

Abstract
Here we characterize the consequences of elevated bone morphogenetic protein (BMP) signaling on neural tube morphogenesis by analyzing mice lacking the BMP antagonist, Noggin. Noggin is expressed dorsally in the closing neural folds and ventrally in the notochord and somites. All Noggin−/− pups are born with lumbar spina bifida; depending on genetic background, they may also have exencephaly. The exencephaly is due to a primary failure of neurulation, resulting from a lack of mid/hindbrain dorsolateral hinge point (DLHP) formation. Thus, as previously shown for Shh signaling at spinal levels, BMP activity may inhibit cranial DLHP morphogenesis. However, the increased BMP signaling observed in the Noggin−/− dorsal neural tube is not sufficient to cause exencephaly; it appears to also depend on the action of a genetic modifier, which may act to increase dorsal Shh signaling. The spinal neural tube defect results from a different mechanism: increased BMP signaling in the mesoderm between the limb buds leads to abnormal somite differentiation and axial skeletal malformation. The resulting lack of mechanical support for the neural tube causes spina bifida. We show that this defect is due to elevated BMP4 signaling. Thus, Noggin is required for mammalian neurulation in two contexts, dependent on position along the rostrocaudal axis.
Keywords: BMP, Noggin, Neural tube, Somites, Neurulation, Exencephaly, Spina bifida
Introduction
Neural tube defects (NTDs) are among the most common types of lethal birth defects in the human population, second only to cardiovascular anomalies, occurring in approximately 1 in 1000 births worldwide (Botto et al., 1999). Anencephaly (failure in closure of the brain or skull vault) and spina bifida (closure defects in the spinal cord) are the most commonly occurring human NTDs. A successful approach for reducing the incidence of human NTDs is consumption of vitamin supplements, especially folic acid, by pregnant mothers (MRC Vitamin Study Research Group, 1991); nevertheless, NTDs remain a serious human health problem.
The recent accumulation of mouse mutants with various NTDs has provided insight into the genetic and molecular mechanisms underlying these birth defects (for a review, see Copp et al., 2003; Harris and Juriloff, 1999; Juriloff and Harris, 2000). The benefits of maternal vitamin supplements in reducing NTDs have also been observed in certain mouse models (Greene and Copp, 2005). Thus, the mouse model system provides a useful paradigm for dissecting the cellular and molecular basis of neurulation and its anomalies.
The formation of the mammalian neural tube is an intricate morphogenetic process (reviewed by Colas and Schoenwolf, 2001). The processes of neural induction and early embryonic morphogenesis result in the formation of the neural ectoderm, distinct from the surface ectoderm, overlying the notochord (a derivative of Hensen’s node/Spemann’s organizer). Signals from the notochord, including Sonic hedgehog (Shh), induce a medially located floor plate in the neural ectoderm (Chiang et al., 1996). The cells of the floor plate constrict at their apical ends, forming a medial hinge point for the initial furrowing of the neural tube (the neural groove).
Further morphogenesis to create a closed neural tube occurs differently in different locations along the anterior–posterior (A–P) axis. In the future spinal cord, the neural tube closes dorsally in a “zippering” action, as the dorsal neural folds are brought into juxtaposition. The final step in formation of the closed neural tube is a change in adhesive properties as the cells of the lateral neural ectoderm/dorsal neural tube must release their contact with the adjacent surface ectoderm in favor of the apposing neural ectoderm to form a distinct, closed neural tube. The formation of the cranial neural tube (future brain regions) is mechanistically different from closure of the caudal neural tube. In addition to the ventral medial hinge point, cells in the dorsolateral neural epithelium also form a hinge: the dorsolateral hinge points (DLHPs). The neural epithelium bends around the dorsolateral hinge points to come into direct apposition and form the closed cranial neural tube.
Genetic loss of function studies support a requirement for bone morphogenetic proteins (BMPs) in mouse neurulation (Solloway and Robertson, 1999; Zhang and Bradley, 1996), although their specific roles are unknown. Indeed, BMP signaling has been implicated in multiple experiments and model systems as a key regulator of dorsal neural tube development. Several BMP ligands and targets of BMP signaling are expressed in dorsal neural tissue (Hemmati-Brivanlou and Thomsen, 1995; Kanzler et al., 2000; Monsoro-Burq et al., 1996; Shimeld et al., 1996; Solloway and Robertson, 1999; Takahashi et al., 1996; Wang et al., 1996; Watanabe and Le Douarin, 1996). Furthermore, studies with an antibody against phosphorylated Smad1 protein to mark areas of active BMP signal transduction show significant pSmad1 accumulation in the dorsal neural tube of chicken (Faure et al., 2002) and mouse embryos (R.M. Anderson and J.K., unpublished observations).
The BMP antagonist Noggin (Nog) is expressed in and around the neural tube, and embryos lacking Noggin show elevated BMP signaling in dorsal tissues during neurulation (Anderson et al., submitted for publication), demonstrating that Nog is an important regulator of BMP signaling in dorsal tissues. Nog−/− homozygotes die perinatally with spina bifida and exencephaly, along with severe defects in all skeletal elements (Brunet et al., 1998; McMahon et al., 1998). However, neither the specific nature of these phenotypes nor their developmental basis has been addressed.
Here, we analyze the mechanistic basis for the NTDs in Nog−/− embryos and assess the ability of nutritional supplements to rescue the mutant phenotypes. Our data indicate the exencephaly and spina bifida defects of Noggin mutants arise by distinct cellular and molecular mechanisms. The exencephaly results from a failure of dorsolateral hinge point formation. In contrast, the spinal defects result from failure to maintain a closed neural tube due to defective paraxial mesoderm; these spinal defects are caused by increased BMP4 activity as a consequence of decreased BMP antagonism. Collectively, our studies reveal the roles of BMP antagonism in regulating closure of the neural tube in mammals. They also provide novel insight into the different morphogenetic causes that can underlie spinal and cranial neurulation defects.
Materials and methods
Mouse maintenance and genotyping
A lacZ insertion at the Noggin locus, resulting in a null allele (Nog9E; McMahon et al., 1998), was maintained on both 129Sv/Ev and C57BL/6 defined genetic backgrounds. To improve fecundity, these strains were also intercrossed and outcrossed to ICR (a random outbred line from Harlan). This mixing of the genetic background did not affect the penetrance or incidence of the spinal NTD. Genotyping of Noggin embryos was done by phenotypic analysis and/or PCR genotyping of extra-embryonic tissues, using previously described primers (McMahon et al., 1998). Adults were PCR genotyped from tail biopsies. A Bmp4-lacZ null allele (Lawson et al., 1999), maintained in a random outbred genetic background, was used both for expression analysis and crossed to Nog+/− heterozygotes to create Nog+/−;Bmp4+/− double heterozygotes. These were further intercrossed to generate Nog−/−;Bmp4+/− animals. New primers were designed for PCR genotyping of the Bmp4-lacZ allele (Bmp4-1: TACATCTTGACCGTCGGTTG; Bmp4-2: CTTCCCGGTCTCAGGTATCA). Wild-type alleles are identified by PCR with Bmp4-1 and Bmp4-2 primers (217 bp product at 39 cycles with an annealing temperature of 65°). Bmp4-lacZ alleles give a 450-bp PCR product with the Bmp4-2 and nog-beta;gal primers (McMahon et al., 1998). Bmp4-lacZ embryos were also identified by patterns of X-gal staining. Pax3Sp/J mice were obtained from The Jackson Laboratories (Bar Harbor, ME) and maintained on the C57BL/6J genetic background. Pax3Sp/J heterozygotes were identified by their characteristic “splotch” of unpigmented abdominal coat and homozygous mutants by lumbosacral rachischisis. Embryos were generated by timed matings with mating assumed at midnight and noon the following day designated as embryonic day (E) 0.5. When appropriate, embryos were further staged by somite count or comparison to established references (Downs and Davies, 1993; Kaufman, 1992).
Gene expression assays and histology
Whole-mount in situ hybridization with digoxygenin (Roche) probes followed the protocol of Belo et al. (1997), with a hybridization temperature of 70°. All probes used in this study are published: Gli1 (Hui et al., 1994), Msx1 (Mackenzie et al., 1991), Msx2 (Liu et al., 1994), Msx3 (Shimeld et al., 1996), Netrin (Serafini et al., 1996), Patched (Goodrich et al., 1996), Pax1 (Deutsch et al., 1988), and Sox9 (Wright et al., 1995). LacZ transgenes were visualized by fixing embryos for 10–15 min at RT with 4% paraformaldehyde, rinsed and stained overnight with X-gal stain (Hogan et al., 1994). Histological analysis was conducted on 8 μm paraffin or cryo-embedded sections using established protocols (Hogan et al., 1994).
Midgestational injections of folic acid, myo-inositol, and pifithrin-α
All compounds were injected intraperitoneally to Nog+/− females used for timed matings with Nog+/− males. Injections were performed at E7.5, E8.5, and E9.5. Embryos were analyzed at E10.5 or E11.5. Folate (Sigma) was injected at 10 mg/kg (Fleming and Copp, 1998), myo-inositol (Sigma) at 400 mg/kg (Greene and Copp, 1997), and pifithrin-α (Calbiochem) at 2.2 mg/kg (Pani et al., 2002). Injection volumes ranged from 150 to 700 μl. Control injections of PBS were with a volume of 400 μl.
Scanning electron microscopy
Embryos were fixed in osmium tetraoxide and electron microscopy was done in the facilities of the Duke University Department of Biology.
Cell death analysis
Cell death was detected by incubating freshly dissected embryos in Lysotracker Red (Molecular Probes) diluted 1:500 in lactated ringers solution in the dark at 37° for 30 min. Embryos were rinsed, fixed overnight, and cleared to benzyl alcohol–benzyl benzoate for confocal microscopy on a Leica LSM510 Meta.
Results
Lack of Noggin leads to exencephaly and spina bifida
An important role for Noggin in neural tube development is suggested by its expression pattern. Noggin is highly expressed in the mouse embryo before neurulation in the anterior mesendoderm emerging from the node, which underlies the medial neural plate; along the entire length of the notochord; and in the margins of the elevating dorsal neural folds (Anderson et al., 2002; McMahon et al., 1998; data not shown). From E8.5, as closure begins, Noggin is expressed in both the dorsal neural tube as well as in the notochord along the length of the embryo. At organogenesis stages, Noggin is also expressed in mesodermal skeletal components surrounding the neural tube (data not shown). Thus, Noggin is expressed in multiple domains relevant to neurulation throughout mouse neural tube formation.
Homozygotes for the Noggin targeted null mutation (McMahon et al., 1998) are perinatal lethal and all have spinal neural tube defects, in addition to severe skeletal malformations (Fig. 1; Table 1). The spina bifida resulting from loss of Noggin is completely penetrant in all genetic backgrounds tested, whether inbred, mixed, or outbred. Nog−/− pups show overgrowth and fusion of axial skeletal elements, but caudal to the forelimb, they lack the dorsal neural arches that normally overlie the spinal cord (Figs. 1E and F). This is a spina bifida phenotype that would be classified as rachischisis in humans. Histological analysis reveals that the neural tube in Nog−/− mutants at the level of the forelimb is only modestly affected but has severe defects more caudally (Figs. 1G–L). Between the limbs, there is no neural tube per se but rather a flattened neural mass beneath edematous tissue. At the hindlimb level, the morphology of the neural tube is less severely affected. Whereas the neural arches of the axial skeleton are certainly bifid (split in two), the surface ectoderm remains intact covering the entire spinal column. Therefore, we classify this defect as spina bifida occulta, not the spina bifida aperta more commonly seen in mouse models with an open neural tube at more caudal positions.
Fig. 1.
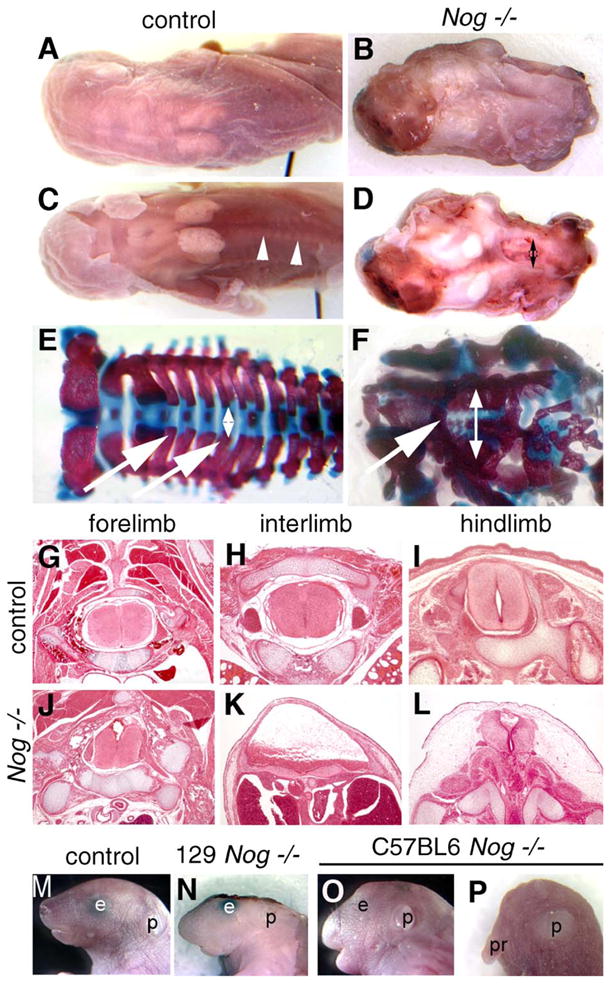
Spina bifida and exencephaly in Nog−/− pups. (A–D) Whole-mount dorsal views of perinatal pups. (A) Wild-type pup Nog+/− intercross, 129/Sv genetic background. (B) Nog homozygous littermate showing exencephaly and spina bifida, covered by skin. (C) Wild-type pup with dorsal skin removed. Arrowheads point out neural arches around spinal cord. (D) Nog homozygote with skin removed, showing open neural tube and gap between neural arches in lumbar region (doubleheaded arrow). (E and F) Skeletal preparations stain bone in red and cartilage in blue. Wild-type pup (E) shows a regular pattern of neural arches (arrows) converging over the neural tube (doubleheaded arrow). (F) Nog−/− pup with dysmorphic neural arches (arrow), and wide distance between spinal sides (doubleheaded arrow). Note general dysplasia of axial skeleton. (G–L) Histological transverse sections of wild-type and Nog late gestation littermates. (G–H) In control fetuses, a closed neural tube and orderly arrangement of skeletal and muscular tissues are seen at all axial levels. (J) Relatively normal neural tube histology in Nog−/− pups at the level of the forelimb. (K) Severe defects in Nog−/− between the limbs, where the neural tissue appears as a thickened ventral plate under a broad lumen overlain by skin and connective tissue: spina bifida occulta. (L) The neural tube at the level of the hindlimb shows less severe defects in the mutant. (M–P) Craniofacial defects in Nog mutants vary with genetic background. Lateral whole-mount views. (M) Wild-type pup. (N) Nog−/− in 129/Sv background with exencephaly. (O) Nog−/− pup in C57BL/6 background with mild facial truncations. (P) Rare Nog−/− pup in C57BL/6 with severe craniofacial truncation. p, pinna (external ear); pr, proboscis.
Table 1.
Penetrance of exencephaly and spina bifida in Nog−/− pups at birth
| Background | Exencephaly | Spina bifida |
|---|---|---|
| 129Sv/Ev, n = 25 | 25 (100%) | 25 (100%) |
| C57Bl6, n = 9 | 0 (0%) | 9 (100%) |
| 129;C57 mixed, n = 17 | 11 (65%) | 17 (100%) |
Nog−/− pups were collected at birth and examined for exencephaly or spina bifida.
The exencephaly defect of Nog−/− pups results in exposure of the midbrain and hindbrain regions directly to the extra-embryonic environment, with no overlying skull or skin tissue (Figs. 1B, D, and N). Exposure of the embryonic brain to extra-embryonic fluid in later stages of human fetal gestation is thought to lead to neural tissue degeneration (Detrait et al., 2005; Greene and Copp, 2005). Such exencephaly in the mouse is thought to correspond to anencephaly in humans, although no rigorous study has been done to substantiate this idea (it is beyond the scope of this manuscript).
In contrast to the spina bifida defects, the exencephaly phenotype of Noggin is dependent on genetic background (Table 1; Figs. 1M–P). For example, there is fully penetrant exencephaly in an inbred 129Sv background, whereas virtually none in a C57Bl/6 background, and a variable phenotype in a mixed or outbred background. This dependence on genetic background suggests the involvement of unidentified genetic modifiers specifically for the exencephaly phenotype. Furthermore, the differential penetrance of the Nog−/− NTDs suggests that the rostral defect (exencephaly) and spinal defect (spina bifida) involve distinct underlying mechanisms.
Known pharmacological suppressors of NTDs do not rescue Nog−/− phenotypes
The spinal and cranial neurulation phenotypes of Nog−/− mutants make this strain another of several mouse genetic models of human neural tube defects. Although the mutant gene responsible is known in most of these models, the ontogeny of the NTDs is unknown in the great majority. A useful means of categorizing mouse NTD models, beyond phenotypic comparisons, is to test their capacity for phenotypic suppression by various agents administered to the pregnant dam (Greene and Copp, 2005). For example, several can be “rescued” by folate injections, others by myo-inositol, although some are not affected by either treatment. Although the molecular mechanisms by which these agents prevent particular NTDs remain unknown, their suppressive effects ultimately reflect similarities and differences among the underlying biochemical defects leading to the NTDs in various mouse mutants.
We have therefore tested the ability of three different pharmacologic agents to rescue the NTDs of Nog−/− pups. The penetrance of NTDs can be reduced (but not eliminated) in several mouse mutants by maternal folate injections, including the Splotch (Pax3) mutant (Epstein et al., 1991). To confirm the efficacy our injection protocol, we injected Splotch heterozygous females crossed to heterozygous males with a folate solution and dissected litters at midgestation. Consistent with previous studies (Fleming and Copp, 1998), this resulted in a 40% decrease in the occurrence of spina bifida in Splotch intercrosses (Table 2). In contrast, examinations of 89 folate-treated embryos from Nog+/− intercrosses did not reveal any significant decrease in the incidence of NTDs. Moreover, all Nog−/− embryos showed NTDs regardless of treatment (7/7 with control PBS injection, 19/19 with folate). We also considered whether Nog−/− might be rescued by myo-inositol injections, as are some other folate-resistant mouse NTD mutants (Greene and Copp, 1997). However, we saw no phenotypic differences in pups from dams injected with myo-inositol (Table 2; 12/12 Nog−/− embryos).
Table 2.
Folate, pifithrin-α, and myo-inositol do not rescue neural tube defects in Nog−/− embryos
| Treatment | Litters | Decidua | Exencephaly | Spinal defect | Resorptions |
|---|---|---|---|---|---|
| Noggin | |||||
| PBS | 3 | 28 | 4 (15%) | 7 (26%) | 1 (4%) |
| Folate | 11 | 89 | 10 (11%) | 19 (21%) | 4 (4%) |
| Pifithrin-α | 11 | 112 | 17 (15%) | 28 (24%) | 4 (4%) |
| Myoinositol | 7 | 54 | 3 (6%) | 12 (22%) | 4 (8%) |
| Splotch | |||||
| PBS | 5 | 34 | 1 (3%) | 8 (24%) | 2 (6%) |
| Folate | 8 | 57 | 4 (7%) | 8 (14%) | 7 (12%) |
| Pifithrin-α | 10 | 75 | 3 (4%) | 7 (9%) | 7 (9%) |
Nog−/− (mixed genetic background) and Splotch−/− embryos were exposed to compounds known to decrease the incidence of some NTD genetic lesions through maternal supplementation. Litters of heterozygous crosses were analyzed for occurrence of NTDs as well number of resorptions. All embryos from noggin crosses were genotyped to assess the penetrance of neural tube defects, regardless of nutritional supplement. Numbers refer to all progeny, 25% of which are homozygous for a given mutation. Chi-square analysis was performed on each treatment. No treatment was significant in the Noggin crosses, whereas for Splotch + folate, P < 0.20 and for Splotch + pifitrin-α, P < 0.05.
Increased cell death is observed in the dorsal neural tube of Nog−/− mutants at E9 and E10 (data not shown; McMahon et al., 1998). Recent work has shown a reduced frequency of spina bifida in Splotch embryos upon maternal injection of pifithrin-α, a cell death inhibitor (Pani et al., 2002). Injections of pifithrin-α did not, however, decrease the incidence of either exencephaly or spinal defects in Noggin mutants (28/28 embryos), although they were indeed able to rescue control Splotch embryos (Table 2). These experiments suggest that known pharmacological suppressors of the NTDs in several mouse models do not rescue Nog NTDs. This in turn implies that the defects caused by a loss of Noggin’s BMP antagonism may disrupt neural tube morphogenesis in ways that are fundamentally distinct from many known NTD lesions.
Exencephaly in Noggin mutants is due to a failure in formation of dorsolateral hinge points
In a 129Sv inbred genetic background, embryos lacking Noggin have an open brain between the diencephalon and myelencephalon, resulting in exencephaly (McMahon et al., 1998; Figs. 1N and 2C). In a C57Bl/6 genetic background, Nog mutants displayed no exencephaly but often showed other craniofacial defects of varying severity (Figs. 1O and P). Embryos containing a mixed C57Bl/6 and 129Sv background, or a random outbred background, showed occasional exencephaly (data not shown).
Fig. 2.
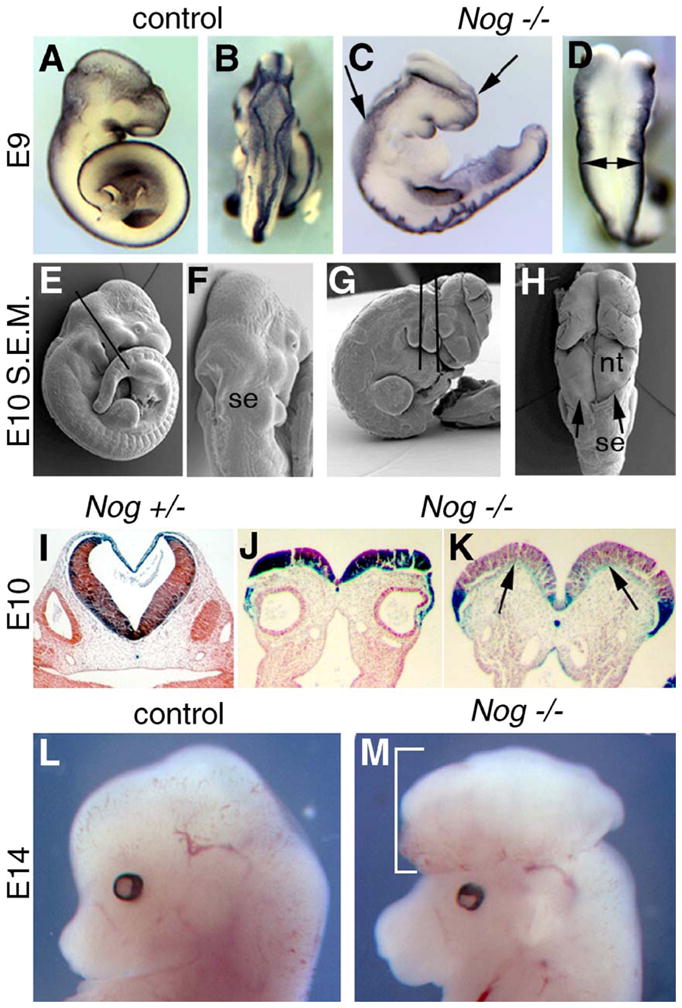
Development of exencephaly in Nog−/− embryos. (A–D) Msx1 expression highlights the dorsal neural tube in control (A and B) and Nog−/− (C and D) embryos at E9. (A) Wild-type embryo, lateral view. (B) Wild-type embryo, dorsal view, showing midbrain, hindbrain, and rostral spinal cord. (C) Nog−/− embryo, lateral view, with exencephaly from caudal forebrain to caudal hindbrain (region between the arrows). Note also the convoluted spinal cord. (D) Nog−/− embryo, dorsal view, midbrain, and hindbrain regions. Note broad distance between the dorsal neural folds (doubleheaded arrow), which should be closed as in the control embryo. (E–H) Scanning electron micrographs (S.E.M.) of E10 littermates. (E) Wild-type embryo, lateral view. Line shows plane of section in panel I. (F) Wild-type embryo, dorsal view, showing closed neural tube covered by surface ectoderm (se). (G) Exencephalic Nog−/− embryo, lateral view, showing planes of section in panels J and K. (H) Dorsal view of Nog−/− mutant, showing surface ectoderm border (arrowheads) and absence over open neural tissue (nt) of hindbrain and midbrain. Note also kinks in neural tissue. (I–K) Frontal sections of E10 littermates from Nog+/− intercross, 129/Sv background, stained for Nog–lacZ activity. (I) Wild-type embryo showing closed neural tube at hindbrain. Lateral lumen is otic vesicle. (J) Mutant showing open neural tube in hindbrain. Note formation of medial hinge point and neural groove. Lumens are otic vesicles. (K) A more rostral section of mutant showing convex curvature of each side of the neural plate due to failure to bend medially at sites where DLHP should form (arrows). (L and M) E14 pups from same cross, lateral views. Further maturation of the open mid/hindbrain results in herniated neural mass in mutant (brackets).
We find that the rostral extent of the open neural tube in the 129Sv background is somewhat variable, but that the midbrain and hindbrain regions are always involved. Defective neurulation is clear by E9.0, when all but the most rostral portion of the cranial neural tube remains splayed open rather than rolled into a closed tube (Figs. 2A–D). Scanning electron microscopy further shows that in contrast to wild type, the open cranial neural tissue is not covered by surface ectoderm (Figs. 2F and H). The mutant neural plate also shows abnormal furrows and kinks, not only in the open cranial portion but also in the closed spinal region (Figs. 2C and H). Histological analysis of Nog−/− mutants reveals that formation of the medial hinge point and neural groove is normal; however, there is a lack of subsequent neural fold bending around the dorsolateral hinge points (DLHP) (Figs. 2I–K). Rather than bending to form concave walls approaching each other at the midline for subsequent fusion, the mutant walls remain relatively flat or even convex. Despite this morphogenetic failure, the neural tissue continues to proliferate in exencephalic Nog−/− embryos, resulting in a large convoluted mass of brain tissue protruding from the head (Figs. 2L and M). This unprotected tissue probably deteriorates perinatally to result in the terminal anencephalic phenotype observed in pups (Figs. 1B and N). Thus, the exencephaly phenotype of Nog pups stems from a failure to form the DLHP of the mid/hindbrain early in neurulation.
Nog shows significant expression in the dorsal neural folds and closed dorsal neural tube and thus is likely acting to attenuate BMP signaling dorsally. We therefore examined the expression of downstream transcriptional targets of BMP signaling in the affected region of the forming neural tube. The transcription factors Msx1 (Mackenzie et al., 1991) and Msx3 (Shimeld et al., 1996) do not show an upregulation in exencephalic tissue (data not shown). However, a third target of BMP signaling, Msx2 (Liu et al., 1994), is expressed at elevated levels in the midbrain and hindbrain of all Nog−/− embryos examined, clearly upregulated as early as E8, before exencephaly is evident (Figs. 3K and M).
Fig. 3.

Ectopic dorsal Shh signaling in exencephalic Nog−/− mutants. (A–I) Whole-mount in situ hybridizations of wild-type (A–C), Nog−/− exencephalic (D–F), and Nog−/− non-exencephalic (G–I) embryos at E9.5. (A, D, and G) Axial Shh expression is not significantly affected in Nog−/− embryos as compared to control embryos (A) regardless of the presence (D) or absence (G) of exencephaly. (B, E, and H) Ptch1 is ectopically expressed around dorsal cranial neural folds in exencephalic (E) but not non-exencephalic (H) mutant embryos. There is some ventral upregulation of Ptch1 in both Nog embryos. Arrows show open neural tube with ectopic expression. (C, F, and I) Gli1 is ectopically expressed in dorsal cranial tissue in exencephalic (F) but not non-exencephalic (I) mutant embryos. (J–M) Msx2 expression (a marker of BMP activity) in E8 and E9 Nog and wild-type embryos. Msx2 is upregulated dorsally in both exencephalic and non-exencephalic mutant embryos at both stages.
An important point is that Msx2 expression is elevated in the mid/hindbrain region of all Nog mutants, regardless of whether they go on to develop exencephaly. Thus, Nog mutants from the 129Sv genetic background as well as other backgrounds show evidence for increased BMP signaling in the dorsal neural folds, although only those of the 129Sv background predictably develop exencephaly. Nonetheless, embryos from Nog+/− inter se crosses in the 129Sv background never show exencephaly unless they are Nog−/− homozygotes. These results indicate that although increased BMP signaling may be necessary for these embryos to develop exencephaly, some other unlinked factor is also involved. Most likely, this is a genetic variant in another gene that alone does not cause exencephaly but can do so in combination with locally decreased BMP antagonism.
Little is known about the molecular mechanisms regulating DLHP formation, although Shh signaling has been shown to inhibit their formation at spinal levels of the neural tube (Ybot-Gonzalez et al., 2002). Although not yet tested, it seems possible that Shh might also inhibit DLHP formation at cranial levels. We therefore assessed the status of Shh signaling in anterior tissues of Nog−/− mutants. Expression of Shh per se showed no difference between wild-type embryos and exencephalic (3/3) or non-exencephalic (3/3) Nog−/− embryos, occurring in the ventral midline in each case (Figs. 3A, D, and G). We then analyzed expression of the direct downstream target gene, Patched-1 (Ptch-1). In all exencephalic Nog−/− mutants examined (6/6), Ptch-1 expression occurred in the dorsal neural folds (Fig. 3E). In contrast, wild-type embryos and Nog−/− mutants with closed cranial neural tubes (2/2) did not show such dorsal Ptch-1 expression (Figs. 3B and H). A separate marker for Shh signaling, the target transcription factor Gli1 (Platt et al., 1997), is also expressed at high levels in the open dorsal neural folds of exencephalic Nog−/− mutants (4/4; Fig. 3F). Nog−/− mutants that did not show signs of exencephaly at E9.0 did not show evidence of increased dorsal Gli1 expression (6/6; Fig. 3I). Thus, there is evidence for ectopic dorsal Shh signal transduction in the mid/hindbrain region of exencephalic Nog−/− mutants, but not in the non-exencephalic mutants.
In sum, the cranial neural tube defect of Noggin mutants results from a failure of DHLP formation in the midbrain region. The morphogenesis defect is brought on by increased BMP signaling in concert with another factor or influence, likely an unlinked modifier allele. This second factor may result in ectopic Shh pathway activation in the dorsal neural folds.
Lumbar neurulation occurs normally but is not maintained in Nog−/− embryos
The spinal NTD of Nog−/− pups occurs at full penetrance in all genetic backgrounds examined (Table 1; data not shown). To clarify the progression of this phenotype, we observed mutant embryos at various stages of neural tube development. Nog−/− spinal cords at E8 and E9 had a “wavy” appearance (Fig. 4B). Surprisingly, however, mutant neural tubes were closed throughout the spinal region (Fig. 4D). Nevertheless, by E10 and E11, Nog−/− mutants show severe disruptions in spinal cord development, with regions where the neural tube appears ruptured (Figs. 4F and H).
Fig. 4.

Spinal neural tube closure occurs in Nog−/− mutants but is not maintained at trunk levels. (A–D) Whole-mount and histological analysis of control (A and C) and Nog−/− (B and D) embryos at E8 (A and B) and E9 (C and D). E8 mutant embryos show characteristic kinking of spinal cord (B), whereas transverse sections of E9 Nog−/− spinal cords (D) show a distinct neural lumen created by closed neural tube. (E–H) Nog+/− (E and G) and Nog−/− (G and H) embryos at E10 (E and F) and E11 (G and H) show the dorsal neural tube beginning to spread apart at multiple sites in the mutants (double headed white arrows; F and H) in the region between the forelimb (f) and hindlimb (h). (I–R) Wild-type (I–M) and Nog mutant (N–R) embryos at E14. Nog−/− mutants show severe neural tube defects caudal to the forelimb and rostral to the hindlimb, with the dorsal neural tube dramatically separated over an enlarged spinal lumen filled with blood (O, arrow). Histological analysis of control embryos (K and L) shows normal neural tube histology (plane of section indicated in panel J). Nog−/− neural tube at the level of the forelimb looks normal (P) but is dysmorphic more caudally (Q). Dorsal neural arches are forming to surround the neural tube in control embryos (arrowhead in panel K). Nog−/− mutants at the level of the forelimb (P) show enlarged axial skeletal elements (arrowhead in panel P), which do not extend as far dorsally (R). At more caudal levels, the Nog−/− axial skeleton is dysplastic with minimal lateral neural arch formation (Q and R). All paired images are shown at identical magnification.
In stillborn Nog−/− pups, the spina bifida is mainly restricted to the lumbar region of the embryo, between the forelimb and hindlimb. The basis for this is clear by E14, when the lumbar spinal region is typically filled with bloody cysts (Fig. 4O) and the spinal cord is widely splayed and dysmorphic (Figs. 4L and Q). The supporting axial skeleton is particularly malformed and deficient in this region (Figs. 4M and R). By contrast, the neural tube remains closed at the forelimb level (Figs. 4K and P). We cannot adequately address neural tube closure at points beyond the hindlimb due to the caudal regression seen in all Nog−/− embryos (McMahon et al., 1998; (Figs. 1, 2, and 4)). These data reveal that the essential role of Noggin in spinal cord morphology is not to facilitate initial neural tube closure, but rather to maintain integrity of the closed neural tube.
Somite differentiation is disrupted in the lumbar region of Nog−/− mutants
Previous work has shown that somitogenesis between the limb buds of Nog−/− embryos is delayed at E9 (McMahon et al., 1998). We continued this analysis at later stages relevant to the onset of the Nog−/− NTD. Expression of Pax1 is a marker for the sclerotomal portion of somites (Wallin et al., 1996) and is required for vertebral development (Wilm et al., 1998). At both E10 and E11, we observed decreased expression of Pax1 in the region between the limbs (Figs. 5E and F). However, expression is detected at levels comparable to wild-type control embryos caudal to the hindlimb bud. This caudal domain is a more recently formed portion of the embryo; therefore, the reduced expression between the limb buds is not simply a delay in onset of expression. Similar results were obtained at E9 with Netrin-1 (Figs. 5I, J, M, and N), a second marker of somitic mesoderm (Serafini et al., 1996), and with Sox9 (Figs. 5C, D, G, and H), a marker of cartilaginous differentiation (Wright et al., 1995). This decrease in expression of paraxial mesoderm genes does not result from increased cell death in this tissue, as we detected no change in levels of apoptosis outside the neural tube, other than in neural crest cells (data not shown; Anderson et al., submitted for publication; McMahon et al., 1998).
Fig. 5.
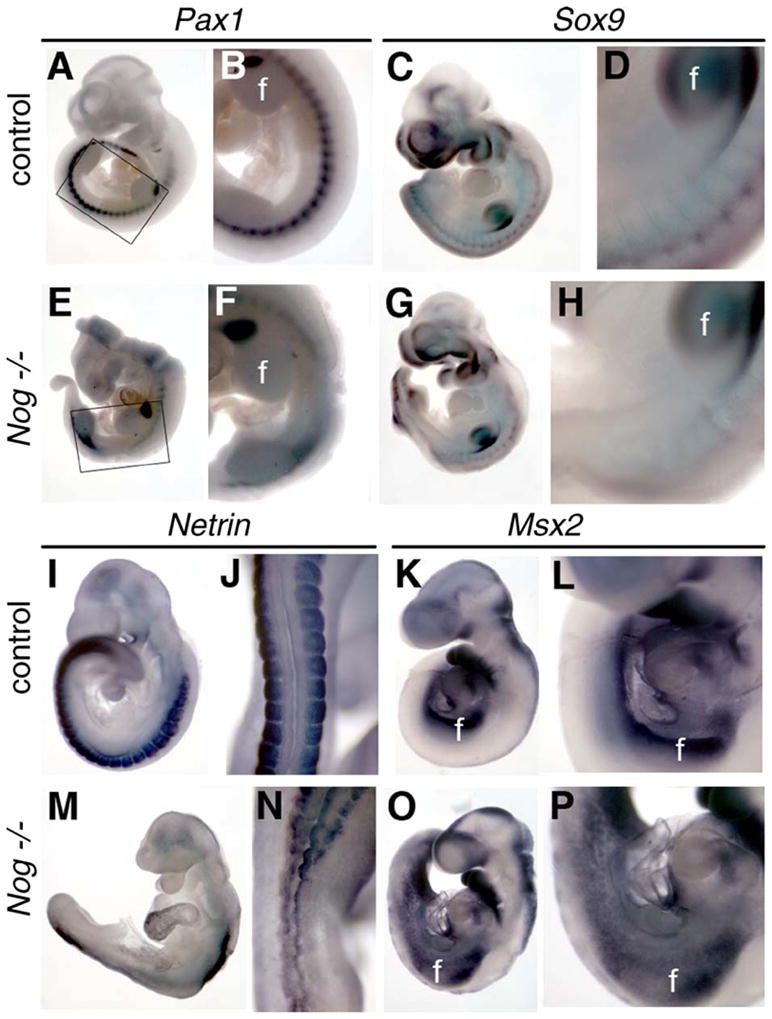
Development of the lumbar sclerotome is disrupted in Nog−/− embryos. Whole-mount in situ hybridization for Pax1 (sclerotome marker; A, B, E, and F), Sox9 (marker of cartilaginous differentiation; C, D, G, and H), and Netrin (somitic mesoderm, I, J, M, and N) was used to characterize somitic development in Nog−/− mutants at E9 and E10. Panels J and N show dorsal views, whereas other views are lateral. Pax1 expression in mutants (E, F) was markedly decreased posterior to the forelimb bud (f) and anterior to the hindlimb bud (h). Boxed region in panels A and E is shown at higher magnification in panels B and F, respectively. Similar high magnification views are shown from the forelimb caudally in each pairwise comparison. Sox9 and Netrin expression were also decreased in mutants between the limb buds (G, H, M, and N). (K, L, O, and P) Msx2 expression at E9 is expanded in Nog−/− mutants (O and P) between the limb buds. All paired images are shown at identical magnification.
Given that Noggin expression extends along the entire A–P axis at relatively uniform levels (McMahon et al., 1998), it seems paradoxical that the most severe spinal neural tube defects are limited to the region between the limb buds. One explanation might be that BMP signaling is perhaps differentially affected in this area by the loss of Noggin. To address this, we assayed the expression of the BMP target gene Msx2. We observed that relative to control embryos, the overall intensity of Msx2 expression between the limb buds was not profoundly affected, but the spatial domain of expression was increased in Nog−/− embryos (Figs. 5K, L, O, and P). Together, these data indicate that BMP antagonism by Noggin is necessary for proper sclerotomal mesoderm development, particularly between the limb buds.
Decreasing Bmp4 dosage in Nog mutants suppresses spinal neural tube defects
The biochemical function of Noggin is to serve as an antagonist of multiple BMPs (Zimmerman et al., 1996), and our evidence for increased BMP signaling in the lumbar region led us to consider whether a particular BMP ligand might be a likely candidate for the BMP most relevant to the Noggin spinal neural tube defects. Neither expression patterns, nor null phenotypes, support Bmp2, Bmp5, or Bmp7 as the likely BMP culprit (Solloway and Robertson, 1999). In contrast, using a lacZ “knock-in” reporter allele to assay BMP4 expression (Lawson et al., 1999), we observed Bmp4 expression at high levels in the mesoderm between the limb buds at E9.5 and E10.5 (Fig. 6; Wijgerde et al., 2005). Therefore, the mesodermal expression of Bmp4 has a spatiotemporal expression domain consistent with its being the BMP ligand related to the lumbar somitogenesis defect in Noggin mutants. Furthermore, BMP4 and Noggin display a strong physical interaction in vitro (Zimmerman et al., 1996).
Fig. 6.
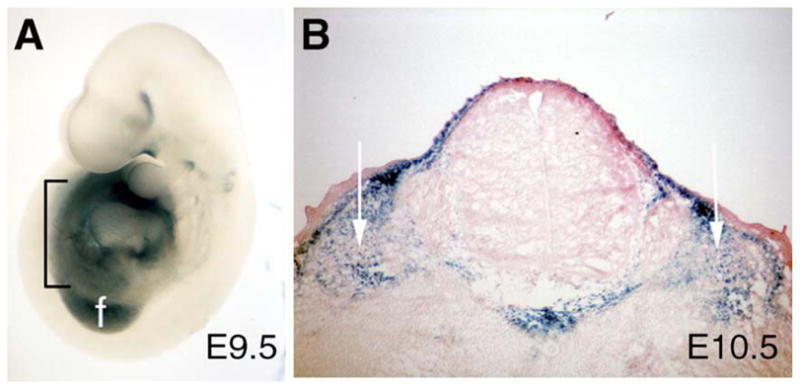
Bmp4-lacZ is expressed in the mesoderm between the limb buds. (A) Lateral view of Bmp4-lacZ embryo at E9.5 shows expression in the mesoderm (indicated by black bracket) between the forelimb bud (f) and hindlimb bud. (B) Transverse section of an E10.5 embryo confirms this expression is in the mesoderm surrounding the neural tube (white arrows).
These considerations led us to hypothesize that the Nog−/− spinal defects result largely from increased activity of BMP4, and to predict that decreasing BMP4 levels might suppress aspects of the Noggin phenotype. Accordingly, we used the Bmp4-lacZ null allele to generate Nog−/−;Bmp4+/− embryos for comparison to Nog−/−;Bmp4+/+ and control embryos. Analysis of offspring from Nog+/− heterozygotes and Bmp4+/− heterozygotes detected no phenotype in Nog+/−;Bmp4+/− double heterozgotes (Table 3).
Table 3.
Survival of embryos generated by Nog+/−;Bmp4+/− intercrosses
| Nog+/+; Bmp4+/− | Nog+/−; Bmp4+/+ | Nog+/−; Bmp4+/− | Nog−/−; Bmp4+/+ | Nog−/−; Bmp4+/− | |
|---|---|---|---|---|---|
| E9–E17, n = 166 | 23 | 20 | 50 | 12 | 30 |
| % Total | 13.9 | 12 | 30 | 7.2 | 18.1 |
| % Expected | 12.5 | 12.5 | 25 | 6.3 | 12.5 |
Progeny of Nog+/−; intercrosses were genotyped for Noggin and Bmp4 alleles. The “expected” percentage refers to the expected ratios predicted by independent segregation of unlinked loci. We saw no reduced representation of any of the genotypes listed here. Note that occurrence of genotypes above “Mendelian” expectation is predictable at these stages, in that Bmp4−/− embryos very rarely survive this late (Winnier et al., 1995); their loss increases the observed frequencies of other genotypes.
Embryos lacking Noggin along with reduced Bmp4 dosage (Nog−/−;Bmp4+/−) display much less severe axial skeleton and neural tube defects than Noggin mutants per se (Figs. 7 and 8). Neural arches are present in these embryos, although still somewhat dysmorphic (Figs. 7C, F, and I). Moreover, lumbar vertebrae are also significantly more developed than in Nog−/−; Bmp4+/+ mutants. At both E10.5 and E11.5, Nog−/−;Bmp4+/− embryos show much less severe NTDs (Figs. 8C and H) than their Nog−/−;Bmp4+/+ littermates (Figs. 8B and G). Histological analysis at E14 further confirms a rescue of the Nog−/− spinal NTD, with a closed neural tube supported by axial skeletal elements (Figs. 8I and J). This phenotypic rescue implies greatly improved sclerotome differentiation in Nog−/−;Bmp4+/− embryos. Indeed, expression of both Netrin and Pax1 indicates that early somite development is largely normal in Nog−/−;Bmp4+/− embryos (Figs. 8K–N, and data not shown). Regarding the exencephaly phenotype, we were unable to accurately assess the consequences of lowering Bmp4 dosage on cranial neurulation due to the dependence of this particular phenotype on a genetic background different from that of our Bmp4 allele. In sum, our data demonstrate that reduction of BMP4 levels suppresses to a large extent the somitic development defects of Noggin mutants, and consequently the axial skeleton deficiency that underlies their spinal neural tube defects.
Fig. 7.

Axial skeletal development is largely rescued with decreased BMP4 dosage in Noggin mutants. Wild-type (A, D, and G), Nog−/−;Bmp4+/+ (B, E, and H), and Nog−/−;Bmp4+/− (C, F, and I) fetuses were collected at E17 and prepared for examination of axial skeletal structures. (A, D, and G) Control fetuses show normal, orderly segmented structure of axial skeleton. (B, E, and H) Nog−/−;Bmp4+/+ fetuses have significant overgrowth and dysplasia of skeletal structures, with loss of dorsal vertebrae in the lumbar region, caudal to the thoracic rib cage. (C, F, and I) Nog−/−;Bmp4+/− animals show well-developed dorsal vertebral structures along the length of the rostrocaudal axis. Dorsal arches present in the Nog−/−;Bmp4+/− embryos (arrows in panels F and I) are larger than control arches (arrow in panels D and E) and also have expanded, intervening cartilaginous elements (double arrowheads in panel F). A higher magnification of the neural arches (D–F) and a lateral view (G–I) are shown for each genotype.
Fig. 8.

Spinal neural tube and somite defects are diminished in Nog−/−;Bmp4+/− embryos. Littermates of the genotypes Nog+/−;Bmp4+/+ (A, D, F, K, and L) Nog−/−; Bmp4+/+ (B and G) and Nog−/−;Bmp4+/− (C, E, H, I, J, M, and N) are shown at E10 (A–E, K–N), E11 (F–H), or E14 (I, J). Nog−/−;Bmp4+/− embryos at E10 show almost no signs of spina bifida (C). Defects are present in Nog−/−;Bmp4+/− embryos at E11 (H) but are much less severe than Nog−/−;Bmp4+/+ (G) embryos. Histological transverse sections also show a closed neural tube at E10 in Nog−/−;Bmp4+/− embryos (E) similar to control embryos (D). At E14, the Nog−/−;Bmp4+/− neural tube is closed at all levels examined and shows few of the neural dysmorphologies seen in similarly staged Nog−/− embryos (compare to Figs. 1K and L), although axial skeletal elements are expanded (I and J, arrows). Pax1 expression in Nog−/−;Bmp4+/− embryos (M and N) is similar to expression in control embryos (K and L) rather than reduced as in Nog−/− mutants (e.g., Figs. 5E and F). f, forelimb bud.
Discussion
In this study, we provide a mechanistic basis for the previous finding that loss of the BMP antagonist Noggin results in neural tube defects (Brunet et al., 1998; McMahon et al., 1998). We find that Noggin promotes cranial and spinal neurulation by distinct mechanisms in different rostral–caudal locations. The spina bifida arises from a failure to maintain a closed neural tube in the lumbar region. This is caused by a defect in sclerotomal differentiation due in turn to increased BMP4 activity. The exencephaly phenotype results from a failure of neurulation in the midbrain and rostral hindbrain region due to a lack of dorsolateral hinge points (DLHPs). This involves not only increased BMP activity in the dorsal neural folds, but also an additional unknown factor, such as a genetic modifier. The second factor, together with increased BMP signaling, may promote ectopic dorsal activation of the Shh pathway—a known inhibitor of DLHP formation at upper spinal levels (Ybot-Gonzalez et al., 2002). In addition to these specific insights about the basis of Noggin phenotypes, this work demonstrates a general principle that different NTDs in a single embryo can arise from fundamentally distinct developmental defects.
Effects of genetic background and pharmacological treatment on Noggin phenotypes
We found that the neural tube defects of Noggin mutants showed markedly different sensitivities to genetic background. The spina bifida defect of Noggin mutants is fully penetrant on the genetic backgrounds we studied, including two inbred strains, as well as hybrid, mixed, and outbred genetic backgrounds. Moreover, although we did not address this issue quantitatively, we saw no evidence that the nature of the spina bifida varied between genetic backgrounds. These considerations suggest that the spinal NTD is a relatively direct consequence of the immediate molecular impact of the Noggin lesion, that is, increased BMP signaling.
In contrast, the exencephaly phenotype of Noggin is profoundly affected by genetic background. We observed fully penetrant exencephaly in a 129/Sv background, but none in a C57BL/6 background. In a mixed background, resulting from breeding of C57;129 F1 hybrids heterozygous for Noggin, we observed about 65% penetrance of exencephaly. This pattern of inheritance suggests that the exencephaly phenotype results from interactions of the Noggin mutant allele with unlinked modifier(s) present in certain genetic backgrounds. For example, this could result from recessive allele(s) present in the 129/Sv background but not in the C57BL/6 background. We further address the basis of the exencephaly phenotype in the next section. This difference in sensitivity of the two Noggin neurulation phenotypes to genetic background is consistent with our finding that these two NTDs occur by distinct mechanisms.
Although Noggin mutants in other backgrounds do not necessarily show exencephaly, their head morphology is not normal. Nog−/− pups show a low-penetrance range of rostral truncations in genetic backgrounds with a BL/6 contribution. The BL/6 background has proven to be susceptible to defects in the anterior of the embryo, and in fact modifiers of anterior defects have been mapped for the Otx2 locus in this background (Hide et al., 2002). Even when heads are neither exencephalic nor truncated, they exhibit craniofacial abnormalities. This is likely to result largely from requirements for Noggin in development of neural crest cells (Anderson et al., submitted for publication), from which much of the skull and face are formed.
Because Noggin NTDs bear resemblance to many other mouse neurulation phenotypes, some of which can be suppressed by maternal vitamin supplements, we expected that the Noggin phenotypes might also be suppressed by such supplements. However, maternal folate injections made no difference for Noggin defects, although they were able to suppress Splotch phenotypes. Because certain mouse NTD models that are resistant to folate can be suppressed by myo-inositol supplements (Greene and Copp, 2005), we tried that treatment as well—again we saw no effect. Although the molecular modes of action by which these vitamins suppress NTDs are unknown, strains that show rescue by a given agent presumably reflect some commonalities in the molecular pathways that underlie their phenotypes. Perhaps, along with other strains resistant to these treatments, the basis of the Noggin NTDs may differ at a biochemical level from the rescuable models.
Maternal injections of pifithrin-α, a cell death inhibitor, reduce the incidence of Splotch NTDs (Pani et al., 2002). Embryos lacking Noggin show increased cell death in the vicinity of the spinal dorsal neural folds as they close (McMahon et al., 1998), primarily in nascent neural crest cells (Anderson et al., submitted for publication). We therefore assessed whether pifithrin-α injections could also suppress Noggin NTDs. However, at least under conditions in which the Splotch phenotype was suppressed, we saw no effect on Noggin mutants. This may indicate that the apoptosis in Noggin involves different molecular regulation than that in Splotch. Alternatively, it is possible that the ectopic cell death observed in Noggin does not contribute substantially to the NTD phenotypes. This seems likely given that the defects stem primarily from hinge-point morphogenesis failures cranially and sclerotomal differentiation problems caudally.
Despite the differences between Noggin and Splotch mutants with respect to suppression by folate and pifithrin-α, there is at least one possible molecular effector downstream of both the Noggin and Splotch lesions. We observed that of three BMP transcriptional targets assessed, Msx2 was upregulated in dorsal cranial tissues and in the interlimb mesoderm in the Noggin embryos, the tissues associated with the exencephaly and spina bifida phenotypes. Msx2 has been shown to be upregulated in Pax3 (Splotch) mutants, which also have spina bifida and exencephaly (Kwang et al., 2002). It appears that Msx2 expression is normally repressed by Pax3, and therefore in the Pax3Sp/Sp mutants, increased Msx2 activity potentially contributes to the NTDs. A human MSX2 transgene expressed in mice also results in exencephaly, further demonstrating a positive correlation between activity of this transcription factor and NTDs (Winograd et al., 1997). It remains to be determined if reduced Msx2 expression on a Noggin background has any effect on NTDs. Is Msx2 downstream of both Pax3 and BMP signaling independently, or do the pathways interact with each other to affect Msx2 levels? To address this, we generated embryos mutant for both Pax3 and Noggin but saw no altered NTD penetrance in any mutant class (our unpublished results). Thus, we conclude that Pax3 and BMP signaling show no genetic interaction, but rather each affects Msx2 expression in an independent manner.
Noggin exencephaly arises from a failure to form dorsolateral hinge points
In Noggin embryos destined to develop exencephaly, histological analysis shows a failure in proper formation of DLHPs. BMP activity is increased in the right spatiotemporal context to be involved, as shown by the increased expression of the direct target, Msx2. However, this increase occurs also in mutant embryos that do form DLHPs and do not develop exencephaly. This indicates that it is not increased BMP activity alone that causes the failure of DLHP formation in the midbrain region.
Further molecular analysis of the future exencephalic embryos shows ectopic dorsal expression of direct Shh signaling targets, Ptch-1 and Gli-1, but not of Shh itself. Previous work has shown that Shh signaling activity inhibits formation of DLHPs in the upper spinal region (Ybot-Gonzalez et al., 2002). Mice mutant for Ptch-1 also have cranial and spinal neural tube defects (Goodrich et al., 1997; Milenkovic et al., 1999). Presumably, this is due to an overactive Shh pathway in the absence of Ptch-1, which ultimately acts as a repressor of the pathway (McMahon et al., 2003). It seems at first paradoxical that increased BMP signaling would coincide with increased Shh signaling in the cranial region of exencephalic Nog embryos because these two pathways can act antagonistically in dorso-ventral (D-V) patterning of the spinal cord (Patten and Placzek, 2002). Perhaps this simultaneous activation of signaling pathways that do not normally coincide in this developmental field leads to the abnormal regulatory events that disrupt morphogenesis in these mutants. On the other hand, there are many context-dependent cases of apparent synergy between BMP and Shh signaling in normal development, even in D-V patterning of the forebrain (Dale et al., 1999). To that end, Nog−/− embryos show increased Ptch-1 expression in the ventral neural tube at all axial levels (McMahon et al., 1998), further suggesting that loss of Noggin can indeed result in increased Shh signaling in some situations.
Our results lead us to propose the following model for the role of Noggin in dorsal development of the mid- and hindbrain region (Fig. 9A). Normally, Noggin keeps dorsal BMP signaling in check to ensure proper development of the neural crest and appropriate D-V patterning of the neural tube. The absence of Noggin protein in mutant embryos leads to locally increased BMP signaling in and around the dorsal neural folds. This alone does not lead to exencephaly. In some genetic backgrounds (e.g., 129/Sv); however, increased BMP signaling creates a permissive environment that sensitizes the tissue to an unknown influence, most likely a genetic modifier, that increases Shh activity dorsally. Such a modifier might be a hypomorphic, recessive allele of a regionally restricted repressor of Shh signaling—insufficient on its own to disrupt neurulation, but together with increased BMP signaling, able to activate the Shh cascade. This ectopic Shh signaling may result in an inhibition of DLHP formation as occurs more caudally (Ybot-Gonzalez et al., 2002), and consequently a morphogenetic failure of neural tube closure. We note, however, that there is not yet direct experimental data demonstrating that Shh can inhibit cranial DLHP formation. Furthermore, our data do not exclude the alternative hypothesis that the increased Shh signaling we observe is directly due to the exencephaly, as opposed to a molecular event that leads directly to the exencephaly. In any case, our Msx2 expression data indicate that although increased BMP signaling is detectable before overt signs of exencephaly are present, this increase in BMP signaling per se is necessary but not sufficient to cause DLHP failure; in addition, some unknown background-dependent influence is also required.
Fig. 9.
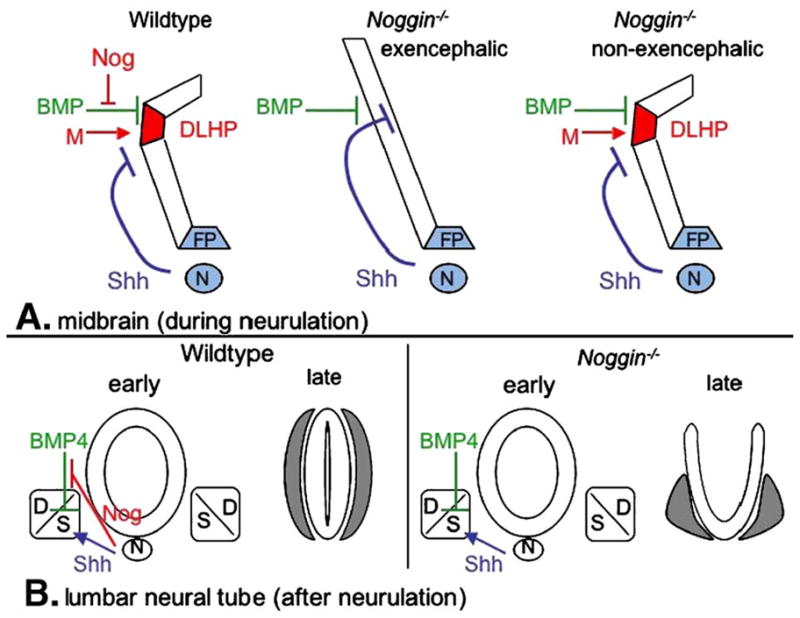
Models for the functions of Noggin in cranial and spinal neural tube morphology. (A) During midbrain neurulation, dorsolateral BMP appears to have an inhibitory effect on dorsolateral hinge point (DLHP) formation. Noggin (Nog) promotes DLHP formation by antagonizing this inhibitory effect of BMP. The source of this Noggin activity is likely the dorsal neural folds but could also include ventral domains. Other factors are also involved in promoting DLHP formation (such as a potential Noggin modifier, M). Shh activity produced by the floorplate (FP) and the notochord (N) may also have an inhibitory effect on DLHP formation, as occurs more caudally. In exencephalic Noggin embryos, the positively acting Nog and genetic modifier influences are deficient, causing DLHP formation to be inhibited by BMP and potentially by Shh as well. The presence of ectopic Shh signaling in the dorsal neural folds of the exencephalic midbrain region raise the possibility that the modifier acts to repress dorsal Shh signal transduction in the midbrain neural folds. In other genetic backgrounds in which the modifier is unaffected, DLHPs form even in the absence of Noggin. As a result, such embryos are non-exencephalic. In these embryos, Shh signaling is limited to its normal ventral extent. (B) At lumbar levels, subsequent to neural tube closure, Noggin antagonizes the inhibitory effects of BMP4 on sclerotomal (s) development. In contrast, sclerotome development is promoted by Shh. The result by late gestation is an encasement of the closed neural tube by the axial skeleton, including the neural arches (gray) that derive from the sclerotome. Resulting sclerotomal proliferation and differentiation lead to the flanking of the closed neural tube by the neural arches (gray). In Noggin mutants, reduced BMP antagonism leads to increased BMP4 activity. This disrupts sclerotome development, ultimately leading to dysmorphic neural arches that fail to support the closed neural tube. Other defects, such as increased dorsal cell death or altered cell adhesion, may also contribute to the spinal NTD. D, dermamyotome.
Noggin spina bifida results from mesodermal defects caused by elevated BMP signaling
Spinal neurulation occurs normally in Nog−/− embryos, but subsequent failures in somite development lead to the lumbar spina bifida observed at birth. This NTD is completely penetrant, regardless of genetic background, suggesting that it results directly from the molecular consequences of Noggin’s absence, expected to be increased BMP ligand availability. Although we have found another axial BMP antagonist, Chordin, to be redundant with Noggin in several contexts (Anderson et al., 2002; Bachiller et al., 2000; Stottmann et al., 2001), there appears to be no redundancy of BMP antagonists in controlling lumbar spinal cord morphogenesis. Thus, we observed increased expression of the BMP target Msx2 in Nog mutants in the region between the limb buds, corresponding to the lumbar region where the spina bifida later occurs. Based on expression pattern, we hypothesized that mesodermal BMP4 is the relevant ligand that is increased. By genetically reducing the dosage of BMP4 signaling in a Nog−/− background, we observed that the axial spine and spinal cord defects were largely rescued. This demonstrates that it is indeed elevated BMP signaling that causes these defects. Although this rescue does not necessarily indicate that elevated BMP4 is solely responsible, it does reveal that any relevant BMPs are among the subgroup with which BMP4 is biochemically redundant (e.g., BMP2).
These considerations lead us to suggest a model for the etiology of the spina bifida defect in Noggin embryos (Fig. 9B). In wild-type embryos, Noggin acts to limit the effects of BMP4 (such as that from lateral plate mesoderm) that normally acts to promote development of the dermamyotome at the expense of more medial sclerotomal tissue (Christ et al., 2004). This together with Shh and perhaps other ventral signals direct proper development of the neural arches and other elements of the axial skeleton that support the close neural tube. In the absence of Noggin, the neural tube still closes correctly at spinal levels. However, enhanced BMP4 activity in the interlimb region penetrates more medially to inhibit medioventral sclerotomal differentiation, in part by reducing expression of Pax1. Reduced Pax1 in turn is associated with reduced proliferation (Wilting et al., 1995). This combines with the effects of abnormal levels of BMP signaling to disrupt the proper patterning and growth of the axial skeletal elements that normally support the neural tube. The lack of structural support for the neural tube results in a failure to maintain the integrity of the lumbar spinal cord. This defect is likely compounded by the patches of increased cell death that occur along the dorsal midline of Noggin mutants (McMahon et al., 1998; Anderson et al., submitted for publication). We cannot exclude that Noggin might also have additional roles that promote the integrity of a closed neural tube, such as effecting cell adhesion between the apposed neural folds. The net result of the loss of Noggin is spina bifida occulta in the lumbar spinal region. Currently, we are unable to address which expression domain(s) of Noggin, either dorsal neural tube or mesodermal, are necessary for BMP antagonism and normal neural tube development. The use of a conditional Noggin allele would allow a further genetic analysis of this process.
A recent study (Wijgerde et al., 2005) has confirmed our findings of rescued axial development in Nog−/−;Bmp4+/− as compared to Nog−/− embryos. This work also confirms our findings that reducing Bmp4 dosage results in improved somitic development and patterning but does not address any effects on neural tube development. Additional support for this model comes from work in the chick embryo in which implants of human BMP2 secreting cells lateral to the notochord resulted in severe disruption in somite development and loss of vertebral elements (Monsoro-Burq et al., 1996). Furthermore, lateral implantation of BMP in the mesoderm led to both a decrease in Pax1 and an increase in Msx2, molecular results mirroring our findings in the Nog−/− mutant. This addition of hBMP2, however, did not lead to spina bifida occulta. The observed effects were constrained to only two to four vertebral elements whereas the Nog−/− phenotype is much more extensive, presumably due to expression of Noggin and interaction with BMPs all along the A/P axis. Thus, a discrete disruption of vertebral elements such as that seen in Monsoro-Burq et al. (1996) may be compensated by other unaffected axial tissue, whereas the more extensive Nog−/− defect results in spina bifida.
Other data also indicates that a neural-specific defect may not be the root cause of the spina bifida in Nog−/− embryos. Transgenic overexpression of a constitutively active BMPRIA (caBMPRIA) in the neural epithelium at E10.5 in the mouse results in a broadened dorsal midline (Panchision et al., 2001). Loss of Noggin presumably results in elevated BMP2/4 signaling in a manner similar to a constitutively active form of BMPRIA. Despite this biochemical similarity, the caBM-PRIA embryos were not observed to have spina bifida when analyzed at later stages of development. This suggests that an elevated BMP signal in only the neural tissue is not sufficient to cause spina bifida, supporting our model of a lack of mesodermal support leading to a structural failure in neural tube development. There is at least one other mouse mutant to date showing spina bifida known to result from a defect in the mesoderm surrounding the neural tube. The spontaneous mutation, Patch, has a deletion containing the Pdgfrα gene (Joosten et al., 1998) and Patch mutants similarly have spina bifida occulta attributed to a mesodermal defect (Payne et al., 1997).
Noggin and human neural tube defects
The NTD phenotypes we observe in Noggin mutants are similar to human anencephaly and rachischisis. Is NOGGIN a candidate locus for human NTDs? Two studies have already addressed this question and have not found a molecular lesion segregating strictly with the NTD (Bauer et al., 2002; Felder et al., 2002). In the former case, a NOGGIN mutation was identified in a patient with an NTD but was also detected in an unaffected parent. This could mean that the mutation was irrelevant but is also consistent with the possibility that NOGGIN mutation causes an NTD phenotype of incomplete penetrance. The general conclusion is that although these mutations do not alone result in spina bifida cases, they may contribute to the apparently polygenic nature of the defects. Whereas a homozygous NOGGIN mutation has not been found in patients with NTDs, these fetuses likely will not be represented in a clinical population but more likely in will result in terminated pregnancies, either natural or elective. Thus, screening aborted fetuses with NTDs would be a more appropriate way to determine the relevance of human NOGGIN mutations to neural tube malformations. Although much remains to be resolved about the causation of neural tube defects in both humans and mice, our results implicate regulation of BMP signaling by Noggin as a relevant component of the underlying mechanisms.
Acknowledgments
We thank our laboratory colleagues for helpful discussions and comments on the manuscript, as well as C. Williams and J. Goodwin for help with mouse husbandry and genotyping. We are grateful to B. Hogan for sharing her Bmp4-lacZ mouse line and to A. Joyner, A. McMahon, M. Scott, M. Tessier-Lavigne, and P. Gruss for plasmids. We appreciate the help of K. Smith and her colleagues with SEM imaging. RWS was supported by the Duke Neonatal-Perinatal Research Institute and by an NRSA fellowship from NINDS. This research was supported by NIH awards to JK (R01DE13674 and P01HD39948).
References
- Anderson RM, Lawrence AR, Stottmann RW, Bachiller D, Klingensmith J. Chordin and noggin promote organizing centers of forebrain development in the mouse. Development. 2002;129:4975–4987. doi: 10.1242/dev.129.21.4975. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Stottmann RW, Choi M, Klingensmith J. Endogenous BMP antagonists regulate mammalian neural crest generation and survival. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachiller D, Klingensmith J, Kemp C, Belo J, Anderson RM, May SR, McMahon J, McMahon A, Harland R, Rossant J, et al. The organizer factors Chordin and Noggin are required for mouse forebrain development. Nature. 2000;403:658–661. doi: 10.1038/35001072. [DOI] [PubMed] [Google Scholar]
- Bauer KA, George TM, Enterline DS, Stottmann RW, Melvin EC, Siegel D, Samal S, Hauser MA, Klingensmith J, Nye JS, et al. A novel mutation in the gene encoding noggin is not causative in human neural tube defects. J Neurogenet. 2002;16:65–71. [PubMed] [Google Scholar]
- Belo JA, Bouwmeester T, Leyns L, Kertesz N, Gallo M, Follettie M, De Robertis EM. Cerberus-like is a secreted factor with neutralizing activity expressed in the anterior primitive endoderm of the mouse gastrula. Mech Dev. 1997;68:45–57. doi: 10.1016/s0925-4773(97)00125-1. [DOI] [PubMed] [Google Scholar]
- Botto LD, Moore CA, Khoury MJ, Erickson JD. Neural-tube defects. N Engl J Med. 1999;341:1509–1519. doi: 10.1056/NEJM199911113412006. [DOI] [PubMed] [Google Scholar]
- Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton. Science. 1998;280:1455–1457. doi: 10.1126/science.280.5368.1455. [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Yong KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Christ B, Huang R, Scaal M. Formation and differentiation of the avian sclerotome. Anat Embryol (Berl) 2004;208:333–350. doi: 10.1007/s00429-004-0408-z. [DOI] [PubMed] [Google Scholar]
- Colas JF, Schoenwolf GC. Towards a cellular and molecular understanding of neurulation. Dev Dyn. 2001;221:117–145. doi: 10.1002/dvdy.1144. [DOI] [PubMed] [Google Scholar]
- Copp AJ, Greene ND, Murdoch JN. The genetic basis of mammalian neurulation. Nat Rev, Genet. 2003;4:784–793. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- Dale K, Sattar N, Heemskerk J, Clarke JD, Placzek M, Dodd J. Differential patterning of ventral midline cells by axial mesoderm is regulated by BMP7 and chordin. Development. 1999;126:397–408. doi: 10.1242/dev.126.2.397. [DOI] [PubMed] [Google Scholar]
- Detrait ER, George TM, Etchevers HC, Gilbert JR, Vekemans M, Speer MC. Human neural tube defects: developmental biology, epidemiology, and genetics. Neurotoxicol Teratol. 2005;27:515–524. doi: 10.1016/j.ntt.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch U, Dressler GR, Gruss P. Pax 1, a member of a paired box homologous murine gene family, is expressed in segmented structures during development. Cell. 1988;53:617–625. doi: 10.1016/0092-8674(88)90577-6. [DOI] [PubMed] [Google Scholar]
- Downs KM, Davies T. Staging of gastrulating mouse embryos by morphological landmarks in the dissecting microscope. Development. 1993;118:1255–1266. doi: 10.1242/dev.118.4.1255. [DOI] [PubMed] [Google Scholar]
- Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell. 1991;67:767–774. doi: 10.1016/0092-8674(91)90071-6. [DOI] [PubMed] [Google Scholar]
- Faure S, de Santa Barbara P, Roberts DJ, Whitman M. Endogenous patterns of BMP signaling during early chick development. Dev Biol. 2002;244:44–65. doi: 10.1006/dbio.2002.0579. [DOI] [PubMed] [Google Scholar]
- Felder B, Stegmann K, Schultealbert A, Geller F, Strehl E, Ermert A, Koch MC. Evaluation of BMP4 and its specific inhibitor NOG as candidates in human neural tube defects (NTDs) Eur J Hum Genet. 2002;10:753–756. doi: 10.1038/sj.ejhg.5200875. [DOI] [PubMed] [Google Scholar]
- Fleming A, Copp AJ. Embryonic folate metabolism and mouse neural tube defects. Science. 1998;280:2107–2109. doi: 10.1126/science.280.5372.2107. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Greene ND, Copp AJ. Inositol prevents folate-resistant neural tube defects in the mouse. Nat Med. 1997;3:60–66. doi: 10.1038/nm0197-60. [DOI] [PubMed] [Google Scholar]
- Greene ND, Copp AJ. Mouse models of neural tube defects: investigating preventive mechanisms. Am J Med Genet C Semin Med Genet. 2005;135:31–41. doi: 10.1002/ajmg.c.30051. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. Mini-review: toward understanding mechanisms of genetic neural tube defects in mice. Teratology. 1999;60:292–305. doi: 10.1002/(SICI)1096-9926(199911)60:5<292::AID-TERA10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Hemmati-Brivanlou A, Thomsen GH. Ventral mesodermal patterning in Xenopus embryos: expression patterns and activities of BMP-2 and BMP-4. Dev Genet. 1995;17:78–89. doi: 10.1002/dvg.1020170109. [DOI] [PubMed] [Google Scholar]
- Hide T, Hatakeyama J, Kimura-Yoshida C, Tian E, Takeda N, Ushio Y, Shiroishi T, Aizawa S, Matsuo I. Genetic modifiers of otocephalic phenotypes in Otx2 heterozygous mutant mice. Development. 2002;129:4347–4357. doi: 10.1242/dev.129.18.4347. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo. Cold Spring Harbor Press; New York: 1994. [Google Scholar]
- Hui CC, Slusarski D, Platt KA, Holmgren R, Joyner AL. Expression of three mouse homologs of the Drosophila segment polarity gene cubitus interruptus, Gli, Gli-2, and Gli-3, in ectoderm- and mesoderm-derived tissues suggests multiple roles during postimplantation development. Dev Biol. 1994;162:402–413. doi: 10.1006/dbio.1994.1097. [DOI] [PubMed] [Google Scholar]
- Joosten PH, Hol FA, van Beersum SE, Peters H, Hamel BC, Afink GB, van Zoelen EJ, Mariman EC. Altered regulation of platelet-derived growth factor receptor-alpha gene-transcription in vitro by spina bifida-associated mutant Pax1 proteins. Proc Natl Acad Sci U S A. 1998;95:14459–14463. doi: 10.1073/pnas.95.24.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. Mouse models for neural tube closure defects. Hum Mol Genet. 2000;9:993–1000. doi: 10.1093/hmg/9.6.993. [DOI] [PubMed] [Google Scholar]
- Kanzler B, Foreman RK, Labosky PA, Mallo M. BMP signaling is essential for development of skeletogenic and neurogenic cranial neural crest. Development. 2000;127:1095–1104. doi: 10.1242/dev.127.5.1095. [DOI] [PubMed] [Google Scholar]
- Kaufman MH. The Atlas of Mouse Development. Academic Press, Inc; San Diego: 1992. [Google Scholar]
- Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, Ishii M, Sangiorgi FO, Rauchman M, Sucov HM, et al. Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129:527–538. doi: 10.1242/dev.129.2.527. [DOI] [PubMed] [Google Scholar]
- Lawson KA, Dunn NR, Roelen BA, Zeinstra LM, Davis AM, Wright CV, Korving JP, Hogan BL. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13:424–436. doi: 10.1101/gad.13.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, Ma L, Wu LY, Luo W, Kundu R, Sangiorgi F, Snead ML, Maxson R. Regulation of the Msx2 homeobox gene during mouse embryogenesis: a transgene with 439 bp of 5′ flanking sequence is expressed exclusively in the apical ectodermal ridge of the developing limb. Mech Dev. 1994;48:187–197. doi: 10.1016/0925-4773(94)90059-0. [DOI] [PubMed] [Google Scholar]
- Mackenzie A, Ferguson MWJ, Sharpe PT. Hox-7 expression during murine craniofacial development. Development. 1991;113:601–611. doi: 10.1242/dev.113.2.601. [DOI] [PubMed] [Google Scholar]
- McMahon JA, Takada S, Zimmerman LB, Fan CM, Harland RM, McMahon AP. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube somite. Genes Dev. 1998;12:1438–1452. doi: 10.1101/gad.12.10.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- Milenkovic L, Goodrich LV, Higgins KM, Scott MP. Mouse patched1 controls body size determination and limb patterning. Development. 1999;126:4431–4440. doi: 10.1242/dev.126.20.4431. [DOI] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Duprez D, Watanabe Y, Bontoux M, Vincent C, Brickell P, Le Douarin N. The role of bone morphogenetic proteins in vertebral development. Development. 1996;122:3607–3616. doi: 10.1242/dev.122.11.3607. [DOI] [PubMed] [Google Scholar]
- MRC Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338:131–137. [PubMed] [Google Scholar]
- Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, McKay RD. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev. 2001;15:2094–2110. doi: 10.1101/gad.894701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorigenesis. Genes Dev. 2002;16:676–680. doi: 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten I, Placzek M. Opponent activities of Shh and BMP signaling during floor plate induction in vivo. Curr Biol. 2002;12:47–52. doi: 10.1016/s0960-9822(01)00631-5. [DOI] [PubMed] [Google Scholar]
- Payne J, Shibasaki F, Mercola M. Spina bifida occulta in homozygous Patch mouse embryos. Dev Dyn. 1997;209:105–116. doi: 10.1002/(SICI)1097-0177(199705)209:1<105::AID-AJA10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Platt KA, Michaud J, Joyner AL. Expression of the mouse Gli and Ptc genes is adjacent to embryonic sources of hedgehog signals suggesting a conservation of pathways between flies and mice. Mech Dev. 1997;62:121–135. doi: 10.1016/s0925-4773(96)00648-x. [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/s0092-8674(00)81795-x. [DOI] [PubMed] [Google Scholar]
- Shimeld SM, McKay IJ, Sharpe PT. The murine homeobox gene Msx-3 shows highly restricted expression in the developing neural tube. Mech Dev. 1996;55:201–210. doi: 10.1016/0925-4773(96)00505-9. [DOI] [PubMed] [Google Scholar]
- Solloway MJ, Robertson EJ. Early embryonic lethality in Bmp5;Bmp7 double mutant mice suggests functional redundancy within the 60A subgroup. Development. 1999;126:1753–1768. doi: 10.1242/dev.126.8.1753. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Anderson RM, Klingensmith J. The BMP antagonists Chordin and Noggin have essential but redundant roles in mouse mandibular outgrowth. Dev Biol. 2001;240:457–473. doi: 10.1006/dbio.2001.0479. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Tonegawa A, Matsumoto K, Ueno N, Kuroiwa A, Noda M, Nifuji A. BMP-4 mediates interacting signals between the neural tube and skin along the dorsal midline. Genes Cells. 1996;1:775–783. doi: 10.1111/j.1365-2443.1996.tb00017.x. [DOI] [PubMed] [Google Scholar]
- Wallin J, Eibel H, Neubuser A, Wilting J, Koseki H, Balling R. Pax1 is expressed during development of the thymus epithelium and is required for normal T-cell maturation. Development. 1996;122:23–30. doi: 10.1242/dev.122.1.23. [DOI] [PubMed] [Google Scholar]
- Wang W, Chen X, Xu H, Lufkin T. Msx3: a novel murine homologue of the Drosophila msh homeobox gene restricted to the dorsal embryonic central nervous system. Mech Dev. 1996;58:203–215. doi: 10.1016/s0925-4773(96)00562-x. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Le Douarin NM. A role for BMP-4 in the development of subcutaneous cartilage. Mech Dev. 1996;57:69–78. doi: 10.1016/0925-4773(96)00534-5. [DOI] [PubMed] [Google Scholar]
- Wijgerde M, Karp S, McMahon J, McMahon A. Noggin antagonism of BMP4 signaling controls development of the axial skeleton in the mouse. Dev Biol. 2005;286:149–157. doi: 10.1016/j.ydbio.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Wilm B, Dahl E, Peters H, Balling R, Imai K. Targeted disruption of Pax1 defines its null phenotype and proves haploinsufficiency. Proc Natl Acad Sci U S A. 1998;95:8692–8697. doi: 10.1073/pnas.95.15.8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilting J, Ebensperger C, Muller TS, Koseki H, Wallin J, Christ B. Pax-1 in the development of the cervico-occipital transitional zone. Anat Embryol (Berl) 1995;192:221–227. doi: 10.1007/BF00184746. [DOI] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BLM. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–2116. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Winograd J, Reilly MP, Roe R, Lutz J, Laughner E, Xu X, Hu L, Asakura T, vander Kolk C, Strandberg JD, et al. Perinatal lethality and multiple craniofacial malformations in MSX2 transgenic mice. Hum Mol Genet. 1997;6:369–379. doi: 10.1093/hmg/6.3.369. [DOI] [PubMed] [Google Scholar]
- Wright E, Hargrave MR, Christiansen J, Cooper L, Kun J, Evans T, Gangadharan U, Greenfield A, Koopman P. The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat Genet. 1995;9:15–20. doi: 10.1038/ng0195-15. [DOI] [PubMed] [Google Scholar]
- Ybot-Gonzalez P, Cogram P, Gerrelli D, Copp AJ. Sonic hedgehog and the molecular regulation of mouse neural tube closure. Development. 2002;129:2507–2517. doi: 10.1242/dev.129.10.2507. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bradley A. Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development. 1996;122:2977–2986. doi: 10.1242/dev.122.10.2977. [DOI] [PubMed] [Google Scholar]
- Zimmerman LB, De Jesus-Escobar JM, Harland RM. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein-4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]