


Abstract
With the aid of next-generation sequencing technology, researchers can now obtain millions of microbial signature sequences for diverse applications ranging from human epidemiological studies to global ocean surveys. The development of advanced computational strategies to maximally extract pertinent information from massive nucleotide data has become a major focus of the bioinformatics community. Here, we describe a novel analytical strategy including discriminant and topology analyses that enables researchers to deeply investigate the hidden world of microbial communities, far beyond basic microbial diversity estimation. We demonstrate the utility of our approach through a computational study performed on a previously published massive human gut 16S rRNA data set. The application of discriminant and topology analyses enabled us to derive quantitative disease-associated microbial signatures and describe microbial community structure in far more detail than previously achievable. Our approach provides rigorous statistical tools for sequence-based studies aimed at elucidating associations between known or unknown organisms and a variety of physiological or environmental conditions.
INTRODUCTION
The biosphere contains an estimated 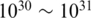 microbial cells, at least
microbial cells, at least 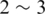 orders of magnitude larger than the number of plant and animal cells combined (1). These microbes play an essential role in processes as diverse as maintenance of human health and biogeochemical activities critical to all life. However, the diversity and the community structure of complex microbial communities are still poorly understood, historically due to our inability to culture most microorganisms using standard microbiological techniques. While there are likely millions of bacterial species, only a few thousand have been formally described to date (2). Accordingly, researchers lack basic information to compare microbial communities under different physical–chemical conditions, and to model dynamic microbe–microbe and environment–microbe interactions.
orders of magnitude larger than the number of plant and animal cells combined (1). These microbes play an essential role in processes as diverse as maintenance of human health and biogeochemical activities critical to all life. However, the diversity and the community structure of complex microbial communities are still poorly understood, historically due to our inability to culture most microorganisms using standard microbiological techniques. While there are likely millions of bacterial species, only a few thousand have been formally described to date (2). Accordingly, researchers lack basic information to compare microbial communities under different physical–chemical conditions, and to model dynamic microbe–microbe and environment–microbe interactions.
The recent development of massively parallel pyrosequencing technology allows researchers to study genetic materials recovered directly from environmental samples, by eliminating the need of laboratory isolation and cultivation of individual species, and thus opens a new window to probe the hidden world of microbial communities (2–4). In recognition of the role of marine microbes in biogeochemical processes, the International Census of Marine Microbes (ICoMM) consortium has launched an international effort to catalog the diversity of microbial populations in the oceanic, coastal and benthic waters. Microbes associated with human health are intensely studied through two large-scale initiatives: the Human Microbiome Project (HMP) sponsored by National Institutes of Health and MetaHIT sponsored by the Europen Union, which seek to establish a correlation between the composition of the human microbiome and various diseases (5). These studies leverage the power of deep sequencing that allows for the rapid and cost-effective surveying of complex microbial communities to reveal the presence of known and currently unknown species alike. However, as emphasized specifically by the NIH HMP working group, computational methods for analyzing massive sequence data generated by these initiatives are still in their infancy, and consequently new computational algorithms and strategies are urgently needed to maximize research yields in these efforts (5).
This article presents a novel computational strategy specifically designed to address the challenges of analyzing large collections of 16S rRNA pyrosequencing data for various biological and ecological inquires. The key idea is to use taxonomy-independent analysis to transform the information encoded in the nucleotide domain into the numerical domain, and then use various advanced machine learning and statistical methods to quantify and visualize the associations between altered microbial community composition with physiological or environmental conditions of interest. We demonstrate the viability of the proposed analytical strategy on a previously published massive human gut 16S rRNA data set generated by Turnbaugh et al. (9) to investigate correlations between the human gut microbiota and obesity. The work by Turnbaugh et al. and other papers mostly by the same group have reported that an obese phenotype is associated with broad, phylum-level changes in the gut community structure (6,7). More specifically, obese individuals appear to have a lower proportion of Bacteroidetes and a higher proportion of Firmicutes compared with lean individuals. This pattern was initially reported only in a small cohort of 12 subjects (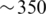 sequences at each sampling point), likely too small to develop a good indicator for the overall population (8). A subsequent study involving a much larger number of samples suggested that it was the ratio between Bacteroidetes and Actinobacteria, not Firmicutes, that differed in the obese group compared with the lean group (9). It is well established that Firmicutes and Bacteroidetes are the two largest phyla in the human gut flora, consisting of over 250 and 125 genera, respectively (10). It is possible that the compositions of only a few genera within these phyla are altered in obesity. Hence, it would be valuable to examine differences in microbial composition at more resolved phylogenetic levels. To this end, we performed a series of data analyses that correlated community structures in the gut with respect to physiological state. Our study showed that while several genus-level operational taxonomic units (OTUs) classified as belonging to Bacteroidetes were all negatively correlated with obesity, there exist both negatively and positively correlated OTUs within Firmicutes, which in part explained some conflicting results observed in previous studies. Through discriminant and topology analyses, we further showed that despite individual diverse gut microbial compositions, common microbial signatures exist that can be used to accurately stratify obese and lean individuals. Our study brought new light onto this human microbiome question that previous methods have been unable to resolve. Our approach is broadly applicable to other sequence-based microbial studies.
sequences at each sampling point), likely too small to develop a good indicator for the overall population (8). A subsequent study involving a much larger number of samples suggested that it was the ratio between Bacteroidetes and Actinobacteria, not Firmicutes, that differed in the obese group compared with the lean group (9). It is well established that Firmicutes and Bacteroidetes are the two largest phyla in the human gut flora, consisting of over 250 and 125 genera, respectively (10). It is possible that the compositions of only a few genera within these phyla are altered in obesity. Hence, it would be valuable to examine differences in microbial composition at more resolved phylogenetic levels. To this end, we performed a series of data analyses that correlated community structures in the gut with respect to physiological state. Our study showed that while several genus-level operational taxonomic units (OTUs) classified as belonging to Bacteroidetes were all negatively correlated with obesity, there exist both negatively and positively correlated OTUs within Firmicutes, which in part explained some conflicting results observed in previous studies. Through discriminant and topology analyses, we further showed that despite individual diverse gut microbial compositions, common microbial signatures exist that can be used to accurately stratify obese and lean individuals. Our study brought new light onto this human microbiome question that previous methods have been unable to resolve. Our approach is broadly applicable to other sequence-based microbial studies.
MATERIALS AND METHODS
Figure 1 presents the schematic diagram of the proposed analytical strategy. We detail each module in the following subsections.
Figure 1.

Schematic diagram of the proposed analytical strategy.
Taxonomy-independent analysis
Providing a detailed description of microbial populations, including high, medium and low abundance components, is frequently the first step to perform in microbial community analysis (10,11). PCR-based techniques for selectively generating 16S rRNA amplicons followed by DNA sequencing are currently the most commonly used approach to characterizing microbial communities, and have been successfully used in numerous applications [see e.g. (12,13) for excellent review]. Existing algorithms for microbial classification using 16S rRNA sequences can be generally categorized into taxonomy-dependent or taxonomy-independent analyses. In the former methods, query sequences are first compared against a database and then assigned to the organism of the best-matched reference sequences (e.g. BLAST). Since most microbes have not been formally described yet, these methods are inherently limited by the lack of completeness of reference databases (12). Taxonomy-dependent analysis is performed generally for the purpose of sequence annotation. In this article, we primarily focus on taxonomy-independent analyses, where sequences are compared against each other to form a distance matrix, based on which hierarchical clustering is performed to group sequences into OTUs of specified sequence variations. Typically, sequences with 1–3% dissimilarity are assigned to the same species, while those with <5% dissimilarity are assigned to the same genus (9,14,15), although these distinctions are controversial. Various ecological metrics can then be estimated from the clustering information to characterize a microbial community. The analysis does not rely on any reference database, and hence is able to enumerate characterized organisms as well as novel pathogenic and uncultured microbes.
We recently developed a new algorithm, referred to as ESPRIT, for large-scale taxonomy-independent analysis (16). The algorithm consists of four modules: (i) filtering out low-quality sequence reads on the basis of multiple criteria, (ii) computing pairwise distances between input sequences, (iii) performing hierarchical clustering to group sequences into OTUs at different distance levels and (4) performing statistical inferences to estimate various ecological metrics. In contrast to many existing 16S rRNA-based studies, ESPRIT uses the Needleman–Wunsch algorithm (17), instead of multiple sequence alignment, to optimally align each pair of 16S rRNA sequences, and the quickdist algorithm (3) to compute pairwise distances. More specifically, each pairwise distance equals mismatches, including indels, divided by a sequence length. To avoid overestimating distances between sequences from rapidly diverging variable regions, end gaps are ignored and gaps of any length are treated as a single evolutionary event or mismatch. Through a benchmark study, we demonstrated that global pairwise alignment provided a much more accurate estimate of microbial richness than multiple sequence alignment. Interested reader may refer to the Supplementary Data for a detailed discussion. Within the ESPRIT framework, we also developed a new clustering algorithm, referred to as Hcluster, to handle large-scale hierarchical clustering analysis. Unlike conventional methods that load a distance matrix directly into memory, Hcluster groups sequences into OTUs on-the-fly while keeping track of linkage information, which overcomes memory limitations. The complete-linkage method was used to ensure that the maximum pairwise genetic distance of the sequences grouped into the same cluster is smaller than the specified distance level defining an OTU. ESPRIT has been used extensively by the research community. Two versions of ESPRIT, one for personal computers and one for computer clusters, are freely available at http://plaza.ufl.edu/sunyijun/ESPRIT.htm.
Constructing profile data matrix
One of the major obstacles of using sequence data to query a biological/ecological hypothesis is that most statistical approaches reported in the literature were designed solely for analyzing numerical-valued data. To overcome this difficulty, we applied taxonomy-independent analysis to transform the information encoded in the nucleotide domain (i.e. A, T, C and G) into the numerical domain. More specifically, we used ESPRIT to hierarchically group sequences into OTUs at various distance levels to form a tree-like structure. Using a barcode labeling system for each sample, the origin of each sequence was retrieved and the number of sequences from each sample within each OTU was counted and recorded in a data matrix. Each column of the data matrix represents a sample, and each row represents an OTU. The data matrix was then normalized along the row direction so that each column vector represents a percentage profile of OTUs in each sample. Analogous to microarray technology that enables researchers to 'simultaneously' monitor the expression levels of all genes in a cell or tissue (18,19), the so-obtained profile data matrix provides microbiologists with a ‘global’ view of how microbial compositions change across individuals or between groups with different physiological states at various phylogenetic levels. Alternatively, a profile data matrix can be generated using taxonomy-dependent analysis. However, a massive amount of query sequences would be grouped into the unknown or uncultured category regardless their origins, and the uncertainties in sequence annotation would propagate to the entire downstream data analyses. Once we obtain a profile data matrix, various advanced computational methods can be applied to analyze massive, high-dimensional data. In this article, we mainly focused on discriminant and topology analyses. In conclusion section, we presented a brief discussion of how to use nucleotide sequence data to infer microbial interaction networks.
Discriminant analysis
The main purpose of discriminant analysis is to identify a list of OTUs containing the most discriminant information that can be used to characterize microbial communities under different conditions. From clinical perspectives, identifying the pathogenic phylotypes stratifying diseased patients from healthy individuals could be used for disease diagnosis and to help physicians make informed decisions to prescribe personalized antibiotics, rather than broad-spectrum antibiotics, to maximize the treatment efficacy (20). Note that the primary goal of the recently launched HMP Project is to determine whether there are associations between changes in the microbiome and various diseases and thus to pave the way for future large-scale human epidemiological studies (5). Discriminant analysis is probably one of the most rigorous analyses one can perform to quantify such associations.
One major characteristic of a profile data matrix is that the number of OTUs is several orders of magnitude larger than the number of samples. For instance, in the case study we present in Result section, at the 0.05 distance level, the number of observed OTUs is 40 765 while there are only 101 samples. In the statistical literature, this is called a `small N and large P' problem (21,22), where N is the number of samples and P is the number of OTUs. In this situation, special care must be taken to avoid overfitting problems. A commonly used practice is to select a small feature subset so that the performance of a learning algorithm is optimized (21–23). For the purpose of this article, we used 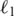 regularized logistical regression to perform feature selection and classification simultaneously (23). Since the objective function optimized by the algorithm is not differentiable, fast implementation of
regularized logistical regression to perform feature selection and classification simultaneously (23). Since the objective function optimized by the algorithm is not differentiable, fast implementation of 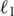 regularized learning has long been considered a challenging problem in the machine-learning community. We recently developed a new gradient descent based algorithm for large-scale
regularized learning has long been considered a challenging problem in the machine-learning community. We recently developed a new gradient descent based algorithm for large-scale 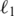 regularized learning (23) (http://plaza.ufl.edu/sunyijun/DGM.htm). The new algorithm makes large-scale studies (e.g. permutation tests) computationally tractable. Due to the small sample size, the leave-one-out cross validation (LOOCV) method was adopted to estimate the prediction performance. In each iteration, one sample was held out for test, and the remaining samples were used for training. The regularization parameter of a logistical regression model was estimated through 10-fold cross validation using the training data, and then a predictive model was trained using the estimated parameter and ‘blindly’ applied to the held-out sample. The experiment was repeated until each sample had been tested. Test samples were not involved in any stage of training process (see Figure 2 for details). A receiver operating characteristic (ROC) curve obtained by varying a decision threshold was then used to visualize how a prediction model performed at different sensitivity and specificity levels. The area under ROC curve (AUC) provides a quantitative assessment of the predictive value of constructed classifiers (AUC = 1: perfect ability to discriminate and AUC = 0.5: random guess) (24).
regularized learning (23) (http://plaza.ufl.edu/sunyijun/DGM.htm). The new algorithm makes large-scale studies (e.g. permutation tests) computationally tractable. Due to the small sample size, the leave-one-out cross validation (LOOCV) method was adopted to estimate the prediction performance. In each iteration, one sample was held out for test, and the remaining samples were used for training. The regularization parameter of a logistical regression model was estimated through 10-fold cross validation using the training data, and then a predictive model was trained using the estimated parameter and ‘blindly’ applied to the held-out sample. The experiment was repeated until each sample had been tested. Test samples were not involved in any stage of training process (see Figure 2 for details). A receiver operating characteristic (ROC) curve obtained by varying a decision threshold was then used to visualize how a prediction model performed at different sensitivity and specificity levels. The area under ROC curve (AUC) provides a quantitative assessment of the predictive value of constructed classifiers (AUC = 1: perfect ability to discriminate and AUC = 0.5: random guess) (24).
Figure 2.

Experimental protocol. Due to the small sample size, the LOOCV method was used to estimate the prediction performance. In each iteration, one sample was held out for test and the remaining samples were used for training. The regularization parameter of a logistical regression model was estimated through 10-fold cross-validation using the training data, and then a predictive model was trained using the estimated parameter and blindly applied to the held-out sample. The experiment was repeated until each sample had been tested.
A typical 16S rRNA-based microbial study involves only tens or at most hundreds of samples. With a small data size, it is possible that the outcomes of discriminant analysis are due to some random confounding factors of no interest to investigators. We performed a permutation test to estimate the P-value of predictive performance. For computational reasons, in this article, the permutation test was repeated 1000 times. In each iteration, the class labels were randomly shuffled, the above-described experimental protocol was executed and the area under the resulting ROC curve was recorded. The P-value was computed as the occurrence frequencies of the iterations where the resulting AUCs outperformed that obtained using the original class labels.
Topology analysis
Topology analysis was preformed that enables microbiologists to visualize and study the global topology structure of a complex microbial community. In this analytical strategy, each sequence is regarded as a data point in a high-dimensional nucleotide space, with each coordinate corresponding to a nucleotide base taking values from set {A, T, C, G}. We used the Isomap algorithm (25) to map sequences into a two-dimensional numerical space that optimally preserves the intrinsic geometry or distribution of the data (i.e. two sequences that have a small genetic distance between each other should stay together in a two-dimensional numerical space). In order to make computation feasible, in this article, we considered only the clusters generated by ESPRIT at the 0.10 distance level, and removed small clusters containing less than 10 sequences. However, we should emphasize that the analysis can be performed at all distance levels. We then randomly selected 100 sequences from each cluster (if a cluster contained less than 100 sequences, all sequences were used.), and computed the pairwise inter-cluster distances as 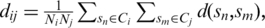 where
where 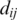 is the distance between clusters
is the distance between clusters 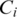 and
and 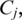
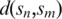 is the pairwise distance between two globally aligned sequences
is the pairwise distance between two globally aligned sequences 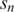 and
and 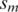 , and
, and 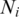 and
and 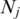 are the numbers of sequences from the two clusters that were used in distance computation. The pairwise inter-cluster distances were then fed into the Isomap algorithm to generate a two-dimensional mapping of massive sequence data. The code is available at http://waldron.stanford.edu/isomap/. The only free parameter of the algorithm is the number of the nearest neighbors used to construct a neighborhood graph, which was set to 10.
are the numbers of sequences from the two clusters that were used in distance computation. The pairwise inter-cluster distances were then fed into the Isomap algorithm to generate a two-dimensional mapping of massive sequence data. The code is available at http://waldron.stanford.edu/isomap/. The only free parameter of the algorithm is the number of the nearest neighbors used to construct a neighborhood graph, which was set to 10.
Sequence annotation
We used the RDP classifier (26) to annotate all the sequences due to its computational efficiency. We also used BLAST search against the RDP-II (27) and greengenes (28) databases to phylogenetically classify the sequences within the top ranked OTUs. A query sequence was assigned to the organism of the best-matched reference sequence if the E-value 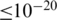 and the identity percentage
and the identity percentage 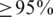 . The analysis was performed on the RAST web application (29). Both the RDP classifier and RAST do not classify sequences below the genus level.
. The analysis was performed on the RAST web application (29). Both the RDP classifier and RAST do not classify sequences below the genus level.
RESULTS
We conducted an intensive computational study on a publicly available human gut microbiota data set to demonstrate the viability of the proposed computational strategy. The data set was originally used to study the connection between obesity and altered composition of the human gut flora (9). It contains 1 119 519 sequences with an average length of 219 nucleotides, covering the V2 hyper-variable region of 16S rRNAs collected from the stool samples of 154 individuals from 54 families. Each sample is labeled as obese, lean or overweight, based on the corresponding body mass index. This is by far the most comprehensive 16S rRNA-based survey of the human gut flora available to date. To reduce random sequencing errors, we applied a trimming procedure similar to those used in (9) to remove reads that (i) contain at least one mismatch in the primers, (ii) contain ambiguous bases or (iii) have a length <200 bp. We performed a taxonomy-independent analysis of the data using the ESPRIT tools described in Taxonomy-independent analysis section, and generated profile data matrices at various distance levels from 0.03 to 0.18 using the approach outlined in constructing profile data matrix section.
Microbial signatures associated with obesity
We first applied unsupervised learning techniques to visualize the distributions of the samples. In order to reduce the effect of confounding factors such as antibiotics usage and sampling depth, we removed the samples that (i) were obtained from the individuals who were on antibiotics within 6 months of stool sample collection, (ii) have less than 3000 sequences and (iii) have ambiguous class labels (i.e. overweight). This resulted in a total of 101 samples with 26 in the lean group and 75 in the obese group. We then performed a correlation analysis of OTUs with respect to physiological state. The heat map of the top 50 ranked OTUs defined at the 0.08 distance level plotted in Figure 3 reveals that obese individuals have a distinguishing pattern of microbial profiles compared with lean individuals. Unsupervised hierarchical clustering clearly partitions the samples into two groups, and this pattern was observed over a wide range of phylogenetic levels (Supplementray Figures S2–S4).
Figure 3.

Heatmap of the top 50 ranked gut microbiota OTUs (rows) defined at the 0.08 distance level. Lean individuals (l) have a distinguishing pattern of microbial composition profiles compared to obese individuals (o). The OTUs were ranked based on their corresponding correlation coefficients with respect to the weight status.
For a more rigorous analysis, we then applied supervised machine-learning techniques to quantify how the predictive value of microbial profiles varies at different phylogenetic levels. We used 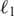 regularized logistical regression to estimate the posteriori probability of a sample belonging to the obese or lean group [see (23) and ‘Materials and Methods’ section for details]. The AUCs obtained at different distance levels ranging from 0.03 to 0.18 are presented in Figure 4 (left panel). We observe that the microbial profile-based predictive models perform very well over a wide range of distance levels. For example, at the 0.08 distance level, the AUC equals 0.88 (P-value <0.001 obtained by a permutation test; Supplementary Figure S5). At the 80% sensitivity level, the model correctly classified 83 out of 101 samples (82%), including 61 obese and 22 lean individuals (Figure 4, right panel). The data set under analysis came from a twin study (9), and it was reported that members within the same family had similar gut microbial community structures. In order to avoid information leakage, we also performed a LOOCV where all the samples from the same family were held out and classified by the predictive model constructed using the samples from `other' families. The classification result had no statistical difference from that obtained using LOOCV (P-value >0.30 based on a Student's t-test; Figure 4, left panel). This experiment demonstrates that despite the fact that each individual has diverse gut microbial compositions (9,10) and that members within the same family have similar overall gut community structures independent of obesity status, there exists a `common microbial signature' that can be used to accurately distinguish obese from lean individuals. Interestingly, the AUC analysis reveals that the discriminant information is contained over a wide range of phylogenetic levels (Figure 4, left panel). This finding extends previous studies by quantifying the association between changes in the microbiome and obesity and pinpointing OTUs that may have a connection with obesity at more resolved phylogenetic levels.
regularized logistical regression to estimate the posteriori probability of a sample belonging to the obese or lean group [see (23) and ‘Materials and Methods’ section for details]. The AUCs obtained at different distance levels ranging from 0.03 to 0.18 are presented in Figure 4 (left panel). We observe that the microbial profile-based predictive models perform very well over a wide range of distance levels. For example, at the 0.08 distance level, the AUC equals 0.88 (P-value <0.001 obtained by a permutation test; Supplementary Figure S5). At the 80% sensitivity level, the model correctly classified 83 out of 101 samples (82%), including 61 obese and 22 lean individuals (Figure 4, right panel). The data set under analysis came from a twin study (9), and it was reported that members within the same family had similar gut microbial community structures. In order to avoid information leakage, we also performed a LOOCV where all the samples from the same family were held out and classified by the predictive model constructed using the samples from `other' families. The classification result had no statistical difference from that obtained using LOOCV (P-value >0.30 based on a Student's t-test; Figure 4, left panel). This experiment demonstrates that despite the fact that each individual has diverse gut microbial compositions (9,10) and that members within the same family have similar overall gut community structures independent of obesity status, there exists a `common microbial signature' that can be used to accurately distinguish obese from lean individuals. Interestingly, the AUC analysis reveals that the discriminant information is contained over a wide range of phylogenetic levels (Figure 4, left panel). This finding extends previous studies by quantifying the association between changes in the microbiome and obesity and pinpointing OTUs that may have a connection with obesity at more resolved phylogenetic levels.
Figure 4.

Results of discriminant analysis. (left panel) The AUC curves obtained at various distance levels ranging from 0.03 to 0.18; (right panel) The ROC curve obtained at the 0.08 distance level. The sensitivity and specificity are defined as the rate of correctly predicting obese and lean individuals, respectively.
It is interesting to note that the AUC versus distance level plot has a bell shape (Figure 4). This makes intuitive sense. When the distance for defining OTUs is large, sequences are grouped into large clusters where discriminant and non-discriminant informations are mixed. On the other hand, when the distance level is small (say 0.03 and 0.05), deep sequencing is required to obtain accurate estimates of microbial composition profiles (3,30). For the gut microbiota data we considered, the average number of sequences in each sample was 7799 with one standard deviation of 5953. This level of coverage may not be sufficient to fully catalog the microbial species resident in the gut, and it is likely that more exhaustive surveys can lead to derivation of a more accurate microbial signature at the genus or even species phylogenetic levels.
Topology structure of human gut microbiota
We next applied topology analysis to the data to visualize the community structure of the human gut microbial community. We used the Isomap algorithm (25) to map the sequences into a two-dimensional numerical space that optimally preserves the geometry of the data (see `Materials and Methods' section for details). Figure 5 presents the output of the analysis. Each circle represents an OTU defined at the 0.10 distance level, and the diameter represents the number of sequences within the OTU divided by the total number of sequences. We used the RDP classifier (26) to annotate the sequences in each cluster. The face color of each circle represents the percentage of the sequences within that cluster that can be annotated by RDP at the genus level with a confidence level >80%. This figure reveals the following points: (i) Bacteroidetes and Firmicutes phyla are the two largest groups within the human gut flora, and a large proportion of sequences (>60%) are unclassifiable at the genus level. These results are consistent with the findings reported in (10). (ii) There are clearly two subgroups within Firmicutes, supporting a recent suggestion that this phylum is likely to be redefined (31).
Figure 5.

Topology analysis performed on the human gut flora. The OTUs were defined at the 0.10 distance level. The face color of each circle represents the percentage of the sequences within an OTU that can be annotated by RDP at the genus level with a confidence level >80%. The circle diameter is proportional to the number of sequences in each OTU divided by the total number of sequences (in log scale).
We next used Isomap to analyze the top ranked OTUs correlated with obesity. Among the 7491 OTUs defined at the 0.10 distance level, only 266 (<3.6%) OTUs had a significant correlation with weight status with a P-value <0.05. The results of topology analysis are presented in Figure 6. Unlike the previous results, the face color of each circle represents the magnitude of the corresponding correlation coefficient with obesity. BLAST search against the RDP-II (27) and greengenes (28) databases was used to phylogenetically classify the 52 227 sequences within the 266 top ranked OTUs (Supplementary Tables S1–S3). For ease of presentation, each cluster was labeled with the name of the phylum it was assigned to. From the analysis, we observed that: (i) The compositions of most OTUs within Bacteroidetes and Firmicutes phyla have little or no correlation with the disease states. (ii) The OTUs within Bacteroidetes tend to have a negative correlation with obesity, which is concordant with previous results, suggesting that obese individuals have a lower proportion of Bacteroidetes in the gut (6,7,9). (iii) As we observed in the total gut topology structure analysis in Figure 5, Firmicutes is partitioned into two subgroups. Interestingly, one subgroup contains more OTUs that have a positive correlation with obesity, while the other group contains more negatively correlated OTUs. This, together with the first observation, may explain why previous studies did not find a significant connection between Firmicutes and obesity since analyses were largely restricted to the phylum level and treated Firmicutes as a single group (9).
Figure 6.

Topology analysis performed on the 266 top ranked OTUs (P-value <0.05) defined at the 0.10 distance level. The face color of each circle represents the magnitude of the corresponding correlation coefficient with obesity.
The full annotation results of the sequences within the 266 top ranked obesity-associated OTUs are reported in Supplementary Tables S1–S3. Notably, as many as 40 000 (>77%) sequences were classified as unknown at the genus level, suggesting that many potentially important gut microbes have yet to be characterized. As this is one of the deepest interrogations of the gut microbiota to date, it is not surprising that there is no prior information available on the association of many OTUs revealed here with obesity or any other human diseases. However, previous reports of phylum level associations and analysis of models using cultivatable species from representative genera provide pointers to potential roles for phylotypes in obesity.
Our analysis revealed that several OTUs classified as belonging to Bacteroidetes were all negatively correlated with obesity (Figure 6). There have been conflicting results with regard to the relationship of Bacteroidetes and obesity in human studies. In a study using FISH probes, Duncan et al. found no relationship between obesity and Bacteroides populations in individuals on controlled weight maintenance diets (32). Zhang et al. also found no difference between the fraction of Bacteroidetes in obese and non-obese individuals in a sequence-based study (14). Conversely, Nadal et al. demonstrated an increase in Bacteroides proportions in adolescents on a weight-loss regimen (33), and studies by Ley et al. proposed that a reciprocal relationship between Bacteroidetes and Firmicutes was evident in obese individuals (7). While the total abundance of microbes within this phylum may not be an accurate biomarker of obesity in itself as shown above, analysis at the genus level may reveal significant associations between specific members of Bacteroidetes and weight status. The two genera from this phylum that were most associated with weight status were Bacterioides and Rikenella (P-value <0.0001). It has been proposed that Bacteroides populations could contribute to the generation of propionate, which may favor a lean phenotype by inhibiting lipid synthesis from acetate (34).
A novel finding derived from applying our new analytical tools is that while some OTUs classified as Firmicutes were correlated positively with obesity, others showed a negatively correlation (Figure 6). The large majority of OTUs in Firmicutes were comprised of the class Clostridia and the order Clostridiales. Notably, unclassified Clostridiales, Clostridiaceae and Lachnospiraceae were the most prevalent components in Clostridiales. The classified genera from this phylum that were most associated with a decrease of abundance in obesity were Megasphaera, Phascolarctobacterium and Erysipelothrix. Megaspheara and Phascolarctobacterium are genera of the Acidaminococcaceae family, anerobic Gram-negative diplococci that use amino acids as their sole energy source. These genera are routinely found in the gut of mammals, but no direct link between energy extraction efficiency or host physiology has been reported to date. The genera from Firmicutes that were increased in obesity included Roseburia, Sporobacter and Faecalibacterium. Faecalibacterium is a major component of the gut flora and members are thought to influence colonic health in a number of ways (35).
CONCLUSIONS
Advances in next-generation DNA sequencing technology allow researchers to obtain millions of DNA sequences rapidly and economically. Consequently, large-scale DNA sequencing is increasingly used as a primary research tool in environmental and human epidemiological studies. Advanced computational algorithms are crucial to efficiently extract pertinent information from massive nucleotide data collections to maximize research yields. While many 16S rRNA-based studies were mainly designed to catalog the diversity of microbial populations (12), we report here a novel analytical strategy that enables researchers to deeply investigate the hidden world of microbes beyond basic microbial diversity estimation. We applied the proposed strategy to derive specific microbial signatures associated with obesity and describe microbial community structures in far more detail than previously achievable. Although we still cannot determine the cause/effect relationship between the human gut microbiota and obesity, we have clearly shown that our approach partially addresses the needs of analyzing the HMP data. Whether the association we identified is direct or indirect is a subject of large-scale population studies, and is outside the scope of this method article. However, the strategy for analyzing the data from population studies largely remains the same.
We herein mainly focused on taxonomy-independent analysis, discriminant analysis and topology analysis. The ultimate goal of a microbial community analysis is to establish a microbial interaction network. Since only a small fraction of microbes can be cultivated in laboratories under current technologies, it would be difficult to use a cultivation-based method to perform such studies. Accordingly, little work has been done in this direction (36). Profile data matrices generated through taxonomy-independent analysis contain sufficient statistical information to study dynamic microbe–microbe and environment–microbe interactions. The results of our ongoing network analyses will be reported elsewhere.
The above bioinformatics analysis can be applied to query multiple research questions. For example, clinical microbiologists may want to derive microbial signatures to characterize microbially caused diseases such as bacterial pneumonia and inflammatory bowel disease; they may also want to perform time series analyses to study how antibiotics usage affects the dynamics of microbial communities over time (4) (in this case, each column of a data profile matrix represents a time point at which a sample is collected.); in our study, we found that Bacteroidetes is the second largest phylum present in the human gut. A recent study showed that in elderly individuals, it is Actinobacteria that is the second most abundant gut phylum (37). It would be interesting to study how microbial composition changes over time by collecting gut samples from individuals of different ages. The above are just a few possible applications. We are currently developing a web application that will provide researchers with a complete package of computational tools for microbial community analysis. We hope that the web application will be of high utility for the microbiology community and beyond.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Conflict of interest statement. None declared.
REFERENCES
- 1.Whitman WB, Coleman DC, Wiebe WJ. Prokaryotes: the unseen majority. Proc. Natl Acad. Sci. USA. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisen JA. Environmental shotgun sequencing: its potential and challenges for studying the hidden world of microbes. PLoS Biol. 2007;5:e82. doi: 10.1371/journal.pbio.0050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. Microbial diversity in the deep sea and the underexplored ``rare biosphere''. Proc. Natl Acad. Sci. USA. 2006;103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc. Natl Acad. Sci. USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 8.Tschöp MH, Hugenholtz P, Karp CL. Getting to the core of the gut microbiome. Nat. Biotechnol. 2009;27:344–346. doi: 10.1038/nbt0409-344. [DOI] [PubMed] [Google Scholar]
- 9.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huse SM, Dethlefsen L, Huber JA, Mark Welch D, Relman DA, Sogin ML. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008;4:e1000255. doi: 10.1371/journal.pgen.1000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabrice A, Didier R. Exploring microbial diversity using 16S rRNA high-throughput methods. J. Comput. Sci. Syst. Biol. 2009;2:74–92. [Google Scholar]
- 13.Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19:1141–1152. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, DiBaise JK, Zuccolo A, Kudrna D, Braidotti M, Yu Y, Parameswaran P, Crowell MD, Wing R, Rittmann BE, et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl Acad. Sci. USA. 2009;106:2365–2370. doi: 10.1073/pnas.0812600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005;71:1501–1506. doi: 10.1128/AEM.71.3.1501-1506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y, Cai Y, Liu L, Yu F, Farrell ML, McKendree W, Farmerie W. ESPRIT: estimating species richness using large collections of 16S rRNA pyrosequences. Nucleic Acids Res. 2009;37:e76. doi: 10.1093/nar/gkp285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 18.van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, Goodison S, Li J, Liu L, Farmerie W. Improved breast cancer prognosis through the combination of clinical and genetic markers. Bioinformatics. 2007;23:30–37. doi: 10.1093/bioinformatics/btl543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarridge JE. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin. Microbiol. Rev. 2004;17:840–862. doi: 10.1128/CMR.17.4.840-862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Y. Iterative RELIEF for feature weighting: algorithms, theories, and applications. IEEE Trans. Pattern Anal. Mach. Intell. 2007;29:1035–1051. doi: 10.1109/TPAMI.2007.1093. [DOI] [PubMed] [Google Scholar]
- 22.Sun Y, Todorovic S, Goodison S. Local learning based feature selection for high dimensional data analysis. IEEE Trans. Pattern Anal. Mach. Intell. 2010;32:1610–1626. doi: 10.1109/TPAMI.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Y, Sun Y, Cheng Y, Li J, Goodison S. Procedings of 10th SIAM International Conference on Data Mining. Columbus, Ohio: 2010. Fast implementation of regularized learning algorithms using gradient descent methods; pp. 862–871. [Google Scholar]
- 24.Duda RO, Hart PE, Stork DG. Pattern Classification. New York: Wiley; 2001. [Google Scholar]
- 25.Tenenbaum JB, de Silva V, Langford JC. A global geometric framework for nonlinear dimensionality reduction. Science. 2000;290:2319–2323. doi: 10.1126/science.290.5500.2319. [DOI] [PubMed] [Google Scholar]
- 26.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole JR, Chai B, Farris RJ, Wang Q, Kulam-Syed-Mohideen AS, McGarrell DM, Bandela AM, Cardenas E, Garrity GM, Tiedje JM. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007;35:D169–D172. doi: 10.1093/nar/gkl889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber JA, Mark Welch DB, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. Microbial population structures in the deep marine biosphere. Science. 2007;318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- 31.Wolf M, Müller T, Dandekar T, Pollack JD. Phylogeny of Firmicutes with special reference to Mycoplasma (Mollicutes) as inferred from phosphoglycerate kinase amino acid sequence data. Int. J. Syst. Evol. Microbiol. 2004;54(Pt 3):871–875. doi: 10.1099/ijs.0.02868-0. [DOI] [PubMed] [Google Scholar]
- 32.Duncan SH, Lobley GE, Holtrop G, Ince J, Johnstone AM, Louis P, Flint HJ. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. (Lond) 2008;32:1720–1724. doi: 10.1038/ijo.2008.155. [DOI] [PubMed] [Google Scholar]
- 33.Nadal I, Santacruz A, Marcos A, Warnberg J, Garagorri M, Moreno LA, Martin-Matillas M, Campoy C, Marti A, Moleres A, et al. Shifts in clostridia, bacteroides and immunoglobulin-coating fecal bacteria associated with weight loss in obese adolescents. Int. J. Obes. (Lond) 2009;33:758–767. doi: 10.1038/ijo.2008.260. [DOI] [PubMed] [Google Scholar]
- 34.Wolever TM, Spadafora PJ, Cunnane SC, Pencharz PB. Propionate inhibits incorporation of colonic [1,2-13C]acetate into plasma lipids in humans. Am. J. Clin. Nutr. 1995;61:1241–1247. doi: 10.1093/ajcn/61.6.1241. [DOI] [PubMed] [Google Scholar]
- 35.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermdez-Humar LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl Acad. Sci. USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuhrman JA. Microbial community structure and its functional implications. Nature. 2009;459:193–199. doi: 10.1038/nature08058. [DOI] [PubMed] [Google Scholar]
- 37.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyren P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.