


Abstract
Tissue-specific alternative splicing is a key mechanism for generating tissue-specific proteomic diversity in eukaryotes. Splicing regulatory elements (SREs) in pre-mature messenger RNA play a very important role in regulating alternative splicing. In this article, we use mouse RNA-Seq data to determine a positive data set where SREs are over-represented and a reliable negative data set where the same SREs are most likely under-represented for a specific tissue and then employ a powerful discriminative approach to identify SREs. We identified 456 putative splicing enhancers or silencers, of which 221 were predicted to be tissue-specific. Most of our tissue-specific SREs are likely different from constitutive SREs, since only 18% of our exonic splicing enhancers (ESEs) are contained in constitutive RESCUE-ESEs. A relatively small portion (20%) of our SREs is included in tissue-specific SREs in human identified in two recent studies. In the analysis of position distribution of SREs, we found that a dozen of SREs were biased to a specific region. We also identified two very interesting SREs that can function as an enhancer in one tissue but a silencer in another tissue from the same intronic region. These findings provide insight into the mechanism of tissue-specific alternative splicing and give a set of valuable putative SREs for further experimental investigations.
INTRODUCTION
In higher eukaryotes, protein coding genes are transcribed as precursors of messenger RNAs, in which exons are separated from each other by intervening introns that have to be spliced out to produce a mature mRNA. A gene may generate different mature mRNA isoforms by selectively including different combinations of exons. This kind of alternative splicing (AS) is a key mechanism for regulating gene expression and for generating proteomic diversity. Recent studies indicate that >90% of human genes undergo alternative splicing (1,2).
In addition to the core splicing signals at the 5′ splice site, the 3′ splice site and the branch point, other splicing regulatory elements (SREs) are pivotal to ensure that splicing events occur accurately and efficiently (3,4). These SREs are classified as exonic splicing enhancers (ESEs) or silencers (ESSs) if they promote or inhibit the inclusion of the exon where they reside, and as intronic splicing enhancers (ISEs) or silencers (ISSs) if they enhance or inhibit the inclusion of the exon adjacent to the intron where they reside. Experimental approaches such as systematic evolution of ligands by exponential enrichment (SELEX) (5), UV crosslinking and immunoprecipitation (CLIP) (6) and splicing reporter system (7), have been employed to identify SREs.
Computational approaches also provide a means of identifying putative SREs that can be validated experimentally. A number of SREs including RESCUE-ESE (8) and PESE/PESS (9) have been identified from constitutively spliced exons using computational methods and some of them have been demonstrated in experiments to function as predicted. Some ESSs were also identified from pseudo exons by computational methods (10). For a detailed review, see (11).
Alternative splicing plays an important role in generating tissue specificity. Recent high-throughput studies based on microarray have shown that 42% cassette exons examined are differently expressed in at least 1 of 48 human tissues (12). This percentage even reaches 72% in a recent RNA-Seq study (1). Tissue-specific alternative splicing is thought to be largely regulated by tissue-specific splicing factors and tissue-specific expression of constitutive splicing factors (2,13). Therefore, it is important to identify SREs that are the targets of these splicing factors.
Brudno et al. (14) identified brain-specific intronic SREs from a relatively small data set that includes 25 brain-specific cassette exons. More recently, Castle et al. (12) measured the expression level of a large number of exons and exon-exon junctions in 48 human tissues using microarray, and then determined up- and down-regulated cassette exons in each tissue. From these cassette exons, they identified 143 tissue-specific motifs. Wang et al. (15) determined the ratio of expression level of cassette exons in different pairs of human tissues from exon arrays and used a linear regression model to identify tissue-specific SREs.
The key technique used in all computational methods for identifying SREs is to find short nucleotide sequences (typically hexamers or octamers) that are over-represented in a positive data set relative to a background data set. For example, constitutive RESCUE-ESEs (8) are hexamers that are over-represented in constitutive exons with weak splice sites comparing to introns and constitutive exons with strong splice sites. In another example (12,14), tissue-specific SREs were identified by contrasting the frequencies of hexamers in a positive data set including cassette exons and their flanking intronic region to the frequencies of hexamers in a background set including sequences neighboring to the cassette exons. However, if a more reliable negative data set, where SREs over-represented in the positive data are most unlikely present, is used, such a discriminative approach will significantly improve the power of detecting SREs as already demonstrated in identifying transcriptional factor binding sites (16–18).
In this article, we used mouse RNA-Seq data (19) to determine a positive and a negative data set for each type of SREs in a specific tissue. For example, the positive data set for ESEs contains cassette exons that are included in the dominant isoforms of genes, whereas the negative data set consists of the cassette exons that are excluded in the dominant isoforms of genes. We then employed a discriminative approach to identify putative SREs. Since the expression level of each mRNA isoform can be calculated from the RNA-Seq data more accurately than from exon microarray data used in previous work (20,21), our method can reliably determine the positive and the negative data sets, which enables our discriminative approach to identify SREs more reliably.
MATERIALS AND METHODS
Data sets
Mouse RNA-Seq data of Mortazavi et al. (19) for three tissues (brain, liver and skeletal muscle) were selected in our study. The mouse genome and the KnownGene table were also downloaded from the University of California Santa Cruz genome database (UCSC) Mouse July 2007 (mm9). The mouse RNA-Seq data set contains 140 millions reads of 25 nt. Mortazavi et al. have mapped these reads against the expanded mouse genome, which consists of the standard UCSC mm9 genome and the 42 nt splice-crossing sequence for each exon junction documented in the UCSC KnownGene table. Reads that could be mapped to multiple loci of the genome were excluded, and 30–40 million uniquely mapped reads for each tissue from two replicates were used in our analysis.
We also selected 922 distinct hexamers from the database SpliceAid (22) as experimentally validated SREs for comparison purpose. SpliceAid is the latest database that collects experimentally assessed target RNA sequences bound by splicing proteins in humans. However, some sequences in SpliceAid are relatively long, and thus part of such long sequences may not be core splicing motifs. In fact, if we take hexamers from all the sequences in SpliceAid, we can get a total of 2321 distinct hexamers that may contain many false SREs. To get more reliable SREs, we only took all sequences assessed by SELEX from the SpliceAid. Since the length of the randomized sequences used in SELEX was usually larger than the length of a protein binding site, multiple alignment of the selected sequences was performed to locate the imbedded consensus sequences (5). These consensus sequences were manually checked and extracted, which gave 922 distinct hexamers.
Overview of computational strategy
Our goal is to identify short motifs that are over-represented in the region flanking alternative splicing sites in a specific tissue. Alternative splicing usually occurs at weak splicing sites with highly conservative flanking sequences (23). So these over-represented motifs most likely function as enhancers or silencers to assist the spliceosome to make a splicing decision.
Our analysis could be divided into several steps depicted in Figure 1a. We first identified cassette exons that are also referred to as alternatively spliced exons (ASEs) from the UCSC mouse KnownGene table. For a specific tissue, we then divided the set of ASEs into an inclusion set and an exclusion set as follows. Using the RNA-Seq data, we calculated the expression level of each isoform of genes that contain ASE(s). If a majority (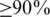 ) of the isoforms of a gene include an ASE, then the ASE is in the inclusion set; on the other hand, if only a minority (
) of the isoforms of a gene include an ASE, then the ASE is in the inclusion set; on the other hand, if only a minority (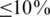 ) of the isoforms of a gene include an ASE, then the ASE belongs to the exclusion set. The inclusion and exclusion sets also include 400 intronic nucleotides upstream and 400 intronic nucleotides downstream of the selected ASEs. For each tissue, we compared the frequency of each hexamer in the inclusion set with the frequency of the same hexamer in the exclusion set to determine if the hexamer is over-represented. The hexamers that are over-represented in one tissue but not over-represented in the other tissue are identified as putative tissue-specific SREs, while the hexamers over-represented in both tissues are identified as SREs common in both tissues. Finally, annotations in three tissues were integrated and similar putative SREs were clustered to form a motif.
) of the isoforms of a gene include an ASE, then the ASE belongs to the exclusion set. The inclusion and exclusion sets also include 400 intronic nucleotides upstream and 400 intronic nucleotides downstream of the selected ASEs. For each tissue, we compared the frequency of each hexamer in the inclusion set with the frequency of the same hexamer in the exclusion set to determine if the hexamer is over-represented. The hexamers that are over-represented in one tissue but not over-represented in the other tissue are identified as putative tissue-specific SREs, while the hexamers over-represented in both tissues are identified as SREs common in both tissues. Finally, annotations in three tissues were integrated and similar putative SREs were clustered to form a motif.
Figure 1.

(a) Schematic flow chart for the identification of tissue-specific SREs. (b) Example of genes with more than two isoforms that were selected or excluded in our analysis. The left one was selected for further analysis since the ASE was either included or skipped in each isoform. The right one was not selected in our analysis because isoform 3 does not strictly skip the ASE.
ASE selection
We selected ASEs and some intronic nucleotides flanking the ASEs to identify SREs for the following reasons. First, AS predominantly generates ASE events in both human and mouse (1,19). Second, other AS events may not generate sequence data compatible to those generated by ASE events. For example, alternative 5′ or 3′ splice site usage lacks an alternative 3′ or 5′ splice site (24). ASEs were selected from the KnownGene table with a strict criterion. An ASE was selected if at least one isoform include the ASE and at least one other isoform do not include any part of the ASE and only these two types of isoforms exist. For example, the right AS event in Figure 1b was not selected in our analysis because although an ASE was included in isoform 1 and skipped in isoform 2, this exon and its flanking regions might also contain SREs governing alternative 3′ splice site since isoform 3 included this ASE partially. Different genes with overlapped open reading frame were also excluded for the simplicity and accuracy of calculating gene expression levels.
Calculation of expression level and inclusion ratio
Expression level for each transcript isoform of a gene was calculated with the algorithm of Jiang et al. (25). This algorithm modeled the count of RNA-Seq reads falling into a region of each gene as a Poisson's variable with a mean proportional to the length of the region. For an exon of length l, Jiang et al. used the effective exon length l−r in the mean of the Poisson's random variable, where r is the read length, since l−r is the number of possible loci of the exon that a read could be mapped to. However, since we only kept uniquely mapped reads and excluded the ambiguous reads that could be mapped to multiple places of the genome, we used an effective exon length l−r−m, where m is the number of 25-nt subsequences of the exon mapped by multiple-mapped reads. To find out these multi-mappable regions, we re-mapped all possible 25-nt subsequences of candidate ASEs and splice junctions against the same expanded genome described above using Bowtie (version 0.9.9.3), an ultrafast and memory-efficient program for the alignment of short DNA sequences to a large genome (26).
After the expression level of each isoform of genes with ASEs were calculated, the inclusion ratio of an ASE in a specific tissue was calculated as the ratio of the expression level of the isoform with the ASE to the total expression level of all isoforms of the gene.
SRE searching
For each tissue, all ASEs with an inclusion ratio ≥0.9 were put together as the exonic inclusion set, and 400 intronic nucleotides upstream or downstream of the ASEs were put together as the intronic inclusion set. All ASEs with inclusion ratio 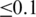 were selected as the exonic exclusion set, and 400 intronic nucleotides upstream or downstream of the ASEs were selected as the intronic exclusion set. The 15-nt-long splicing acceptor site consensus Y10NCAG/G and the 9-nt-long donor site consensus MAG/GURAGU (27) were not included in corresponding exonic and intronic sequences.
were selected as the exonic exclusion set, and 400 intronic nucleotides upstream or downstream of the ASEs were selected as the intronic exclusion set. The 15-nt-long splicing acceptor site consensus Y10NCAG/G and the 9-nt-long donor site consensus MAG/GURAGU (27) were not included in corresponding exonic and intronic sequences.
To identify ESEs and ESSs, we calculated the frequencies of each of 4096 possible hexanucleotides, fTI and fTS, in the exonic inclusion set and exclusion set of tissue T. The z-score (8,9) of the hexamer in tissue T was then given by
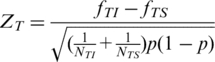 |
where NTI and NTS are the total number of hexamers in the inclusion and exclusion sets, respectively, and p = (NTIfTI + NTSfTS)/(NTI + NTS). Tissue-specific ESEs were identified as over-represented hexamers in the exonic inclusion set of tissue T1 but not over-represented in the exonic inclusion set of tissue T2. To test the statistical significance of over-representation under the null hypothesis of fT1I − fT1S = 0, we considered hexamers with ZT1 > 2.1701 (P < 0.03, two-tail test) as being over-represented. The 0.03 cutoff value for the P-value was selected based on the distribution of P-values as will be described in ‘Discussion’ section. To test the statistical significance of non-over-representation, we assumed the null hypothesis of over-representation as ZT2 = 2.1701 and considered hexamers with ZT2 < −1.8808 + 2.1701 = 0.2893 (P < 0.03, one-tail test) as non-over-represented hexamers.
For each pair of tissues (three pairs in total), we compared the z-score of each hexamer as shown in Figure 2. Hexamers with 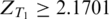 in tissue T1 but
in tissue T1 but 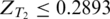 in tissue T2 were considered as tissue T1-specific ESEs (P < 0.032 ). Hexamers with both ZT1 and
in tissue T2 were considered as tissue T1-specific ESEs (P < 0.032 ). Hexamers with both ZT1 and 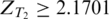 (P < 0.032 ) were identified as ESEs common to both tissues. The interval (0.2893, 2.1701) of a z-score corresponds to the unsure region where we do not have statistical evidence to decide whether a hexamer is over-represented or not.
(P < 0.032 ) were identified as ESEs common to both tissues. The interval (0.2893, 2.1701) of a z-score corresponds to the unsure region where we do not have statistical evidence to decide whether a hexamer is over-represented or not.
Figure 2.

z-scores for all hexamers in liver and muscle. (a) z-scores in exons. ESEC, ESEL and ESEM stand for common ESE, liver-specific ESE and muscle-specific ESE, respectively. (b) z-scores in 400 nt intronic sequences upstream of the exons. (c) z-scores in 400 nt intronic sequences downstream of the exons.
Similarly, tissue-specific ESSs were identified as hexamers over-represented in the exonic exclusion set of tissue T1, but not over-represented in the exonic exclusion set of tissue T2. Therefore, hexamers with  in tissue T1 but
in tissue T1 but 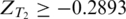 in tissue T2 were considered as tissue T1-specific ESSs (P < 0.032). Hexamers with both ZT1 and
in tissue T2 were considered as tissue T1-specific ESSs (P < 0.032). Hexamers with both ZT1 and  (P < 0.032) were identified as ESSs common to both tissues.
(P < 0.032) were identified as ESSs common to both tissues.
This searching process was repeated for upstream intronic sequences of 400-nt-long and downstream intronic sequences of 400-nt-long. The z-score of each hexamer was computed for each pair of tissues as depicted in Figure 2. Tissue-specific and common ISEs and ISSs in upstream and downstream introns were then identified based on the z-scores in the same way as ESEs and ESSs were identified.
Integration and clustering
After implementing the above steps, we got six classes of SREs, which include ESE, ESS, us' ISE, us' ISS, ds' ISE and ds' ISS, where us' and ds' stand for upstream and downstream, respectively. We integrated all of them into one table (Supplementary Table S1) to make their relationship more clear with the following annotation rule. Every SRE is associated with three characters to indicate its role in brain, liver and muscle. The first character can be ‘B’, ‘−’ or `?' to indicate that the SRE is present, absent or unsure in brain. Similarly, the second character can be `L', `−' or `?' and the third character can be `M', `−' or `?'. Note that tissue specificity is a relative concept. For example, an ESE can be present in brain but not in other tissues. We annotated this type of ESE as ESEB−−. An ESE can also be present in both brain and liver but not in muscle. We represented this type of ESE as ESEBL−. If an SRE was present in all three tissues, we referred to it as a common SRE.
We also clustered similar SREs using the hierarchical clustering algorithm (8) for each of six classes of SREs to determine splicing motifs. The Hamming distance was used in the clustering algorithm as the dissimilarity metric between any two SREs. We say that two SREs have incompatible annotations if any character associated with the SREs is a letter (B, L or M) in one SRE but a `−' in the other SRE. For example, two SREs, annotated as `BL−' and ‘BLM’, respectively, are incompatible for their annotation in muscle, but two SREs annotated as `BL−' or `BL?' are compatible. Since we do not want to put those incompatible SREs into the same cluster, we add a sufficiently large value (>6) to the dissimilarity distance between any two incompatible SREs. Therefore, each cluster only contains compatible SREs including SREs annotated with `?'. The dissimilarity cutoff for each cluster was chosen to be 2.0, which was relatively small to make the clustering result more reliable.
Position bias test
The chi-square goodness of fit test was adopted to determine if an SRE is uniformly distributed in a selected region or is biased toward certain specific locations. The selected region includes introns or exons in which the SRE was predicted. For example, for SREs annotated as ESS−LM, the exonic exclusion sets of liver and muscle were used to test position bias. Since the exons have different lengths, we only chose exons of ≥110 nt, and took 55 nt from each end of the exon. For introns, we took first 395 nt upstream or downstream of the exon. An SRE which is a hexamer can be mapped to 390 positions of an intronic sequence or 100 positions of an exonic sequence. We, therefore, divided each exonic or intronic sequence into 10 or 39 intervals, each with 10 nucleotides. A significance level of 0.01 was used to reject the null hypothesis that an SRE uniformly appears in all intervals. Since for a uniform distribution, the chi-square test is robust when the average number of the SREs falling into an interval is ≥2 for a significance level as small as 0.01 (28), only SREs with ≥78 counts in intronic sequences or ≥20 counts in exonic sequences were chosen in the analysis.
Comparison with constitutive data
We collected two sets of sequences from the KnownGene table and put them together as the data set for constitutive exons. We took 49 649 internal exons of genes with only one isoform as the first data set. The second set consists of 34 403 exons locating in alternatively spliced genes but included by all isoforms. In addition to exons, intronic sequences of 400 nt upstream or downstream of the constitutive exons were also collected as the intronic constitutive data set. Note that these data sets from constitutive exons and their flanking intronic regions are similar to the inclusion set of ASEs, since all exons in the data sets are constitutively included in the mature mRNA. We compared the frequency of an SRE in the constitutive data with the frequency in its corresponding positive data. The frequency of an SRE in the negative data was also compared with that in the positive data. For example, when we examined ESEBLM, the constitutive data were constitutive exons; the positive data were exonic inclusion set of brain, liver and muscle; and the negative data were the exclusion sets of these three tissues. If we examined ds' ISS−L−, the constitutive data were constitutive downstream intronic sequences; the positive data were downstream intronic exclusion set of liver; and the negative data were downstream intronic inclusion set of liver. Frequency comparison was only performed for clusters annotated without `?' (369 SREs in total).
RESULTS
Putative enhancers and silencers
As shown in Table 1, we got 300–400 ASEs in the inclusion and exclusion sets of three tissues. The average length of ASEs is 123 nt, and the average length of upstream and downstream introns is 5900 nt and 6116 nt, respectively. This is consistent with the observation that ASEs are generally short and flanked by long introns (23).
Table 1.
Number of ASEs used in SRE searching
| Brain | Liver | Muscle | |
|---|---|---|---|
| Inclusion set | 399 | 369 | 411 |
| Exclusion set | 372 | 454 | 408 |
The z-scores for hexamers in liver and muscle data sets and the regions defining each type of the SREs are plotted in Figure 2. The z-scores for hexamers in other two pairs of tissues (brain versus liver and brain versus muscle) are included in Supplementary Figure S1 and S2. It is seen from these figures that most hexamers are not over-represented in any tissue, and thus are not an SRE, as expected.
After integrating all the SREs identified in these figures, we predicted 456 putative enhancers and silencers which are listed in Supplementary Table S1. The statistics of these 456 SREs are summarized in Table 2. The second row annotated with `BLM' contains 45 SREs common to all three tissues. The next three rows consist of SREs annotated with `BL?', `B?M' and `?LM', which are common to two tissues but may or may not be an SRE in the third tissue. The next 12 rows contain a total of 221 tissue specific SREs. Note that only 18, 8 and 15 SREs are unique to brain, liver and muscle, respectively.
Table 2.
Number of common and tissue-specific SREs
| Anno. | ESE | ESS | us' ISE | us' ISS | ds' ISE | ds' ISS | Total |
|---|---|---|---|---|---|---|---|
| BLM | 15 | 11 | 6 | 3 | 4 | 6 | 45 |
| BL? | 17 | 15 | 7 | 11 | 13 | 13 | 76 |
| B?M | 12 | 14 | 9 | 9 | 9 | 11 | 64 |
| ?LM | 21 | 10 | 10 | 13 | 6 | 7 | 67 |
| B–? | 2 | 2 | 5 | 6 | 7 | 4 | 26 |
| B?– | 6 | 1 | 2 | 8 | 6 | 4 | 27 |
| –L? | 3 | 2 | 10 | 8 | 4 | 4 | 31 |
| ?L– | 10 | 3 | 2 | 4 | 8 | 8 | 35 |
| –?M | 6 | 4 | 6 | 5 | 2 | 4 | 27 |
| ?–M | 6 | 2 | 4 | 2 | 5 | 7 | 26 |
| BL– | 0 | 0 | 1 | 0 | 1 | 0 | 2 |
| B–M | 0 | 1 | 0 | 1 | 1 | 0 | 3 |
| –LM | 0 | 0 | 0 | 2 | 0 | 1 | 3 |
| B– – | 8 | 1 | 4 | 2 | 3 | 0 | 18 |
| –L– | 0 | 0 | 1 | 1 | 2 | 4 | 8 |
| – –M | 2 | 2 | 2 | 2 | 2 | 5 | 15 |
| TSSE | 43 | 18 | 37 | 41 | 41 | 41 | 221 |
The last row contains the total number of tissue-specific splicing elements for each type of SREs, which equals to the sum of rows with annotation `–'.
We compared our results with constitutive exonic splicing enhancers RESCUE-ESE in mouse (29,30). Among 508 mouse RESCUE-ESEs, only 43 (8%) are included in the SREs we identified, 20 of which are also ESEs in our analysis. Note that our SREs include 108 ESEs and <20% (20/108) are also RESCUE-ESE. This shows that most of our SREs are different from RESCUE-ESEs possibly due to their tissue-specificity.
In addition, we compared our results with tissue-specific SREs in human identified recently by other two groups (1,12), as shown in Figure 3. Using human RNA-Seq data, Wang et al. (1) identified 362 SREs in 15 tissues and cell lines, of which 51 distinct elements were identified as SREs specific to brain, liver and muscle. Among these 51 SREs, 13 (25.5%) are also included in the SREs we identified. To compare our SREs with the results of Castle et al. (12), we extracted all the hexamers significantly over-represented (P < 10−3) in up-regulated or down-regulated cassette exons in samples related to brain, liver and muscle. This gave 783 distinct hexamers in total, of which 89 (11.4%) are also included in the SREs we identified. As shown in Figure 3, the number of SREs identified by any two studies is relatively small. Overall, 20% (93/456) of our SREs are in the SREs of Wang et al. and/or Castle et al.
Figure 3.

Venn diagram for the number of SREs identified in three studies.
We also compared our results with two tissue-specific motifs in mouse identified by Sugnet et al. (43) from their microarray data. The first CU-rich motif with consensus sequence UGYUUUC was identified by Sugnet et al. in upstream of brain-included exons. The most similar SREs in our results are UGAUUU (us’ ISE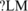 ) and UGAUUG (us’ ISEBL?). The second motif with consensus sequence UACUAAC was identified by Sugnet et al. in downstream intron of muscle-included exons. We can find two hexamers in our ds’ ISE consistent with this motif, which are CCAAAC (ds’ ISEB?M) and CGCUAA (ds’ ISEB?M).
) and UGAUUG (us’ ISEBL?). The second motif with consensus sequence UACUAAC was identified by Sugnet et al. in downstream intron of muscle-included exons. We can find two hexamers in our ds’ ISE consistent with this motif, which are CCAAAC (ds’ ISEB?M) and CGCUAA (ds’ ISEB?M).
We further compared our results with 992 hexamers selected from the SpliceAid databases (22) (see ‘Materials and Methods’ section for the selection of 992 hexamers). About 26% (118/456) of our SREs are among these 992 hexamers (see Supplementary Table S1 for details). On the other hand, 20% (10/51) of the SREs identified by Wang et al. (1) and 22% (173/783) of the SREs of Castle et al. (12) are also included in the 992 hexamers. This shows that our study gives slightly higher portion of experimentally validated SREs than other two studies.
To systematically examine the quality of our SREs and choose reliable candidate SREs for further analysis, we ranked all the SREs without `?' in their annotations by their final P-values (product of P-values in three tissues). The top 15 SREs and the relevant experimental evidence reported in the literature are listed in Table 3. Some of the 15 SREs may actually come from the same motif; for example, CCUGCC, CUGCCU and GCCUGC may come from the CCUG repeat. We examined some well-studied SREs by comparing studies in the literature and their annotations in our result. Some of our annotations match previous studies very well. For example, UCUCUC and CUCUCU are both identified as us' ISS−LM in our analysis. Previous experimental study has identified the conserved CUCUCU sequence within intron regions as splicing silencer in non-neuronal cells, since it is responsible for repressing splicing of neuron-specific N1 exon of mouse c-src transcript in nonneuronal cells (34), possibly by interacting with PTB proteins (44). We will discuss several interesting SREs in the following sections.
Table 3.
Fifteen SREs with most significant P-values
| SREs | Annotation | P-value | Reference | Related experimental results |
|---|---|---|---|---|
| CCUGCC | ESSBLM | 2.43e-18 | (31) | CCUG repeats specifically interact with MBNL1. |
| UCUAUC | ds' ISS−−M | 7.53e-13 | (32,33) | Downstream ISE UCUAUC, bound by protein HRP-2, regulates alternative splicing of exon 16 in unc-52 gene of C. elegans. |
| CUCUCU | us' ISS−LM | 1.23e-12 | (34–36) | Within polypyrimidine tract, interact with PTB, responsible for the skipping of N1 exon of mouse c-src. |
| CUGCCU | ESSBLM | 1.51e-10 | Same as CCUGCC. | |
| CUAUCU | ds' ISS−−M | 1.64e-10 | May be from the same motif as UCUAUC. | |
| GCGCGC | ds' ISS−−M | 2.20e-10 | ||
| GCCUGC | ESSBLM | 2.78e-10 | Same as CCUGCC. | |
| AAAUAA | ESSBLM | 3.16e-10 | ||
| UGCAUG | ds' ISE−−M | 3.95e-10 | (37–39) | When bound by tissue-specific factor, Fox-1 Protein family, it acts as splicing enhancer. |
| UGCAUG | ds' ISS−L− | 1.15e-09 | ||
| ACACAC | us' ISS−−M | 1.58e-09 | (40) | Intronic CA repeat could function as enhancers or silencers, depending on its proximity to the 5′ ss. |
| UGGAGC | ESEBLM | 2.79e-09 | ||
| UUCUUC | ds' ISSBLM | 2.94e-09 | (41) | It is the second pyrimidine-rich(PY) elements in the three PY elements downstream of CFTR exon 9. |
| AUCUAU | ds' ISEBL− | 7.24e-09 | ||
| GCAGCA | us' ISEBLM | 7.40e-09 | (42) | splicing factor CUGBP1 interacts with GCA repeats located within the MEF2A mRNA. |
Position bias of SREs
Since splicing factors function primarily in the vicinity of a splice site (45), it is possible that positions of some SREs are biased towards certain locations, while non-functional sequences may tend to locate more randomly. To test if SREs have position bias, we adopted chi-square goodness of fit test as described in ‘Materials and Methods’ section. Under the selection criterion described in ‘Materials and Methods’ section, 156 SREs were selected for position bias test (Supplementary Tables S2). About 46% (71/156) of SREs show significant position bias at a significance level of 0.01. We ranked these SREs according to their P-values, and visually examined the position distribution of top 30 SREs with most significant P-values. We found that 12 SREs' positions show significant position bias, eight of which were depicted in Figure 4. It is interesting to see that not all the SREs are biased towards a splice site. The common us' ISS AAGAUU and the common us' ISE UUGUAC occupy the position near 200-nt upstream of the acceptor site as their preferred location. The common ds' ISS UUCUUC is abundant almost evenly in the region <170-nt downstream of the ASE but is less abundant in the region further away from the splice site. The common ESS UUAAAG prefers the interval between 22 and 31-nt downstream of the 5′ end of the ASE. We also checked position distributions of other SREs with 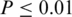 , but did not find any general pattern for position bias.
, but did not find any general pattern for position bias.
Figure 4.

Position distribution of top eight SREs with smallest P-values in the position bias test. Each bar represents the average number of SREs falling into a region of 10 nt divided by the number of intron or exon sequences used in analysis.
Two tissue-specific SREs, CUCUCU (P = 1.33e-16) and UCUCUC (P = 2.59e-14), which were identified as upstream ISS−LM, showed most significant P-values. They were clustered with other two SREs UCUCUU (us' ISS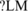 , P = 1.58e-10) and CUCUUU (us' ISS
, P = 1.58e-10) and CUCUUU (us' ISS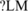 , P = 3.50E-06) in the clustering result described later. The position distributions of CUCUCU, UCUCUC and UCUCUU were shown in Figure 4. We also compared the distribution of CUCUCU in three tissues' exclusion sets with that in inclusion sets as shown in Figure 5. The SRE UCUCUC has a very similar position distribution that is not shown here. We can see from Figure 5 that in the exclusion set of liver and muscle, CUCUCU is not only abundant, but also shows a significant position bias towards the acceptor site while in the inclusion set, it is less abundant and almost evenly distributed. Since the region between 15 and 30 nt upstream of a 3′ splice site coincides with the location of the polypyrimidine tract, it is highly possible that CUCUCU and UCUCUC are part of the polypyrimidine tract. Note that Castle et al. (12) found that UCUCU is enriched in the region from 35 to 110-nt upstream of tissue-regulated ASEs in human tissues.
, P = 3.50E-06) in the clustering result described later. The position distributions of CUCUCU, UCUCUC and UCUCUU were shown in Figure 4. We also compared the distribution of CUCUCU in three tissues' exclusion sets with that in inclusion sets as shown in Figure 5. The SRE UCUCUC has a very similar position distribution that is not shown here. We can see from Figure 5 that in the exclusion set of liver and muscle, CUCUCU is not only abundant, but also shows a significant position bias towards the acceptor site while in the inclusion set, it is less abundant and almost evenly distributed. Since the region between 15 and 30 nt upstream of a 3′ splice site coincides with the location of the polypyrimidine tract, it is highly possible that CUCUCU and UCUCUC are part of the polypyrimidine tract. Note that Castle et al. (12) found that UCUCU is enriched in the region from 35 to 110-nt upstream of tissue-regulated ASEs in human tissues.
Figure 6.

Comparison of frequencies of different SREs in different data sets. The first bar in each group stands for the ratio of the frequency in constitutive data to the frequency in the positive data. The second bar stands for the ratio of frequency in the negative data to the frequency in the positive data. Number of SREs used in each comparison is shown in parenthesis.
Figure 5.

position distribution comparison for the us' ISS−LM CUCUCU in upstream introns of the exclusion set and the inclusion set of brain, liver and muscle. Each bar represents the average number of SREs falling into a region of 10 nt normalized by the number of intronic sequences used in analysis.
This result may imply the main position where this SRE takes effect, since it is consistent with the finding that polypyrimidine-tract binding protein (PTB; also known as hnRNP I) silence splicing by binding to the polypyrimidine tract and blocks the binding of U2AF (35,36). Interestingly, this SRE was annotated as us' ISS−LM, which implies that it is specific to liver and muscle but not a us' ISS in brain. We also checked its position distribution in the brain data set, but no position bias was found as shown in Figure 5. Our annotation is consistent with the experimental evidence showing that skipping of neuron-specific N1 exon of mouse c-src in non-neuronal cells requires conserved CUCUCU elements within polypyrimidine tract and downstream intron (34).
Comparing with the results of Castle et al. (12) and Wang et al. (1), we got some identical and some different findings for the SRE CUCUCU. Both our result and the result of Castle et al. (12) indicate that CUCUCU is an upstream ISS in liver, but Wang et al. (1) did not identify it as an SRE in liver. In muscle, we identify CUCUCU as an upstream ISS, but the data of Castle et al. (12) indicate that it is an upstream ISE, and Wang et al. (1) did not identify it as an SRE. In brain, the data of Castle et al. (12) show that CUCUCU is over-represented in the upstream of up-regulated ASEs (which is equivalent to our upstream intronic inclusion set) but not in the upstream of down-regulated ASEs (which is equivalent to our upstream intronic exclusion set). Based on this observation, we may identify CUCUCU as an upstream ISE in brain. However, data of Castle et al. (12) also show that the expression of PTBP1, whose target motif is CUCUCU, is down-regulated in brain. Hence, if CUCUCU is an ISE, there must be another unknown SF that binds to it. Another possibility is that PTBP1 is the only SF that can bind to CUCUCU, and CUCUCU is always a silencer; however, its silencing function is lost in brain due to the low level of PTBP1. The RNA-Seq data of both Mortazavi et al. (19) that are used in our study and Wang et al. (1) indicate that CUCUCU is over-represented in both upstream intronic inclusion and exclusion sets of brain. This is in conflict with the microarray data of Castle et al. (12). Nevertheless, combining the RNA-Seq data of Wang et al. (1) and Mortazavi et al. (19) and the expression level of PTBP1 reported by Castle et al. (12), we can eliminate the possibility that CUCUCU is an ISE, but predict it to be an upstream ISS with lost silencing function in brain. Note that if we only use the information of CUCUCU without using the information about the expression level of PTBP1, the data of Castle et al. (12) will predict CUCUCU to be an ISE, which is likely wrong; the non-discriminative method will predict CUCUCU to be both upstream ISS and ISE, which conflict with each other; on the other hand, our discriminative method does not identify CUCUCU to be an SRE in brain. Our result in this complicated case seems most reasonable, because the most reasonable prediction is that CUCUCU is an ISS generally but not function in brain, as we discussed earlier. These studies indicate that CUCUCU may play an important and complicated role in tissue-specific splicing, particularly in brain and worth further experimental investigation.
Clustering results
Some of the 456 SREs are very similar to each other. These similar SREs may come from the same motif that is bound by the same splicing factor. Our clustering process resulted in 247 clusters as shown in Table 4 and Supplementary Table S3. Relatively large number of clusters is due to the fact that we used a relatively small cutoff value (2.0) for the dissimilarity distance between any two SREs in the same cluster.
Table 4.
Number of common and tissue-specific splicing motifs
| Anno. | ESE | ESS | us'ISE | us'ISS | ds'ISE | ds'ISS | Total |
|---|---|---|---|---|---|---|---|
| BLM | 15 (44) | 13 (36) | 9 (18) | 7 (14) | 9 (25) | 10 (23) | 63 (160) |
| BL? | 2 (3) | 3 (3) | 3 (3) | 2 (3) | 4 (4) | 3 (3) | 17 (19) |
| B?M | 0 (0) | 3 (4) | 1 (1) | 2 (2) | 0 (0) | 1 (1) | 7 (8) |
| ?LM | 3 (4) | 2 (3) | 0 (0) | 1 (2) | 1 (1) | 1 (1) | 8 (11) |
| B–? | 0 (0) | 1 (1) | 2 (2) | 3 (4) | 3 (4) | 2 (4) | 11 (15) |
| B?– | 2 (2) | 0 (0) | 0 (0) | 4 (5) | 3 (3) | 2 (2) | 11 (12) |
| –L? | 2 (2) | 1 (1) | 4 (4) | 2 (2) | 1 (1) | 2 (2) | 12 (12) |
| ?L– | 4 (5) | 1 (2) | 1 (1) | 0 (0) | 1 (1) | 3 (4) | 10 (13) |
| –?M | 0 (0) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (2) | 5 (6) |
| ?–M | 1 (1) | 1 (1) | 1 (3) | 0 (0) | 2 (3) | 1 (2) | 6 (10) |
| BL– | 5 (11) | 1 (3) | 2 (5) | 3 (7) | 4 (10) | 2 (6) | 17 (42) |
| B–M | 4 (8) | 2 (4) | 3 (7) | 4 (9) | 2 (3) | 3 (8) | 18 (39) |
| –LM | 4 (10) | 1 (2) | 5 (13) | 8 (20) | 0 (0) | 2 (4) | 20 (49) |
| B– – | 7 (12) | 1 (1) | 4 (5) | 2 (4) | 3 (7) | 0 (0) | 17 (29) |
| –L– | 0 (0) | 0 (0) | 1 (2) | 1 (2) | 3 (7) | 3 (8) | 8 (19) |
| – –M | 3 (6) | 3 (6) | 3 (5) | 1 (2) | 3 (4) | 4 (8) | 17 (31) |
| TSSM | 32 (57) | 13 (22) | 27 (48) | 29 (56) | 26 (44) | 25 (50) | 152 (277) |
The last row contains the total number of tissue-specific splicing motifs (TSSM) for each type of motifs. The number of hexamers in each type of motifs is shown in parenthesis.
After the clustering process, we re-annotated each cluster to eliminate some `?' annotation. For example, one cluster consists of sequences with annotation `BL?' and `BL−', then we re-annotate all the elements in the cluster as ‘BL−’. The high ratio between the number of tissue-specific SREs and common SREs (221/45) observed in Table 2 was decreased to 152/63 in Table 4, but the ratio is still significantly large, implying that tissue-specific motifs may play a very important role in splicing regulation. The average number of SREs per cluster is 160/63 = 2.54 for common SREs and 277/152 = 1.82 for tissue-specific SREs, which implies that tissue-specific motifs may be more conservative than common motifs.
Frequencies of identified SREs in constitutive exons
The 456 SREs were identified based on their frequencies in the inclusion and exclusion sets of the ASEs. We also wished to know the frequencies of these SREs in the constitutively spliced exons and their flanking intronic regions to gain more insight of the role of these SREs. Using the constitutive data described in ‘Materials and Methods’ section, we compared the frequency of SREs in different data sets, as depicted in Figure 6. For the clarity of comparison, frequencies in the constitutive data and the negative data have been normalized by the frequencies in the positive data.
First, let us look at the frequencies of the enhancers. We would expect the constitutive exon data set to have abundant enhancers to assist splicing. However, it is seen from Figure 6 that enhancers we identified have lower frequencies in the constitutive exon data set than in the inclusion set of ASEs. This may be due to the following two reasons. First, most of the tissue-specific enhancers may be different from the enhancers present in the constitutively spliced exons and flanking introns. This may also explain why most of enhancers we identified are not RESCUE-ESEs. Second, tissue-specific enhancers are more abundant in ASEs than in constitutively spliced exons. We also compared the frequencies of constitutive RESCUE-ESEs in constitutive exons and our inclusion sets of brain, liver and muscle, but no frequency difference was found.
The frequencies of silencers are expected to be lower in the constitutive data set than in the exclusion set of the ASEs. Indeed, this is observed in Figure 6. Comparing the relative frequencies of ESS with the frequencies of other silencers and enhancers, we see that the relative frequencies of ESS are generally the lowest. This may imply that ESSs play a stronger role in AS than ISSs and ISEs.
Another observation from Figure 6 is that the frequencies of all SREs in the constitutive data set are higher than the frequency in the corresponding negative data set (exclusion set for enhancers and inclusion set for silencers) of ASEs. Therefore, if we use the constitutive data as the negative control data as did in (8,10,14,46) to identify SREs, we would lose some detection power. To verify this, we calculated the z-score and the corresponding P-value of each of the 369 SREs by replacing the negative data set used in the previous analysis with the constitutive data set. We found that 179 SREs have a 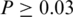 , implying that 48.5% (179/369) of these SREs would not be identified if we have used constitutive data set as the negative data set. Among these 179 SREs, 34% (60/179) SREs could be found in the 992 hexamers selected from SpliceAid; whereas among the remaining 190 SREs, only 22% (42/190) can be found in the 992 hexamers. This indicates that more percentage of true positive SREs can be lost if the non-discriminative approach is employed.
, implying that 48.5% (179/369) of these SREs would not be identified if we have used constitutive data set as the negative data set. Among these 179 SREs, 34% (60/179) SREs could be found in the 992 hexamers selected from SpliceAid; whereas among the remaining 190 SREs, only 22% (42/190) can be found in the 992 hexamers. This indicates that more percentage of true positive SREs can be lost if the non-discriminative approach is employed.
Special SREs that can be both enhancer and silencer
Among 456 SREs we identified, two SREs are special because they were identified as an enhancer in one tissue but a silencer in another tissue. These two SREs are UGCAUG and UCUAUC, whose z-score are shown in Figure 2c.
UGCAUG was annotated as a downstream ISE−−M and downstream ISS−L−. Our annotation ISE−−M (muscle-specific ISE) of UGCAUG is consistent with the computational result (14) and experimental observation (37,38), as well as with the results of Wang et al. (1) and Castle et al. (12). Our annotation ds' ISS−L− is also consistent with the result of Castle et al. (12), but Wang et al. (1) did not predict UGCAUG to be an SRE in liver. To the best of our knowledge, this putative role of downstream ISS in liver has not been reported in any experimental results, although it was experimentally verified to be an upstream ISS (47). Further experimental investigations worth being carried out to see if it is a liver-specific ISS as Castle et al. and we predicted. If this is true, new splicing factors binding to this hexamer may be identified, given the fact that Fox-1 is not expressed in liver (47).
We did not identify UGCAUG to be an upstream ISE in brain as previous computational work did (14,37). The data of Wang et al. (1) also indicate that UGCAUG is an ISE in brain, but enrichment in brain is not so significant as in heart and muscle. The data of Castle et al. (12) are more complicated, because UGCAUG is over-represented in the downstream intronic region of both up- and down-regulated ASEs in several brain cells including medulla oblongata, thalamus and in fetal brain, but is over-represented in the downstream intronic region of only up-regulated ASEs in other brain cells including cerebellum and hippocampus. Hence, if we use the non-discriminative method, we would predict UGCAUG to be both ISE and ISS in the same downstream region and in the same type of cell, which is obviously a conflictive result. To find out why our method using the data of Mortazavi et al. (19) did not predict UGCAUG to be an ISE in brain, we rechecked the data and found that UGCAUG is moderately abundant in both the inclusion and exclusion sets of brain. The z-score of our discriminate approach is lower than the critical value at the 0.03 significance level. Generally, for those sequences that are abundant in both inclusion and exclusion sets, our discriminative approach will not predict them to be an SRE, but the non-discriminative will give a conflictive prediction: such sequences are both an enhancer and a silencer. Given the different results in different studies and the fact that UGCAUG is a binding target of Fox-1 protein family specifically expressed in brain (47,48), more carefully designed experiment is needed to investigate the role of UGCAUG in brain, especially in different brain cell types.
Another hexamer UCUAUC was predicted as a downstream ISS−−M and a downstream ISE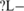 . No corresponding experimental result in mammals was found for this hexamer, but it was found to be over-represented in both flanking introns of ASE 16 of the unc-52 gene of Caenorhabditis elegans. Using an unc-52 splicing reporter trans-gene containing alternative exons 15 through 19, it was reported that alternative splicing is regulated by the hexamer UCUAUC in the intron downstream of exon 16 (32). It was also reported that this hexamer was bound by protein HRP-2 with high affinity (33) and was concluded that UCUAUC could enhance the inclusion of exon 16 in the muscle-expressed reporter trans-gene, which seems inconsistent with our annotation ISS−−M. Since half of C. elegans introns are of <60 nt, which are too short to be spliced in mammals, the role of UCUAUC in mammals needs to be investigated by further experimental and computational approaches.
. No corresponding experimental result in mammals was found for this hexamer, but it was found to be over-represented in both flanking introns of ASE 16 of the unc-52 gene of Caenorhabditis elegans. Using an unc-52 splicing reporter trans-gene containing alternative exons 15 through 19, it was reported that alternative splicing is regulated by the hexamer UCUAUC in the intron downstream of exon 16 (32). It was also reported that this hexamer was bound by protein HRP-2 with high affinity (33) and was concluded that UCUAUC could enhance the inclusion of exon 16 in the muscle-expressed reporter trans-gene, which seems inconsistent with our annotation ISS−−M. Since half of C. elegans introns are of <60 nt, which are too short to be spliced in mammals, the role of UCUAUC in mammals needs to be investigated by further experimental and computational approaches.
DISCUSSION
Reads from RNA-Seq give information about how exons are connected, which can be explored in the investigation of AS. RNA-Seq also provides more accurate measurement of expression levels of transcripts and their isoforms across a very broad dynamic range than other methods such as microarray (20). Capitalizing on these two advantages of RNA-Seq, we identified ASEs from the mouse RNA-Seq data set (19) and calculated the expression levels of isoforms of the genes containing the selected ASEs. This enabled us to determine reliable positive and negative data sets for SREs and then to employ a powerful discriminative approach to identify enhancers and silencers regulating alternative splicing. We chose the RNA-Seq data for three mouse tissues (19) rather than more comprehensive RNA-Seq data for 15 human tissues and cell lines (1) due to the following two reasons. First, unlike the human RNA-Seq data (1), the mouse RNA-Seq data (19) have not been explored to predict any SREs. Second, as demonstrated in (19), the RNA-Seq reads generated from the protocol using RNA fragmentation provide more uniform coverage along the transcripts than those generated from the protocol using cDNA fragmentation (1), and thus, the mouse RNA-Seq data can be used to calculate the expression level of each isoform of each gene more accurately.
As shown in (16–18), a discriminative approach using reliable positive and negative data can significantly increase the power of detecting motifs that are over-represented in the positive data set relative to the negative data, without increasing the false positive rate. However, most computational methods for identifying SREs do not employ the discriminative approach. These include the ones used to identify RESCUE-ESEs from constitutively spliced exons (8) and tissue-specific SREs from microarray data (12) as we discussed in ‘Introduction’ section. Similar to the method used to identify RESCUE-ESEs, intronic sequences flanking constitutively spliced exons were used as background data to identify brain-specific SREs (14). The putative ESEs and ESSs (PESEs/PESSs) were identified by comparing the frequencies of octamers in constitutively spliced non-protein-coding exons with those in a negative control set including the pseudo exons and 5′ untranslated regions of intronless gene (9). Although this negative set may be more reliable than the one used in identifying RESCUE-ESEs, it may not be as reliable as the negative data in our method due to the following arguments. Pseudo exons are good negative sequences for identifying ESEs because they are never spliced. However, although the ASEs in our exclusion set are also not spliced in a tissue or under certain condition, they are spliced in other tissue(s) or under other conditions. This is a stronger indication that these ASEs in our exclusion set may lack the ESEs that assist the splicing of ASEs in the positive data. Similar arguments hold for other enhancers or silencers. In the identification of ESS from pseudo exons (10), constitutively spliced exons and their flanking intronic regions were used as the negative data set, which is again not as reliable as the ASEs and their flanking intronic regions in our inclusion set because these ASEs can also be skipped under different conditions.
Another advantage of our discriminative approach is that it can identify both common and tissue-specific SREs. This is an important feature because both tissue-specific splicing factors and tissue-specific expression of constitutive splicing factors may play a role in regulating alternative splicing. If we use constitutively spliced exons as the negative data as used in (8,10,14,46), we would not only lose detection power as shown in the ‘Results’ section, but also miss those common SREs present in constitutively splice exons. As a side note, similar to the method used to identify PESE/PESS (9), our method do not have problem of sequence bias such as codon or CpG bias, since our positive and negative data sets have similar sequence composition. If a sequence is abundant in both inclusion and exclusion sets, our discriminative approach generally will not predict it as an SRE, but the non-discriminative approach will likely predict it to be both an enhancer and a silencer, which obviously is a conflictive and confusing result. On the other hand, if an SRE is abundant in both the data set from which we try to identify the SRE and the background data set, non-discriminative approach cannot identify such an SRE, but our discriminative approach using negative data set is very likely able to identify it.
To reduce the false positive rate without losing detection power, we used a validating process to determine the cutoff P-value, which was chosen to be 0.03. Specifically, we first used a cutoff P-value equal to 0.05. This gave 799 SREs, 200 of which could be found at least one match in the 992 hexamers selected from SpliceAid (22) containing experimentally identified SREs. We plotted the distribution of the P-values of these 200 SREs and of the remaining 599 SREs, as shown in Figure 7. It is seen that at a P < 0.03, the probability of experimentally validated SREs is generally higher than the probability of SREs without experimental validation, and that this trend is reversed at P > 0.03. Therefore, we selected 0.03 to be the cutoff P-value.
Figure 7.

Probability density of P-values of the SREs with or without experimental validation. SREs are computationally identified at a significance level of 0.05.
About 26% (118/456) of 456 SREs we identified can be found in database with experimentally validated SREs. This percentage is slightly higher than that for the SREs identified by Wang et al. (1) and Castle et al. (12) from human tissues. About 48% (221/456) of our SREs are tissue-specific, which shows that tissue-specific SREs play an important role in regulating alternative splicing as observed early. Although only 10% (45/456) SREs are common to all three tissues in this study, it does not imply that common SREs are less important, because 45% (207/456) SREs were common to two tissues but unsure to the other tissue. If more data are available, we may identify these SREs as common or tissue-specific SREs. Only 18% (20/108) of our ESEs are included in RESCUE-ESE identified from constitutively spliced exons, and only 14% (15/108) of our ESEs are annotated as common to three tissues. This shows that much more tissue-specific ESEs are involved in regulating tissue-specific splicing than constitutive ESEs.
It worths some discussions on three SREs: CUCUCU (us' ISS−LM), UGCAUG (ds' ISE–M and ds' ISS−L−) and UCUAUC (ds' ISS−−M and ds' ISE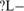 ). The first two have been repeatedly identified as an SRE in both experimental and computational approaches (12,34–39), but our study reveals some new information. Specifically, our position analysis showed that CUCUCU appears at 15–30 nt upstream of the ASE skipped in liver and muscle but not brain with much higher frequency than any other locations. Since these locations are in the polypyrimidine tract, CUCUCU most likely functions in the polypyrimidine tract as a tissue-specific silencer. While previous results showed that an SRE can be an enhancer or silencer depending on its location. For example, UGCAUG can be a ds' ISE or a us' ISS. Our analysis showed that UGCAUG and UCUAUC can function as an enhancer in one tissue but a silencer in another tissue from the same intronic region downstream of the ASE, which calls further investigation about the mechanism that these two SREs function.
). The first two have been repeatedly identified as an SRE in both experimental and computational approaches (12,34–39), but our study reveals some new information. Specifically, our position analysis showed that CUCUCU appears at 15–30 nt upstream of the ASE skipped in liver and muscle but not brain with much higher frequency than any other locations. Since these locations are in the polypyrimidine tract, CUCUCU most likely functions in the polypyrimidine tract as a tissue-specific silencer. While previous results showed that an SRE can be an enhancer or silencer depending on its location. For example, UGCAUG can be a ds' ISE or a us' ISS. Our analysis showed that UGCAUG and UCUAUC can function as an enhancer in one tissue but a silencer in another tissue from the same intronic region downstream of the ASE, which calls further investigation about the mechanism that these two SREs function.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Science Foundation (NSF) under NSF CAREER Award No. 0746882. Funding for open access charge: The NSF grant (partial); Institutional fund (partial).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank three anonymous reviewers for their comments that aided us to improve the quality of this work.
REFERENCES
- 1.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- 4.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Djordjevic M. Selex experiments: new prospects, applications and data analysis in inferring regulatory pathways. Biomol. Eng. 2007;24:179–189. doi: 10.1016/j.bioeng.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- 9.Zhang XH, Chasin LA. Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 2004;18:1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sironi M, Menozzi G, Riva L, Cagliani R, Comi GP, Bresolin N, Giorda R, Pozzoli U. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res. 2004;32:1783–1791. doi: 10.1093/nar/gkh341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chasin LA. Searching for splicing motifs. Adv. Exp. Med. Biol. 2007;623:85–106. doi: 10.1007/978-0-387-77374-2_6. [DOI] [PubMed] [Google Scholar]
- 12.Castle JC, Zhang C, Shah JK, Kulkarni AV, Kalsotra A, Cooper TA, Johnson JM. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008;40:1416–1425. doi: 10.1038/ng.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartmann B, Valcárcel J. Decrypting the genome′s alternative messages. Curr. Opin. Cell Biol. 2009;21:377–386. doi: 10.1016/j.ceb.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Brudno M, Gelfand MS, Spengler S, Zorn M, Dubchak I, Conboy JG. Computational analysis of candidate intron regulatory elements for tissue-specific alternative pre-mRNA splicing. Nucleic Acids Res. 2001;29:2338–2348. doi: 10.1093/nar/29.11.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Wang K, Radovich M, Wang Y, Wang G, Feng W, Sanford J, Liu Y. Genome-wide prediction of cis-acting RNA elements regulating tissue-specific pre-mRNA alternative splicing. BMC Genomics. 2009;10:S4. doi: 10.1186/1471-2164-10-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinha S. Discriminative motifs. J. Comput. Biol. 2003;10:599–615. doi: 10.1089/10665270360688219. [DOI] [PubMed] [Google Scholar]
- 17.Smith AD, Sumazin P, Zhang MQ. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc. Natl Acad. Sci. USA. 2005;102:1560–1565. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redhead E, Bailey T. Discriminative motif discovery in DNA and protein sequences using the DEME algorithm. BMC Bioinformatics. 2007;8:385. doi: 10.1186/1471-2105-8-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuperlovic-Culf M, Belacel N, Culf AS, Ouellette RJ. Microarray analysis of alternative splicing. OMICS. 2006;10:344–357. doi: 10.1089/omi.2006.10.344. [DOI] [PubMed] [Google Scholar]
- 22.Piva F, Giulietti M, Nocchi L, Principato G. SpliceAid: a database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics. 2009;25:1211–1213. doi: 10.1093/bioinformatics/btp124. [DOI] [PubMed] [Google Scholar]
- 23.Kim E, Goren A, Ast G. Alternative splicing: current perspectives. BioEssays. 2008;30:38–47. doi: 10.1002/bies.20692. [DOI] [PubMed] [Google Scholar]
- 24.Holste D, Ohler U. Strategies for identifying RNA splicing regulatory motifs and predicting alternative splicing events. PLoS Comput. Biol. 2008;4:e21. doi: 10.1371/journal.pcbi.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang H, Wong WH. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25:1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun H, Chasin LA. Multiple splicing defects in an intronic false exon. Mol. Cell. Biol. 2000;20:6414–6425. doi: 10.1128/mcb.20.17.6414-6425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zar JH. Biostatistical Analysis. 4th edn. New Jersey: Prentice Hall; 1998. [Google Scholar]
- 29.Fairbrother WG, Yeo GW, Yeh R, Goldstein P, Mawson M, Sharp PA, Burge CB. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res. 2004;32:W187–W190. doi: 10.1093/nar/gkh393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeo G, Hoon S, Venkatesh B, Burge CB. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc. Natl Acad. Sci. USA. 2004;101:15700–15705. doi: 10.1073/pnas.0404901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum. Mol. Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 32.Kabat JL, Barberan-Soler S, McKenna P, Clawson H, Farrer T, Zahler AM. Intronic alternative splicing regulators identified by comparative genomics in nematodes. PLoS Comput. Biol. 2006;2:e86. doi: 10.1371/journal.pcbi.0020086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabat JL, Barberan-Soler S, Zahler AM. HRP-2, the C. elegans homolog of mammalian heterogeneous nuclear ribonucleoproteins Q and R, is an alternative splicing factor that binds to UCUAUC splicing regulatory elements. J. Biol. Chem. 2009;284:28490–28497. doi: 10.1074/jbc.M109.023101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan RC, Black DL. Conserved intron elements repress splicing of a neuron-specific c-src exon in vitro. Mol. Cell. Biol. 1995;15:6377–6385. doi: 10.1128/mcb.15.11.6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spellman R, Smith CW. Novel modes of splicing repression by PTB. Trends Biochem. Sci. 2006;31:73–76. doi: 10.1016/j.tibs.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 36.Sauliere J, Sureau A, Expert-Bezancon A, Marie J. The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the -tropomyosin Pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol. Cell. Biol. 2006;26:8755–8769. doi: 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minovitsky S, Gee SL, Schokrpur S, Dubchak I, Conboy JG. The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res. 2005;33:714–724. doi: 10.1093/nar/gki210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ponthier JL, Schluepen C, Chen W, Lersch RA, Gee SL, Hou VC, Lo AJ, Short SA, Chasis JA, Winkelmann JC, et al. Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J. Biol. Chem. 2006;281:12468–12474. doi: 10.1074/jbc.M511556200. [DOI] [PubMed] [Google Scholar]
- 39.Zhou HL, Baraniak AP, Lou H. Role for Fox-1/Fox-2 in mediating the neuronal pathway of calcitonin/calcitonin gene-related peptide alternative RNA processing. Mol. Cell. Biol. 2007;27:830–841. doi: 10.1128/MCB.01015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hui J, Hung LH, Heiner M, Schreiner S, Neumuller N, Reither G, Haas SA, Bindereif A. Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. EMBO J. 2005;24:1988–1998. doi: 10.1038/sj.emboj.7600677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zuccato E, Buratti E, Stuani C, Baralle FE, Pagani F. An intronic polypyrimidine-rich element downstream of the donor site modulates cystic fibrosis transmembrane conductance regulator exon 9 alternative splicing. J. Biol. Chem. 2004;279:16980–16988. doi: 10.1074/jbc.M313439200. [DOI] [PubMed] [Google Scholar]
- 42.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, cugbp1, in mice inhibits myogenesis. J. Biol. Chem. 2004;279:13129–13139. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- 43.Sugnet CW, Srinivasan K, Clark TA, O’Brien G, Cline MS, Wang H, Williams A, Kulp D, Blume JE, Haussler D, et al. Unusual intron conservation near tissue-regulated exons found by splicing microarrays. PLoS Comput. Biol. 2006;2:e4. doi: 10.1371/journal.pcbi.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh R, Valcarcel J, Green MR. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 45.Graveley BR, Hertel KJ, Maniatis T. A systematic analysis of the factors that determine the strength of pre-mRNA splicing enhancers. EMBO J. 1998;17:6747–6756. doi: 10.1093/emboj/17.22.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kechris K, Yang YH, Yeh RF. Prediction of alternatively skipped exons and splicing enhancers from exon junction arrays. BMC Genomics. 2008;9:551. doi: 10.1186/1471-2164-9-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuroyanagi H. Fox-1 family of RNA-binding proteins. Cell Mol. Life Sci. 2009;66:3895–3907. doi: 10.1007/s00018-009-0120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL. Homologues of the caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol. Cell. Biol. 2005;25:10005–10016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.