

Abstract
While studying the brain function of the human partial epilepsy gene, leucine-rich glioma-inactivated 1 (LGI1), a new mechanism of human epileptogenesis was revealed—persistent immaturity of glutamatergic circuitries. LGI1, a novel secreted protein, was found to be increased during the postnatal period; when glutamatergic synapses both downregulate their presynaptic vesicular release probability and reduce their postsynaptic NMDA-receptor subunit NR2B. During this same period, the dendritic arbor and spines are pruned and remodeled. Using bacterial artificial chromosome transgenic mouse techniques, excess wild-type LGI1 was shown to magnify these critical brain developmental events in the hippocampal dentate gyrus; while an epilepsy-associated, truncated, dominant-negative form of LGI1 blocked them. By contrast, the hippocampal dentate granule neuron GABAergic synapses and intrinsic excitability were unaltered. A role for LGI1 in downregulating glutamate synapse function was confirmed by germline gene deletion; this intervention also revealed a selective increase of glutamatergic synaptic transmission with unaltered GABAergic synapses and intrinsic excitability of hippocampal CA1 pyramidal neurons. Interestingly, the role of LGI1 in neurological disease was further expanded when a subset of patients with limbic encephalitis (an autoimmune disorder with memory loss in 100% and seizures in 80% of individuals) were discovered to carry autoantibodies to LGI1.
Epilepsy is one of the most common neurologic disorders worldwide, afflicting approximately 1 in 100 individuals. Partial epilepsies, in which the aberrant discharge begins within a specific brain region (e.g., temporal lobe), are the most common form. While in some cases the cause is readily defined by the onset of an acute event (e.g., trauma, hemorrhage) and/or the finding of a structural abnormality (e.g., cortical dysplasia, tumor, vascular malformation); in many other cases, the cause remains idiopathic. A subset of these idiopathic, partial epilepsies affect multiple individuals within an extended family, and a significant number of the families report prominent auditory auras (and sometimes other sensory perceptions: visual, vertiginous, epigastric, or psychogenic) associated with the seizure event. Based on the aura type and its suspected locus, the familial partial epilepsy disorder was designated either autosomal dominant partial epilepsy with auditory features (ADPEAFs) or autosomal dominant lateral temporal lobe epilepsy (ADLTE) (1–4). Identifying the genes underlying human familial epilepsies is important not only in helping to assess seizure susceptibility in individual family members (estimate at approximately 60% in ADPEAF), but also in providing the opportunity to discover new biological pathways (and therapeutic targets) for epilepsy.
The vast majority of idiopathic epilepsy genes identified to date encode ion channels. Furthermore, current evidence suggests that most of these ion-channel mutations and other non–ion-channel gene mutations, either directly or indirectly, impair GABAergic neuron excitability or GABAergic synaptic transmission to promote epilepsy. For this reason, it was quite exciting when ADLTE patients were found to harbor mutations in a novel, non–ion-channel gene—leucine-rich glioma-inactivated 1 (LGI1)—whose function in the brain was previously completely undefined (2,3). LGI1 was independently identified in a segment of genome that is deleted in glioblastoma, a high-grade astrocytic neoplasm, and was demonstrated to regulate growth, migration, and mitogen-activated protein kinase signaling in these cells (5,6). Thus, the potential cellular basis of this epilepsy disorder could not be predicted from either the protein's sequence or the limited amount of other existing information.
Timing Is Everything
LGI1 was shown to encode a secreted (7), vertebrate-specific protein that localizes to glutamatergic synapses, binds its receptors ADAM22 and ADAM23 (disintegrin and metalloproteinase domain 22 and 23), and copurifies with presynaptic and postsynaptic regulatory molecules—that is, presynaptic Kv1.1 potassium channel (8) and postsynaptic density protein (PSD95) (9). In vitro coexpression studies in oocytes showed that wild-type LGI1 slowed the kinetics of voltage-dependent inactivation of Kv1.1 and epilepsy-associated mutant dominant negatively inhibited this effect of wild-type LGI1 (8). If this Kv1.1 were present in the presynaptic membrane of glutamatergic synapses, LGI1 would be predicted to inhibit excitatory synaptic transmission, while mutant LGI1 would be predicted to enhance excitatory synaptic transmission; a reasonable, but not yet proven mechanism by which mutant LGI1 might magnify circuit excitability to promote epilepsy. Yet, another study found contrasting influences of LGI1 on excitatory synaptic transmission. Exogenous addition of LGI1 to acute brain slices was reported to increase AMPA-receptor currents (however, the conclusions of this finding contrast with multiple other studies that follow) (9). The first clue to LGI1's potential in vivo brain function was the timing of its expression—it markedly increased when glutamatergic synapses are normally, functionally down-regulated and pruned (10). LGI1 increases to adult levels during the third week of postnatal life in mice, probably equivalent to early childhood (1–3 years).
To test the hypothesis that LGI1 mediates the postnatal developmental maturation and pruning of glutamatergic synapses, two new transgenic mice that expressed full-length mouse LGI1 genes were created as follows: (1) a wild-type form that magnified the effects of native LGI1 and (2) a truncated, dominant-negative, epilepsy-associated mutant mouse that inhibited the effects of native LGI1 (10). Using a long genomic DNA fragment containing the native LGI1 promoter sequences meant the LGI1 transgene would express during the same development time period as the native LGI1, permitting an assessment of the impact of excess and dominant-negative mutant LGI1 on neuronal circuit development in vivo.
Consistent with the hypothesis, excess wild-type and dominant-negative mutant LGI1 caused opposite effects on glutamatergic synapse development (10). Mutant LGI1 arrested and excess wild-type LGI1 magnified the decrease of presynaptic release probability and the decrease of the postsynaptic NMDA-receptor subunit NR2B/NR2A ratio that normally occurs during postnatal life. Presumably, the high probability of glutamate neurotransmitter release enables strong synaptic transmission, while high NR2B-receptor subunit composition of the NMDA receptors facilitates synaptic plasticity, which likely facilitates the activity-dependent remodeling of neuronal circuit that is guided by sensory–motor experiences. The results indicated LGI1 mediates this important developmental period of glutamate synapse maturation. Consequently, the glutamate synapses of individuals with ADLTE appear to be stuck in a child-like state (10).
In addition to finding that glutamate synapses remain functionally immature, structural trimming of glutamatergic circuits also failed to occur in dominant-negative, epilepsy-associated LGI1 mutant transgenic mice (10). Synaptic connections between neurons are well known to initially overgrow, but then these contacts are pruned. It was found that in the ADLTE mutant LGI1 mouse, this normal developmental pruning of dendritic branches and spines is impaired. Importantly, the defect in pruning resulted in a marked increase of excitatory synaptic transmission. By contrast, the intrinsic excitability of these same glutamatergic neurons and their GABAergic synaptic transmission was unaffected—a finding distinct from many other human epilepsy disorders (10). Overall, the results established that LGI1 is a key mediator of postnatal glutamatergic circuit development and implicate a novel mechanism for human partial epilepsy: persistent immaturity of excitatory circuits (10).
Immaturity Begets Seizure Susceptibility
Patients with ADLTE experience partial seizures infrequently (a few times a month) and secondarily generalized seizures about once a year. The partial seizures (not detectable by EEG) and the secondarily generalized seizures (detectable by EEG) are often associated with a triggering event (e.g., phone call) or condition (e.g., sleep deprivation or alcohol abuse). Thus, triggering conditions were used to unmask the seizure susceptibility of the ADLTE mutant LGI1 transgenic mice (10). Repeated alternate day injections of low, subconvulsive doses of the GABAergic synapse blocker pentylenetetrazol caused a kindling of seizures in wild-type mice. While no seizures occurred during the first few injections, seizures gradually developed in more and more mice with the same initially nonconvulsive drug dosage. Importantly, ADLTE-associated, dominant-negative mutant LGI1 mice displayed a lower threshold for the development of these seizures.
Figure 1 summarizes the impact of ADLTE mutant LGI1 on the balance between excitatory and inhibitory synaptic transmission that was observed in the hippocampus and that may partially explain the increased seizure susceptibility in ADLTE. Not shown are the suspected aberrant connections of the glutamatergic circuitry (predicted from the persistently widened dendritic arbor and large number of spines) and the potentially enhanced synaptic plasticity (predicted from the increased presynaptic release probability and increased postsynaptic NMDA receptor NR2B subunit). Despite the marked increase of glutamatergic synaptic transmission present in ADLTE-associated LGI1 mutant mice, GABAergic synaptic transmission still vastly exceeded glutamatergic synaptic transmission 20 to 1, when measuring spontaneous synaptic currents (10). Thus, the investigators suspect that circuit excitability may remain in check under most conditions, explaining the low-seizure frequency in ADLTE patients. As shown in the diagram (see Figure 1), conditions that lower inhibition should uncover the seizure propensity of ADLTE patients.
FIGURE 1.
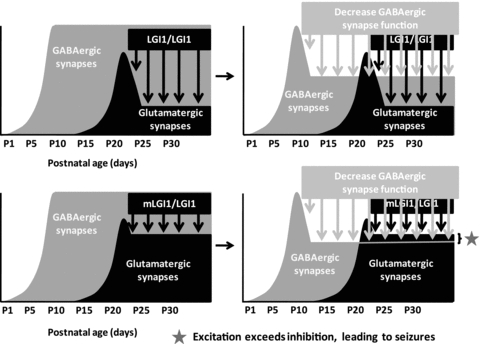
LGI1 down-regulates glutamatergic synapses during postnatal life. Seizure susceptibility in autosomal dominant lateral temporal lobe epilepsy may result, in part, from an imbalance of the excitation and inhibition circuitries. Reducing inhibition would uncover the elevated function of the excitatory circuitry present in ADLTE patients.
The Sounding Board
Many puzzles remain concerning this partial epilepsy disorder. Why do seizures tend to begin in auditory sensory pathways? More sharply posed, might the circuit immaturity (marked by higher transmission efficiency) leave the cortex susceptible to seizure kindling in response to natural auditory stimuli? The presence of multiple different sensory and limbic auras implicates seizure initiation in other sensory and limbic systems at various times; thus, might the kindled site relate to the specific experiences of the individual—potentially explaining heterogeneity of the disease? Once LGI1 has increased during postnatal brain development to promote glutamatergic circuit development, why must it remain elevated throughout adulthood? Would neural circuits otherwise revert to a child-like state, if LGI1 were lost in adulthood? Does LGI1 have additional roles in regulating adult circuit function (e.g., synaptic plasticity, as suggested by its regulation of NMDA receptor composition)? LGI1 does not appear to affect the GABAergic synapses onto hippocampal dentate granule neurons or CA1 pyramidal neurons, as will be discussed in the next section (10,14). Does LGI1 regulate other GABAergic synapses not yet examined? Does LGI1 regulate glutamatergic synapses onto GABAergic neurons in the same way that it regulates glutamatergic synapses onto glutamatergic neurons?
Lose It and Die: Other Recent Discoveries in LGI1 Research
Kunapuli et al. first characterized LGI1 as a gene within a chromosomal segment of DNA deleted in high-grade astrocytic tumors of the brain (5). Interestingly, expressing the protein in a glioma cell line suppressed glioma cell growth, migration, matrix metalloprotease gene expression, and MEK/ERK signaling (6). More recently, Kunapuli et al. performed a genome-wide profiling analysis to assess the impact of LGI1 expression on transcripts in the glioma cell line (11). Intriguingly, they found a marked change in many transcripts, including some involved in axonal development.
Epilepsy-associated LGI1 mutations act in a dominant-negative fashion to antagonize the effects of wild-type LGI1 (8,10). The precise mechanism remains undefined but may relate to the ability of LGI1 to form dimers (9) and to the finding that ADLTE-related mutant LGI1 fails to normally secrete from cultured cells (7). Lesions of the entorhinal cortex reduce LGI1 protein levels in the dentate gyrus molecular layer, suggesting LGI1 may be released from presynaptic sites (8). Direct studies addressing whether LGI1 is released presynaptically or postsynaptically are yet to be performed. Furthermore, is LGI1 released constitutively, as in cultured nonneuronal cells, or is its release neuronal activity-dependent?
Three recent studies reported the effects of a complete loss of LGI1 function through germline gene deletion (12–14). A complete loss of LGI1 function caused severe seizures and premature death during the third postnatal week of life (12–14), at approximately the same time when LGI1 levels increase, and glutamate synapses are matured and pruned (10). This finding contrasts with the relatively mild phenotype found in humans with partial loss of LGI1 function that is because of the haploid insufficiency (50% loss of functional gene dosage) and the dominant negative inhibition of the remaining LGI1 by the mutant gene product (>50% loss of LGI1 function).
At the circuit level, Yu et al. reported that in hippocampal CA1 pyramidal neurons of LGI1 knockout mice, miniature EPSCs (mEPSCs) frequency and evoked AMPA and NMDA currents are increased, while there is no effect on mEPSC amplitude (12). These results closely match the findings of increased mEPSC frequency and evoked AMPA and NMDA currents in hippocampal dentate granule neurons, with no effect on mEPSC amplitude reported in mice carrying the dominant-negative, epilepsy-associated LGI1 mutant (10). Both studies also reported no effects of LGI1 loss on GABAergic synapses or intrinsic excitability (10,12).
However, in contrast to the results of Zhou et al. (10) and Yu et al. (12), Fukata et al. reported that in the same CA1 hippocampal pyramidal neurons studied by Yu et al. at postnatal day 14 to 15 (when mice first begin to die from spontaneous seizures), both the mEPSC amplitude and AMPA/NMDA ratio are decreased, while the mEPSC frequency is unaltered (13). Importantly, unlike the studies of Zhou et al. and Yu et al., the amplitude of evoked AMPA and of NMDA glutamatergic synaptic currents was not assessed by Fukata et al. Although not yet measured, Fukata et al. suggested that LGI1 knockout might also decrease AMPA-mediated synaptic transmission onto GABAergic neurons as a way to explain how decreased glutamatergic synaptic transmission might then promote epilepsy. However, a reduction of AMPA synaptic transmission onto glutamatergic neurons, as they report, would be predicted to dampen circuit excitability and to reduce seizure susceptibility.
The reasons for the conflicting results of Yu et al. (12) and Fukata et al. (13) remain unclear. One possibility is that the LGI1 knockout mice studied by Fukata et al. and Yu et al. may have had different seizure histories. Either a recent seizure or prolonged, repeated seizures may have down-regulated synaptic function in the hippocampal CA1 pyramidal neurons study by Fukata et al. Supporting this possibility, in a third independent study of LGI1 knockout mice, Chabrol et al. reported that LGI1 knockouts display a severe astrogliosis throughout the hippocampus, including in the CA1 region, as well as dentate mossy fiber sprouting (14). Furthermore, during simultaneous hippocampal and cortical field potential recordings in vivo, these authors also found that epileptiform discharges first appear in the hippocampus. Thus, depending on the age of the mice and also possibly on their genetic background and experiences (e.g., stress associated with transportation between institutions), the mice recorded in the Fukata et al. study may have experienced severe seizure-related damage of hippocampal circuits. Histologic analysis of the hippocampal formation in the same mice that were studied using electrophysiology might have revealed seizure-related damage.
By contrast, in the studies of dominant-negative, epilepsy-associated LGI1 mutant transgenic mice, spontaneous seizures were infrequent (i.e., not detected during a 5-day recording) and premature deaths were not observed; therefore, the risk of secondary effects of seizures would not confound the results of the study (10). Importantly, the finding that excess wild-type LGI1 had effects opposite to those of mutant LGI1 provides compelling evidence that LGI1 functionally regulates and structurally down-regulates glutamatergic circuits during postnatal development (10). Future electrophysiology studies may do well to focus on the heterozygous rather than the homozygous LGI1 knockout mice. Interestingly, Chabrol et al. demonstrated that heterozygous LGI1 knockout mice display increased audiogenic kindling of seizures, consistent with the reported susceptibility of humans to auditory-stimulus–induced seizures (e.g., telephone seizures), suggesting these mice represent a good model of the human partial epilepsy disorder (14).
A Diversity of Binding Partners: Consequences Largely Unknown
LGI1 binds ADAM22, ADAM23, ADAM11, and NogoR1 (9,15–17). A recent study using primary neuronal cultures showed that adding exogenous LGI1 promotes axon growth and that this effect depends on ADAM23 (16). Note that this contrasts to the proposed role of LGI1 in dendritic pruning as suggested from the in vivo studies of hippocampal dentate gyrus perforant pathway development (10). As ADAM22 interactions with PSD95 rely on the carboxyl-terminal 4 amino acids of ADAM22, which are missing from ADAM23, ADAM23 may play a role that is distinct from ADAM22 in regulating neuronal processes.
Interestingly, the Fukata et al. study also provided evidence that LGI1 promotes the formation of an ADAM22 and ADAM23 complex (13). ADAM22 and ADAM23 mutually coimmunoprecipitate, but when LGI1 is missing, coimmunoprecipitation fails. Based on this observation, the investigators suggested, but have not yet proven, that LGI1 might provide a transynaptic bridge cross-linking presynaptic ADAM23 to postsynaptic ADAM22. To prove this hypothesis, one would have to independently and selectively purify these proteins from presynaptic and postsynaptic neurons—a technical feat that has never been reported.
A recent study has further expanded the repertoire of potentially important binding sites for LGI1 (17). LGI1 binds NogoR1 (but not NogoR2 or NogoR3) expressed in cultured cells and antagonizes binding of NogoR1 to Nogo66. Like the ADAM23 study, LGI1 facilitated neurite outgrowth on myelin, a substrate that normally inhibits neurite outgrowth via NogoR1. Interestingly, NogoR1 and ADAM22 also copurify from brain; and when NogoR1 and ADAM22 are coexpressed in vitro, LGI1 binding to NogoR1 is reduced, while the binding to ADAM22 is increased.
The LGI1 receptor mediating the in vivo postnatal development of glutamatergic synapses remains undefined (10). But, these recent findings suggest the possibility that distinct receptors may underlie discrete functional and structural developmental processes.
Beyond Genetics
Lai et al. recently discovered that a subset of individuals with limbic encephalitis (which is associated with seizures in 80%, memory loss in 100%, and hyponatremia in 60% of patients and is considered to be an autoimmune disorder) have serum antibodies to LGI1 (18). This finding is very exciting, as it expands the potential role of LGI1 in human neurologic disease to include autoimmune disorders. However, again, many questions remain. Are LGI1 antibodies sufficient to produce limbic encephalitis? Do these antibodies mimic or antagonize LGI1 function? Alternately, do they instead simply trigger the recruitment of immune cells and the release of numerous cytokines, chemokines, and growth factors that could then promote seizures and disrupt normal circuit function (19–21)?
Conclusion
Studies of LGI1 have uncovered a new potential mechanism for human epilepsy—persistent immaturity of glutamatergic circuits. Interestingly, partial epilepsies are common in a variety of neurodevelopmental disorders, including autism and tuberous sclerosis as well as in cortical malformations, suggesting persistent circuit immaturity may explain the epilepsy in a subset of these disorders as well (22,23).
References
- 1.Ottman R, Risch N, Hauser WA, Pedley TA, Lee JH, Barker-Cummings C, Lustenberger A, Nagle KJ, Lee KS, Scheuer ML, Neystat M, Susser M, Wilhelmsen KC. Localization of a gene for partial epilepsy to chromosome 10q. Nat Genet. 1995;10:56–60. doi: 10.1038/ng0595-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, Choi C, Morozov P, Das K, Teplitskaya E, Yu A, Cayanis E, Penchaszadeh G, Kottmann AH, Pedley TA, Hauser WA, Ottman R, Gilliam TC. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, Sáenz A, Poza JJ, Galán J, Gesk S, Sarafidou T, Mautner VF, Binelli S, Staub E, Hinzmann B, French L, Prud’homme JF, Passarelli D, Scannapieco P, Tassinari CA, Avanzini G, Martí-Massó JF, Kluwe L, Deloukas P, Moschonas NK, Michelucci R, Siebert R, Nobile C, Pérez-Tur J, López de Munain A. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119–28. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]
- 4.Ottman R, Winawer MR, Kalachikov S, Barker-Cummings C, Gilliam TC, Pedley TA, Hauser WA. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology. 2004;62:1120–1126. doi: 10.1212/01.wnl.0000120098.39231.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunapuli P, Chitta KS, Cowell JK. Suppression of the cell proliferation and invasion phenotypes in glioma cells by the LGI1 gene. Oncogene. 2003;22:3985–3991. doi: 10.1038/sj.onc.1206584. [DOI] [PubMed] [Google Scholar]
- 6.Kunapuli P, Kasyapa CS, Hawthorn L, Cowell JK. LGI1, a putative tumor metastasis suppressor gene, controls in vitro invasiveness and expression of matrix metalloproteinases in glioma cells through the ERK1/2 pathway. J Biol Chem. 2004;279:23151–23157. doi: 10.1074/jbc.M314192200. [DOI] [PubMed] [Google Scholar]
- 7.Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- 8.Schulte U, Thumfart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, Wangler T, Knaus HG, Fakler B. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 9.Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- 10.Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med. 2009;15:1208–1214. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunapuli P, Lo K, Hawthorn L, Cowell JK. Reexpression of LGI1 in glioma cells results in dysregulation of genes implicated in the canonical axon guidance pathway. Genomics. 2010;95:93–100. doi: 10.1016/j.ygeno.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu YE, Wen L, Silva J, Li Z, Head K, Sossey-Alaoui K, Pao A, Mei L, Cowell JK. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum Mol Genet. 2010;19:1702–1711. doi: 10.1093/hmg/ddq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukata Y, Lovero KL, Iwanaga T, Watanabe A, Yokoi N, Tabuchi K, Shigemoto R, Nicoll RA, Fukata M. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc Natl Acad Sci U S A. 2010;107:3799–3804. doi: 10.1073/pnas.0914537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chabrol E, Navarro V, Provenzano G, Cohen I, Dinocourt C, Rivaud-Pechoux S, Fricker D, Baulac M, Miles R, LeGuern E, Baulac S. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain. 2010;133:2749–2762. doi: 10.1093/brain/awq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4:387–396. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owuor K, Harel NY, Englot DJ, Hisama F, Blumenfeld H, Strittmatter SM. LGI1-associated epilepsy through altered ADAM23-dependent neuronal morphology. Mol Cell Neurosci. 2009;42:448–457. doi: 10.1016/j.mcn.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas R, Favell K, Morante-Redolat J, Pool M, Kent C, Wright M, Daignault K, Ferraro GB, Montcalm S, Durocher Y, Fournier A, Perez-Tur J, Barker PA. LGI1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci. 2010;30:6607–6612. doi: 10.1523/JNEUROSCI.5147-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, Cowell JK, Dalmau J. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: A case series. Lancet Neurol. 2010;9:776–785. doi: 10.1016/S1474-4422(10)70137-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S, Benati D, Chakir A, Zanetti L, Schio F, Osculati A, Marzola P, Nicolato E, Homeister JW, Xia L, Lowe JB, McEver RP, Osculati F, Sbarbati A, Butcher EC, Constantin G. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim JV, Kang SS, Dustin ML, McGavern DB. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature. 2009;457:191–195. doi: 10.1038/nature07591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A. Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med. 2010;16:413–419. doi: 10.1038/nm.2127. [DOI] [PubMed] [Google Scholar]
- 22.Brooks-Kayal A. Epilepsy and autism spectrum disorders: Are there common developmental mechanisms? Brain Dev. 2010;32:731–738. doi: 10.1016/j.braindev.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Jacobs KM, Kharazia VN, Prince DA. Mechanisms underlying epileptogenesis in cortical malformations. Epilepsy Res. 1999;36:165–188. doi: 10.1016/s0920-1211(99)00050-9. [DOI] [PubMed] [Google Scholar]