PAGE analysis followed by Western blotting for Akt1 or GSK3β.
Detection of Phosphorylation Sites by Mass Spectrometry
Recombinant PTEN phosphorylated by His6-Plk3 using the cold kinase assay protocol as described above was fractionated on an SDS-denaturing gel. After Coomassie Brilliant Blue staining, gels were sliced and subjected to mass spectrometric analysis at the Center for Functional Genomics, the Research Foundation of the State University of New York, Albany. The phosphorylation status was confirmed by companion kinase reactions using the hot kinase assay protocol.
RESULTS
PLK3 Ablation Results in Akt Activation
We have previously shown that PLK3 null MEFs contain an elevated level of HIF-1α under hypoxia in comparison with wild-type MEFs (4). As HIF-1α expression is regulated by the PI3K/Akt/GSK3β signaling pathway (28, 29), we analyzed activities and expression of Akt1 and GSK3β. Using a panel of antibodies, including those that recognize phospho-epitopes of targeting proteins, we observed that p-Akt1Ser-473 (associated with activation) was significantly higher in naive PLK3−/− MEFs and that the difference in the level of phosphorylation was more pronounced after treatment with nickel chloride, a hypoxic mimetic (Fig. 1). On the other hand, the total Akt1 levels were not significantly affected either by the Plk3 status or by the hypoxic condition (Fig. 1). Because active Akt1 is known to phosphorylate and inhibit GSK3β (30, 31), we also examined the level of phosphorylation of GSK3β serine 9 (p-GSK3βSer-9), an Akt1 target, before and after the hypoxia mimetic treatment. Similar to that of p-Akt1Ser-473, the p-GSK3βSer-9 signal, but not its total protein level, was much higher in PLK3−/− MEFs than that in wild-type MEFs, in naive MEFs, or in MEFs treated with the hypoxic mimetic for up to at least 8 h (Fig. 1). The mTOR activity (p-mTor) was comparable in both wild-type and PLK3 null MEFs. After treatment with the hypoxia mimetic, the activity of mTOR (p-mTor) gradually increased in PLK3−/− but not in the wild-type MEFs until 16 h post-treatment when both types of MEFs contained similar levels of active mTOR (Fig. 1). Combined, our MEF studies strongly suggest that PLK3 deficiency significantly alters the activities of a panel of important signaling molecules that regulate cell survival pathway.
FIGURE 1.

PLK3 ablation significantly modulates Akt1 and GSK3β activation. Paired wild-type (W) and PLK3−/− (H) MEFs were treated with Ni2+ for various times as indicated. Equal amounts of cell lysates were blotted for phospho-Akt1 (Ser-473), total Akt1, phospho-GSK3β (Ser-9), total GSK3β, phospho-GSK3β, total GSK3β, phospho-mTOR, total mTOR, and β-actin.
Plk3 Does Not Phosphorylate Akt1 or GSK3β
To determine whether Plk3 may directly regulate either Akt1 or GSK3β, we first examined the physical interaction between Plk3 and Akt1 or between Plk3 and GSK3β. Co-immunoprecipitation experiments revealed that both Akt1 and GSK3β antibodies, but not the control IgG, precipitated Plk3 (supplemental Fig. 1), suggesting their physical interaction. As GSK3β migrated at almost the same position as IgGs, we used the pulldown approach to confirm the physical interaction between Plk3 and GSK3β. His6-Plk3 expressed in Sf9 cells was immobilized to Ni-NTA resin. Control resin was generated by incubating with the same amount of Sf9 cell lysates. Both the Plk3 resin and the control resin were incubated with A549 cell lysates. Proteins specifically bound to either resin were analyzed by immunoblotting for Plk3 and GSK3β. The Plk3 resin, but not the control resin, specifically precipitated GSK3β (Fig. 2A). Additional co-immunoprecipitation experiments showed that the Plk3 antibody, but not the control IgG, brought down Akt1 that migrated slightly above IgGs on a denaturing SDS gel (Fig. 2B).
FIGURE 2.
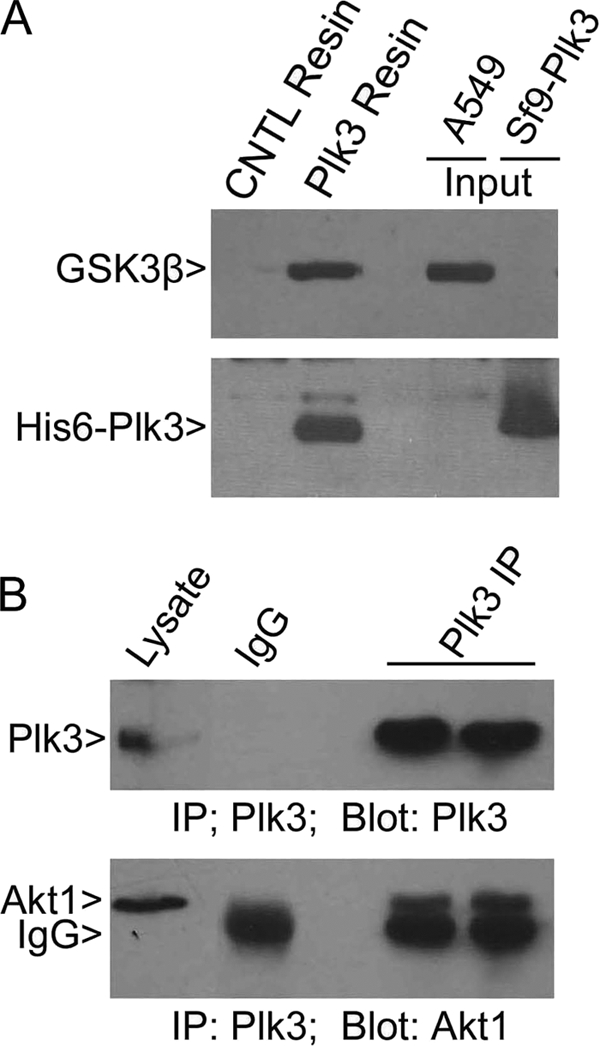
Plk3 interacts with Akt1 and GSK3β. A, His6-Plk3 Ni-NTA resin or control resin was incubated with equal amounts of A549 lysates. After washing, proteins eluted from either resin were blotted for Plk3 and GSK3β. Lysates from A549 and Sf9 cells expressing His6-Plk3 were also used for blotting. B, A549 cell lysates were immunoprecipitated (IP) with Plk3 IgG or with the control IgG. After washing, the immunoprecipitates, along with the lysate inputs, were blotted for Plk3 or Akt1.
Given the observed physical interactions between Plk3 and Akt1 or between Plk3 and GSK3β, we tested the possibility of phosphorylation of Akt1 and/or GSK3β in vitro by Plk3. Ser-473 of Akt1 is one of two sites that are phosphorylated in vivo, and the Akt1Ser-473 phosphorylation is tightly associated with its activation (32). Immunoblotting revealed that Plk3 at two different concentrations failed to generate any p-Akt1Ser-473 signals (Fig. 3A), indicating that Plk3 does not phosphorylate Akt1 on this residue. We further examined whether Plk3 phosphorylated Akt1 at all. Because recombinant Akt1 exhibited potent autophosphorylation (data not shown), we used purified kinase-dead Akt1 in our in vitro kinase assays. Plk3 did not phosphorylate the kinase-defective Akt1, although it was capable of weak autophosphorylation (Fig. 3B). As serine 9 of GSK3β is the primary phosphorylation site that is directly phosphorylated under hypoxia (33, 34), we examined whether Plk3 might phosphorylate GSK3βSer-9. In vitro kinase assays using a GST-GSK3β peptide containing Ser-9 did not reveal significant phosphorylation by Plk3, although it phosphorylated casein efficiently in a wortmannin-sensitive manner (Fig. 3C). Full-length GSK3β also exhibited significant autophosphorylation that was manifested as the incorporation of 32P into the recombinant protein; the addition of Plk3 did not further boost the incorporation of radioactivity (Fig. 3D), suggesting that GSK3β is not an in vitro substrate of Plk3.
FIGURE 3.

Plk3 does not significantly phosphorylate Akt1 or GSK3β. A, His6-Plk3 (0.1 or 0.4 μg) and Akt1 were incubated in the kinase reaction containing cold ATP. After the reaction, the samples were blotted for Akt1, phospho-Akt1 (Ser-437), and Plk3. HeLa cells treated with insulin for 4 h were used as a positive control. B, in vitro kinase assays were carried out in the presence of various components as indicated. The kinase-dead Akt1 protein was used for the assay as Akt1 has a strong autophosphorylation activity (see the supplemental material). His6-Plk3 was also capable of autophosphorylation (p-Plk3 arrowhead). Wort denotes wortmannin. C, Plk3 was assayed for its kinase activity toward a GST-GSK3β peptide [GSK3β(Pep)] containing serine 9, the primary site phosphorylated by Akt1. Then size of the peptide is similar to that of casein on denaturing gel. D, purified Plk3 and GSK3β were assayed for their in vitro kinase activities using 32P-ATP in a buffer containing various components as indicated.
Plk3 Regulates PTEN by Phosphorylation
The observations that Akt1 activity is high in PLK3 null MEFs and that Plk3 does not seem to directly regulate this protein prompted us to search for upstream protein(s) that could be a direct target(s) of Plk3. PTEN dephosphorylates the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate, antagonizing the PI3K/Akt signaling pathway (35). Because the PTEN activity and stability are subjected to the regulation by phosphorylation (16), we performed in vitro kinase assays using purified recombinant PTEN. His6-Plk3 significantly phosphorylated PTEN, as well as casein, as evidenced by the incorporation of 32P; the Plk3-catalyzed incorporation of 32P into these substrates was significantly suppressed by wortmannin (Fig. 4A), suggesting the specificity of the reaction. To eliminate the remote possibility of a contaminating kinase activity in purified recombinant Plk3, we expressed and purified a Plk3 kinase-defective mutant (Plk3-K91R) in the same manner as for Plk3. Protein kinase assays showed that Plk3 but not Plk3-K91R efficiently phosphorylated PTEN as well as casein (Fig. 4B). Immunoblotting confirmed that about equal amounts of Plk3 or Plk3-K91R were used in the kinase assays (Fig. 4B).
FIGURE 4.

Plk3 regulates PTEN by direct phosphorylation. A, recombinant His6-Plk3 was analyzed for its kinase activity toward purified PTEN in the kinase buffer supplemented with [32P]ATP. Casein was used as a positive control for the assays. Wort denotes wortmannin. Representative autoradiogram was shown. B, purified Plk3 and its kinase-defective mutant (Plk3-K91R) were assayed for their kinase activities toward PTEN and casein in the kinase buffer supplemented with [32P]ATP. Representative autoradiogram is shown. Equal amounts of reaction mixtures were also blotted for Plk3. C, schematic representation of PTEN C-tail and Plk3-targeting residues Thr-366 and Ser-370 are highlighted in red and with asterisks. D, purified Plk3 and its kinase-defective mutant (Plk3-K91R) were assayed for their kinase activities toward PTEN in the buffer supplemented with cold ATP. The reaction mixtures were then blotted with antibodies to phosphorylated PTEN (Thr(P)-366/Ser(P)-370) and to Plk3. E, purified Plk3 and Plk3-K91R were assayed for their kinase activities toward dephosphorylated PTEN (PTEN-dp) in the buffer supplemented with cold ATP. The reaction mixtures were then blotted with antibodies to phosphorylated PTEN (p-PTENThr-366/Ser-370) and to Plk3.
Subsequent mass spectrometric analyses identified that Plk3 phosphorylated PTEN in its tail domain on threonine 366 and serine 370 (Fig. 4C), two sites previously known to be phosphorylated by GSK3β (24) and CK2 (29), respectively. To confirm that these sites were the targets of Plk3 in vitro, PTEN samples phosphorylated by Plk3 were blotted with an antibody recognizing phosphorylated Thr-366/Ser-370 residues (p-PTENThr-366/Ser-370). To our surprise, there was a basal level of p-PTENThr-366/Ser-370 signals that interfered with the detection of Plk3-mediated phosphorylation (Fig. 4D). To circumvent the problem, we pretreated PTEN samples with λ phosphatase. PTEN protein was then pulled down with Ni-NTA resin and thoroughly washed. Dephosphorylated PTEN (PTEN-dp) was then used as a substrate in the kinase assay. Incubation with Plk3 but not with Plk3-K91R resulted in the generation of phospho-epitopes, and the phosphorylated signals were greatly diminished after the addition of wortmannin (Fig. 4E). These results thus confirm that Plk3 specifically targets Thr-366 and Ser-370 by phosphorylation.
It has been reported that phosphorylation of Thr-366 and Ser-370 occurs in vivo and that its phosphorylation significantly affects PTEN activity and/or stability (22, 23). Hence, we examined phosphorylation of PTEN on Thr-366 and Ser-370 in paired wild-type and PLK3−/− MEFs using the phospho-antibody. There was a basal level of p-PTENThr-366/Ser-370 signal in both types of MEFs; however, the p-PTENThr-366/Ser-370 signal was significantly lower in PLK3−/− MEFs than that of wild-type MEFs (Fig. 5A), consistent with the in vitro observation that Plk3 phosphorylates these sites. Intriguingly, the total PTEN level was also low in PLK3 null MEFs, which was associated with more active Akt1 (p-Akt1) in these cells in comparison with those of the wild-type MEFs. We then determined p-PTENThr-366/Ser-370 levels in paired MEFs exposed to the hypoxia stress. We observed that the p-PTENThr-366/Ser-370 signal was consistently lower in PLK3 null MEFs than in wild-type MEFs before and after treatment with the hypoxia mimetic Ni2+ (Fig. 5A). Furthermore, the total PTEN level was also low in PLK3−/− MEFs in comparison with the wild-type cells, suggesting that Plk3-mediated phosphorylation of PTEN may regulate its stability in vivo. Consistent with the reduced PTEN level in PLK3 null cells, the Akt1 activity, as revealed by its phosphorylation, was higher in PLK3−/− MEFs than in wild-type MEFs; this increased Akt1 activity resulted in enhanced phosphorylation of GSK3β (p-GSK3β) in PLK3 null cells (Fig. 5A).
FIGURE 5.

Plk3 phosphorylation promotes PTEN stabilization, thereby negatively regulating the PI3K/PDK1/Akt1 signaling axis. A, paired wild-type (W) and PLK3−/− (H) MEFs were treated with the hypoxia mimetic NiCl2 for various times. Equal amounts of cell lysates were blotted for phospho-PTENThr-366/Ser-370, total PTEN, phospho-GSK3βSer-9, total GSK3β, phospho-Akt1Ser-437, total Akt1, and β-actin. B, paired wild-type (W) and PLK3−/− (H) MEFs were treated with NiCl2 for various times. Equal amounts of cell lysates were blotted for phospho-PDK1, total PDK1, and β-actin. C, paired wild-type (W) and PLK3−/− (H) MEFs were treated with or without LiCl for 4 h after which cells were collected for lysate preparation. Equal amounts of cell lysates were blotted with antibodies to phosphorylated PTEN (p-PTENThr-366/Ser-370), total PTEN, and β-actin. The levels of PTEN from each treatment are shown after quantification.
Plk3 Positively Regulates PTEN Stability, Thereby Attenuating PDK1/Akt1 Signaling
The observation that PLK3 disruption causes a reduced PTEN level prompted us to examine its activity because it has been reported that the PTEN level may not always correlate with its activity (22). PDK1 activity correlates inversely with PTEN activity as PDK1 is directly activated by phosphatidylinositol 3,4,5-trisphosphate, a substrate of PTEN. The PDK1 activity can be determined by the phosphorylation status on serine 241 (36). The PDK1 activity reflected by its phosphorylation, but not its protein level, was much higher in PLK3−/− MEFs than that of wild-type MEFs before treatment with Ni2+; the PDK1 activity remained high in PLK3 null MEFs after exposure to the hypoxia mimetic (Fig. 5B). These results are fully consistent with the enhanced Akt1 activity in PLK3 null cells (Fig. 1).
As GSK3β and CK2 are known to phosphorylate Thr-366 and Ser-370, respectively, we examined phosphorylation of PTEN after inhibition of GSK3β and CK2 in vivo. Treatment with LiCl, a potent inhibitor of GSK3β, as well as CK2 to a less extent (37) significantly suppressed p-PTENThr-366/Ser-370 signals in both wild-type and PLK3−/− MEFs (Fig. 5C). A basal level of p-PTENThr-366/Ser-370 signals remained after LiCl treatment, suggesting the presence of an additional kinase(s) that phosphorylates these sites. The p-PTENThr-366/Ser-370 signals were greatly reduced in PLK3−/− MEFs in comparison with wild-type MEFs (Fig. 5C), consistent with the notion that Plk3 phosphorylates these sites.
To further confirm that Plk3 phosphorylation may stabilize PTEN, we obtained individual HA-tagged PTEN mutant constructs in which Thr-366 or Ser-370 was replaced with alanine (T366A or S370A); as control, we also obtained an HA-tagged PTEN mutant construct in which Ser-380, Thr-382, Thr-383, and Ser-385 were replaced with alanine residues (4A) (Fig. 6A). Various mutant PTEN expression constructs, as well as a PTEN wild-type expression construct, were transfected into HEK293T cells for 24 h. To normalize transfection efficiency, GFP expression construct was also used for co-transfection. Immunoblotting revealed that both PTENT366A and PTENS370A mutants were expressed at a reduced level in comparison with the wild-type PTEN (Fig. 6B). The reduced levels of PTENT366A or PTENS370A proteins were correlated with an increased phosphorylation of PDK1. Supporting early observations regarding phosphorylation of C-tail of PTEN and its stability (22, 38), phosphorylation-resistant PTEN mutant 4A was expressed at a greatly reduced level (Fig. 6B) compared with that of wild-type PTEN. It is worthwhile to note that the p-PDK1 level was not tightly correlated with reduced expression of PTEN4A mutant. This is not surprising because it has been shown that the PTEN phosphorylation mutant (4A) is expressed at a lower level, but it exhibits a higher biological activity than that of wild-type PTEN (22).
FIGURE 6.

Plk3 promotes PTEN stability by phosphorylation. A, schematic representation of HA-tagged PTEN and its phospho-mutants used for transfection analyses. B, HEK293T cells were co-transfected with various expression constructs as shown in A and a GFP expression construct for normalization of transfection efficiency for 1 day. Equal amounts of cell lysates were blotted for the HA tag, GFP, p-PDK1, and β-actin. Relative levels of ectopically expressed HA-PTEN are shown after quantification. C, paired wild-type (W) and PLK3−/− (H) MEFs were treated with MG132 for 4 h. Equal amounts of cell lysates were blotted for p-PTENThr-366/Ser-370, total PTEN, and β-actin. Representative data are shown. Relative endogenous PTEN levels are presented after quantification. D, model is proposed that illustrates how Plk3 regulates PTEN and PI3K/Akt signaling pathway. Green arrows denote positive regulation, and red bars denote negative regulation.
To further test the possibility that Plk3 regulates the stability of PTEN in vivo, we treated PLK3−/− and wild-type MEFs with the proteasome inhibitor MG132 for 4 h. Consistent with early observations, PLK3 null MEFs contained reduced levels of p-PTENT366A/Ser-370 and PTEN (Fig. 6C). MG132 treatment significantly enhanced PTEN levels in PLK3−/− MEFs but much less so in wild-type MEFs. Moreover, MG132 treatment did not affect the level of p-PTENT366A/Ser-370 signals in MEFs of either genotype (Fig. 6C). Therefore, these studies strongly support the notion that Plk3-mediated phosphorylation regulates PTEN stability, which is at least partly mediated by the proteasomal pathway.
DISCUSSION
Given the series of new biochemical, molecular, and genetic data obtained, we proposed the following model to explain the molecular mode of action of Plk3 in the regulation of cell survival and proliferation (Fig. 6D): Plk3 has multiple in vivo targets, one of which is PTEN. Plk3 phosphorylates PTEN on Thr-366 and Ser-370, residues that are situated within the C-tail domain. Biochemically, Plk3-mediated phosphorylation facilitates PTEN stabilization via a mechanism that is largely dependent on the proteosome. In the absence of Plk3, the PTEN protein level is negatively affected, leading to enhanced PDK1 and Akt1 activities. Active Akt1 directly phosphorylates GSK3β and positively regulates the mTORC1 pathway as well.
Tumor development and progression are frequently the result of deregulated protein kinases that control cell proliferation. Extensive research in the past has revealed that Polo-like kinases, a family of evolutionarily conserved proteins, are targeted for deregulation in cancer cells (39–41). We have previously shown that Plk3 functions as a negative regulator for cell proliferation in vitro (1, 42, 43) and that it suppresses tumorigenesis in vivo (4). Through a series of biochemical, molecular, and genetic analyses, we now demonstrate for the first time that Plk3 is an important component in the regulation of the PI3K/PTEN/Akt signaling axis during normoxic and hypoxic conditions. Specifically, PLK3−/− MEFs contain reduced PTEN levels and activities, which result in the enhanced activation of PDK1 and Akt1. Up-regulation of Akt1 in PLK3−/− MEFs causes mTOR activation and GSK3β inactivation, respectively. In vitro biochemical studies reveal that Plk3 directly phosphorylates PTEN, a key regulator for cell survival and angiogenesis (20, 44), in a region that is critical for regulating the stability of the phosphatase.
We have demonstrated that Plk3 specifically phosphorylates PTEN in vitro on Thr-366 and Ser-370, two sites previously shown to be phosphorylated by GSK3β and CK2, respectively (24, 29). This study strongly suggests that Plk3 also directly regulates PTEN by phosphorylation in vivo because PLK3 null MEFs contain significantly reduced p-PTENThr-366/Ser-370 signals that are associated with elevated levels of PDK1 and Akt1 activities. The remaining p-PTENThr-366/Ser-370 signals in PLK3−/− MEFs could be due to the presence of compensatory activities, including those of GSK3β and CK2 (24). Indeed, inhibition of GSK3β and CK2 activities by LiCl treatment significantly reduces p-PTENThr-366/Ser-370 signals in MEFs.
PTEN stability is known to be regulated by phosphorylation (10, 22, 38). CK2, a stress response protein kinase, phosphorylates PTEN at multiple sites in the C-terminal domain (29, 38). CK2 contributes to PTEN stabilization because CK2-targeting mutants are less stable (22) and because TNFα-induced cleavage of PTEN by caspase 3 can be suppressed by CK2 phosphorylation at the C-terminal tail (22). The PTEN level is lower in PLK3−/− MEFs than in wild-type MEFs before and after exposure to the hypoxic condition. The reduced level of PTEN correlates with the reduced phosphorylation of PTENThr-366/Ser-370 in these cells. That Plk3 regulates the PTEN stability by phosphorylation is further supported by our molecular studies. PTEN expression is enhanced after treatment with MG132, especially in PLK3 null MEFs. This observation strongly suggests that the steady-state level of PTEN is positively regulated by the inhibition of proteasome and that the regulation is at least partly dependent on Plk3. Supporting this notion, the PTENT366A or PTENS370A mutant is expressed at a reduced level in comparison with the wild-type PTEN, leading to an elevated PDK1 activity.
Both GSK3β and Plk3 phosphorylate Thr-366 of PTEN, whereas CK2 and Plk3 phosphorylate Ser-370. The cellular consequences that various protein kinases regulate PTEN stability and activity remain not entirely clear. However, Plk3-mediated phosphorylation appears to play a more important role than GSK3β or CK2 alone in stabilizing PTEN because it simultaneously phosphorylates Thr-366 and Ser-370. The half-life of PTEN is rather long (>8 h), but a PTEN deletion mutant (amino acids 1–353) has a half-life of about 2 h (38). The stabilization of full-length PTEN is likely due to protection of phosphorylation in the C-tail region mediated by Plk3 and, to a less extent, by GSK3β and CK2. Supporting this notion, PLK3 ablation or inhibition of GSK3β and CK2 by LiCl treatment results in significantly reduced p-PTENThr-366/Ser-370 signals that are coupled with a lower level of PTEN protein than that of controls. This may also explain why MG132 treatment greatly stabilizes PTEN in PLK3 null MEFs but not in wild-type MEFs, although it does not significantly modulate the phosphorylation level in PLK3 null cells.
Supplementary Material
Acknowledgments
We thank Qishan Lin (Center for Functional Genomics, the Research Foundation of the State University of New York, Albany) for assistance in the mass spectrometric study and Dr. Kyung Lee for providing us with recombinant Plk1 as control. We also thank Nedda Tichi for administrative assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants CA074229 and CA113349 from USPHS (to W. D.) and AG009953 (to D. X.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- PTEN
- phosphatase and tensin homolog
- MEF
- murine embryonic fibroblast
- Ni-NTA
- nickel-nitrilotriacetic acid
- mTOR
- mammalian target of rapamycin.
REFERENCES
- 1.Xie S., Wu H., Wang Q., Cogswell J. P., Husain I., Conn C., Stambrook P., Jhanwar-Uniyal M., Dai W. (2001) J. Biol. Chem. 276, 43305–43312 [DOI] [PubMed] [Google Scholar]
- 2.Xie S., Wang Q., Wu H., Cogswell J., Lu L., Jhanwar-Uniyal M., Dai W. (2001) J. Biol. Chem. 276, 36194–36199 [DOI] [PubMed] [Google Scholar]
- 3.Wang L., Gao J., Dai W., Lu L. (2008) J. Biol. Chem. 283, 25928–25935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y., Bai J., Shen R., Brown S. A., Komissarova E., Huang Y., Jiang N., Alberts G. F., Costa M., Lu L., Winkles J. A., Dai W. (2008) Cancer Res. 68, 4077–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li B., Ouyang B., Pan H., Reissmann P. T., Slamon D. J., Arceci R., Lu L., Dai W. (1996) J. Biol. Chem. 271, 19402–19408 [DOI] [PubMed] [Google Scholar]
- 6.Dai W. (2005) Oncogene 24, 214–216 [DOI] [PubMed] [Google Scholar]
- 7.Chizhikov V., Zborovskaya I., Laktionov K., Delektorskaya V., Polotskii B., Tatosyan A., Gasparian A. (2001) Mol. Carcinog. 30, 151–158 [DOI] [PubMed] [Google Scholar]
- 8.Perotti D., Pettenella F., Luksch R., Giardini R., Gambirasio F., Ferrari D., Fossati-Bellani F., Biondi A. (1999) Haematologica 84, 110–113 [PubMed] [Google Scholar]
- 9.Di Cristofano A., Pandolfi P. P. (2000) Cell 100, 387–390 [DOI] [PubMed] [Google Scholar]
- 10.Salmena L., Carracedo A., Pandolfi P. P. (2008) Cell 133, 403–414 [DOI] [PubMed] [Google Scholar]
- 11.Yin Y., Shen W. H. (2008) Oncogene 27, 5443–5453 [DOI] [PubMed] [Google Scholar]
- 12.Trimboli A. J., Cantemir-Stone C. Z., Li F., Wallace J. A., Merchant A., Creasap N., Thompson J. C., Caserta E., Wang H., Chong J. L., Naidu S., Wei G., Sharma S. M., Stephens J. A., Fernandez S. A., Gurcan M. N., Weinstein M. B., Barsky S. H., Yee L., Rosol T. J., Stromberg P. C., Robinson M. L., Pepin F., Hallett M., Park M., Ostrowski M. C., Leone G. (2009) Nature 461, 1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang G., Sun Q., Teng Y., Li F., Weng T., Yang X. (2008) Development 135, 3587–3597 [DOI] [PubMed] [Google Scholar]
- 14.Di Cristofano A., Pesce B., Cordon-Cardo C., Pandolfi P. P. (1998) Nat. Genet. 19, 348–355 [DOI] [PubMed] [Google Scholar]
- 15.Kwabi-Addo B., Giri D., Schmidt K., Podsypanina K., Parsons R., Greenberg N., Ittmann M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 11563–11568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carracedo A., Pandolfi P. P. (2008) Oncogene 27, 5527–5541 [DOI] [PubMed] [Google Scholar]
- 17.Phillips R. J., Mestas J., Gharaee-Kermani M., Burdick M. D., Sica A., Belperio J. A., Keane M. P., Strieter R. M. (2005) J. Biol. Chem. 280, 22473–22481 [DOI] [PubMed] [Google Scholar]
- 18.Zhong H., Chiles K., Feldser D., Laughner E., Hanrahan C., Georgescu M. M., Simons J. W., Semenza G. L. (2000) Cancer Res. 60, 1541–1545 [PubMed] [Google Scholar]
- 19.Emerling B. M., Weinberg F., Liu J. L., Mak T. W., Chandel N. S. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 2622–2627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bárdos J. I., Ashcroft M. (2004) BioEssays 26, 262–269 [DOI] [PubMed] [Google Scholar]
- 21.Rong Y., Durden D. L., Van Meir E. G., Brat D. J. (2006) J. Neuropathol. Exp. Neurol. 65, 529–539 [DOI] [PubMed] [Google Scholar]
- 22.Torres J., Pulido R. (2001) J. Biol. Chem. 276, 993–998 [DOI] [PubMed] [Google Scholar]
- 23.Al-Khouri A. M., Ma Y., Togo S. H., Williams S., Mustelin T. (2005) J. Biol. Chem. 280, 35195–35202 [DOI] [PubMed] [Google Scholar]
- 24.Maccario H., Perera N. M., Davidson L., Downes C. P., Leslie N. R. (2007) Biochem. J. 405, 439–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brauer P. M., Tyner A. L. (2009) Cell Cycle 8, 2728–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yim E. K., Peng G., Dai H., Hu R., Li K., Lu Y., Mills G. B., Meric-Bernstam F., Hennessy B. T., Craven R. J., Lin S. Y. (2009) Cancer Cell 15, 304–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ouyang B., Li W., Pan H., Meadows J., Hoffmann I., Dai W. (1999) Oncogene 18, 6029–6036 [DOI] [PubMed] [Google Scholar]
- 28.Flügel D., Görlach A., Michiels C., Kietzmann T. (2007) Mol. Cell. Biol. 27, 3253–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller S. J., Lou D. Y., Seldin D. C., Lane W. S., Neel B. G. (2002) FEBS Lett. 528, 145–153 [DOI] [PubMed] [Google Scholar]
- 30.Zhao H., Sapolsky R. M., Steinberg G. K. (2006) Mol. Neurobiol. 34, 249–270 [DOI] [PubMed] [Google Scholar]
- 31.Fukumoto S., Hsieh C. M., Maemura K., Layne M. D., Yet S. F., Lee K. H., Matsui T., Rosenzweig A., Taylor W. G., Rubin J. S., Perrella M. A., Lee M. E. (2001) J. Biol. Chem. 276, 17479–17483 [DOI] [PubMed] [Google Scholar]
- 32.Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 33.Jiang B. H., Jiang G., Zheng J. Z., Lu Z., Hunter T., Vogt P. K. (2001) Cell Growth Differ. 12, 363–369 [PubMed] [Google Scholar]
- 34.Schnitzer S. E., Schmid T., Zhou J., Eisenbrand G., Brüne B. (2005) FEBS Lett. 579, 529–533 [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Echeverria C., Sellers W. R. (2008) Oncogene 27, 5511–5526 [DOI] [PubMed] [Google Scholar]
- 36.Casamayor A., Morrice N. A., Alessi D. R. (1999) Biochem. J. 342, 287–292 [PMC free article] [PubMed] [Google Scholar]
- 37.Davies S. P., Reddy H., Caivano M., Cohen P. (2000) Biochem. J. 351, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vazquez F., Ramaswamy S., Nakamura N., Sellers W. R. (2000) Mol. Cell. Biol. 20, 5010–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie S., Xie B., Lee M. Y., Dai W. (2005) Oncogene 24, 277–286 [DOI] [PubMed] [Google Scholar]
- 40.Eckerdt F., Yuan J., Strebhardt K. (2005) Oncogene 24, 267–276 [DOI] [PubMed] [Google Scholar]
- 41.Strebhardt K., Ullrich A. (2006) Nat. Rev. Cancer 6, 321–330 [DOI] [PubMed] [Google Scholar]
- 42.Wang Q., Xie S., Chen J., Fukasawa K., Naik U., Traganos F., Darzynkiewicz Z., Jhanwar-Uniyal M., Dai W. (2002) Mol. Cell. Biol. 22, 3450–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang N., Wang X., Jhanwar-Uniyal M., Darzynkiewicz Z., Dai W. (2006) J. Biol. Chem. 281, 10577–10582 [DOI] [PubMed] [Google Scholar]
- 44.Maynard M. A., Ohh M. (2007) Cell. Mol. Life Sci. 64, 2170–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.