

Summary
Costimulatory receptors and ligands trigger the signaling pathways that are responsible for modulating the strength, course and duration of an immune response. High-resolution structures have provided invaluable mechanistic insights by defining the chemical and physical features underlying costimulatory receptor/ligand specificity, affinity, oligomeric state, and valency. Furthermore, these structures revealed general architectural features that are important for the integration of these interactions and their associated signaling pathways into overall cellular physiology. Recent technological advances in structural biology promise unprecedented opportunities for furthering our understanding of the structural features and mechanisms that govern costimulation. In this review we highlight unique insights that have been revealed by structures of costimulatory molecules from the immunoglobulin and tumor necrosis factor superfamilies, and describe a vision for future structural and mechanistic analysis of costimulation. This vision includes simple strategies for the selection of candidate molecules for structure determination and highlights the critical role of structure in the design of mutant costimulatory molecules for the generation of in vivo structure-function correlations in a mammalian model system. This integrated ‘atoms-to-animals’ paradigm provides a comprehensive approach for defining atomic and molecular mechanisms.
Keywords: T-cell costimulation, structure, immunoglobulin superfamily, TNF/TNFR superfamily
Introduction
Cell surface receptors and adhesion molecules are gatekeepers of cellular function, as they are responsible for the detection and integration of signals arising from the extracellular milieu. In adaptive and innate immunity, these molecules underlie the initial recognition and ultimate destruction of foreign pathogens and malignancies, and at the same time are critical components of the tolerance mechanisms that protect the host from harmful autoimmune responses. Soluble versions of these receptors and their cognate ligands, as well as monoclonal antibodies (mAbs) targeted against these proteins represent a major class of protein therapeutics for the manipulation of immune responses to treat a wide range of infectious diseases, autoimmune diseases, and malignancies. Central to understanding the mechanisms that control normal, pathological, and therapeutic immune responses is a detailed structural description of the complexes, higher-order assemblies, and the long-range organizational properties that characterize costimulatory receptors and ligands.
As in all areas of modern biology, high resolution crystallographic approaches are playing increasingly widespread and important roles, and have been instrumental in defining the chemical and physical features responsible for the receptor-ligand recognition events and adhesive interactions that are central to adaptive and innate immunity. Indeed, the science of crystallography has matured to the point where structure is no longer a luxury, but is now essential for our continued understanding of the basic workings of complex biological processes. The remarkable advances that have been made in the practice of crystallography can be put in historic perspective by considering Max Perutz's assessment in 1948 of his own work on the structural characterization of hemoglobin:
“Due largely to the absence of any direct method for obtaining the atomic positions from the observed intensities of the diffracted rays, a detailed analysis of an organic compound of comparatively moderate size, such as sucrose or cholesterol, takes two or more man-years to complete. On the face of it, therefore, an attempt to analyze the crystal structure of haemoglobin, or of any crystalline protein for that matter, looks about as promising as a journey to the moon (1).”
Perutz thus demonstrated almost clairvoyant abilities. He published the heroic result of the full atomic structure of haemoglobin in 1968 (2); Apollo 11 landed on the moon the following year. This situation has of course dramatically changed. The advent of new X-ray detector and synchrotron technologies, and associated software now allows for the collection of X-ray diffraction data, and, in favorable cases, complete structure determination in a matter of hours or days instead of months and years. One of the greatest impacts on structural biology has been the remarkable advances in protein expression technologies that now allow almost any protein or multi-component assembly to be targeted, or at least considered, for structure determination. Indeed, based on these advances, a number of national and international ‘Structural Genomics’ initiatives have been established for the purpose of developing additional technologies and infrastructure for high throughput structure discovery. For example, the US National Institutes of Health is currently supporting the Protein Structure Initiative (PSI), which is tasked with the generation of thousands of new structures, so as to increase our knowledge of the range of protein folds present in Nature and to provide useful structural models for the majority of primary amino acid sequences, while at the same time making a direct contribution to problems of biomedical relevance. These efforts have launched a number of programs that are particularly noteworthy in terms of biological importance and magnitude. For example, one program is systematically evaluating the structures of all human protein phosphatases and protein phosphatases from relevant human pathogens (3). These structures are providing insights into a wide range of normal and disease processes, including transcriptional control, the regulation of major signaling pathways, neural development, and autoimmune disease, and these efforts promise to provide an exceptional resource for the structure-guided development of inhibitors for all classes of protein phosphatases. A second illustrative example is the program to generate complete structural coverage of the entire Thermotoga maritima genome (i.e., provide structural models for every gene product in the genome). These efforts have energized the entire international Thermotoga community by fostering numerous multi-disciplinary collaborations between biochemists, cell biologists, geneticists, informaticists, and structural biologists (4).
Ultimately one would like to apply this new philosophy and infrastructure to the study of immunity by targeting and determining the structures of all relevant cell surface receptors and adhesion molecules, and in particular the complexes they form with cognate ligands and regulatory proteins. It is estimated that the human genome encodes ∼7,000 secreted and integral membrane proteins, of which several superfamilies of molecules make substantial contributions to immunity, including the immunoglobulin (Ig), tumor necrosis factor (TNF), TNF receptor (TNFR), G-protein coupled receptor (GPCR), chemokine, and lectin superfamilies (Fig. 1).
Fig. 1. Structures and folds of immunological significance.
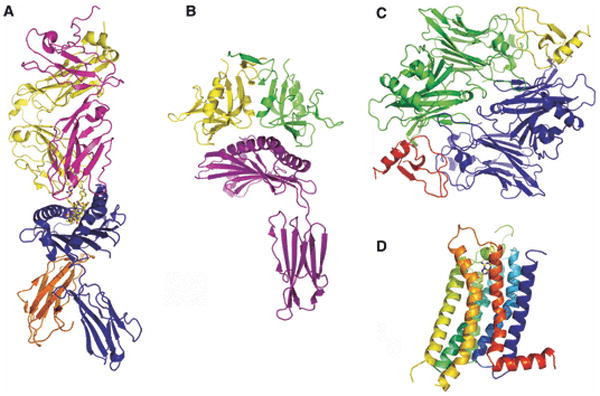
(A) T-cell receptor (yellow and magenta):MHC-peptide (blue and orange with peptide in stick representation) complex from mouse, PDB Code 1G6R; (B) Human MICA (an MHC class-I-related molecule; magenta) in complex with NK cell receptor NKG2-D (a member of the lectin superfamily; yellow and green), PDB Code 1HYR; (C) Herpesvirus M3 decoy receptor (blue and green) in complex with the CC-chemokine MCP-1 (red and yellow), PDB Code 1ML0; and (D) human GPCR A2a adenosine receptor; this structure provides a model for the many GPCRs relevant to immunity, PDB Code 3EML.
Structural analysis of antibody:antigen complexes have revealed the atomic mechanisms utilized for antigen recognition and highlighted the significance of gene rearrangements leading to the enormous diversity of an individual's antibody repertoire. Structures of peptide-major histocompatibility complex (MHC): T-cell receptor (TCR) complexes have revealed details of another remarkable antigen recognition strategy used by CD4+ and CD8+ T cells. By presenting linear peptides in the context of class-I and class-II MHC molecules, Nature has significantly reduced the complexity of this process from a three-dimensional problem to a one-dimensional recognition problem. Similarly, structures of natural killer (NK) cell surface receptors bound to their cognate MHC-family molecules have begun to reveal the recognition elements involved in the initiation of innate immune responses. The structures of costimulatory receptor:ligand pairs from the Ig and TNF/TNFR superfamilies and associated adhesive molecules have given unique insights into the recognition and signaling processes that modulate the strength, course, and duration of an immune response. Structural studies are also revealing the molecular and atomic strategies that are exploited by a wide range of pathogens and malignancies for evasion of the host immune system. Importantly, as these molecular interactions, and their associated signaling pathways, are primary therapeutic targets for immune-modulation, these structures provide the basis for the rational engineering of soluble agents with enhanced therapeutic efficacy.
Structural analyses not only reveal the chemical and structural features responsible for the specificity, kinetics and thermodynamics of these interactions, but place these interactions into a larger cellular context by providing insights into the mechanisms responsible for molecular partitioning, colocalization, and assembly processes that are essential for the formation and function of the ‘immunological synapse’ formed between T cells and antigen presenting cells (APCs). For example, the oligomeric state and overall organization of cell surface receptors and ligands impose constraints on the stoichiometries of specific receptor:ligand complexes, which in turn impact the types of multi-component signaling assemblies that can be formed. Furthermore, the molecular organization of a receptor:ligand complex may contribute to the specific localization of assemblies within the immunological synapse, as it has been proposed that molecular pairs are segregated and compartmentalized within the synapse on the basis of overall linear dimension (5). Thus, beyond simply generating high resolution pictures of macromolecules, structure can directly drive biological investigation through the generation of testable hypotheses regarding oligomeric state, receptor:ligand stoichiometry, and the overall organization and cellular localization of signaling assemblies.
Despite the remarkable technical advances that have been made and the tremendous insights arising from high resolution structural approaches, it is not yet feasible for an individual laboratory to express and determine the structures of all proteins and assemblies relevant to adaptive and innate immunity. Nonetheless, the opportunity does exist to exploit these technologies by developing approaches to identify those proteins and complexes for which a structure would be most likely to provide new functional and mechanistic insights. In this review we describe the strategies we are developing and applying for the study of cell surface receptors and ligands in general, and highlight the unique functional insights we have gained from the structural analyses of a number of costimulatory molecules belonging to the Ig and TNF superfamilies.
Strategies for identifying candidate proteins for structure determination
High-resolution structures provide an invaluable scaffold on which to map mutagenesis, biochemical, and biophysical data, so as to identify functionally important features, including ligand recognition sites, regulatory sites and oligomerization interfaces. In some cases, the structures themselves provide novel and unanticipated insights into function and mechanism. These are the structures that are most desired, as they directly translate into new biological understanding. It is thus a major challenge to develop strategies that identify proteins and complexes for which a structure would be particularly informative. Such considerations form the basis for hypothesis-driven structural biology, where structure not only allows for retrospective analysis, but provides the driving force for the discovery of new biology and mechanisms. Several strategies provide valuable insights into the identification of such targets, including (1) biological considerations based on functional and biochemical properties, and (2) genomic considerations that rely on the identification of unique primary amino acid sequence signatures for the prediction of unique structural features, which are in turn responsible for unique biological function.
Biological considerations
Without question, the most powerful rationale for structure determination is biological relevance, which may come in the form of (1) an important biological function, (2) the possession of a unique biological activity, or (3) a critical position within a complex biological network.
Functional considerations
A powerful example is offered by one of the most heavily studied families of costimulatory molecules, the CD28/cytotoxic T lymphocyte antigen-4 (CTLA-4)/ inducible costimulator (ICOS) family. As detailed below, even though CTLA-4 and CD28 were predicted to possess similar structural and organization features, the explicit structural analysis of these two related molecules revealed important differences that result in distinct modes of association with their B7 ligands, which ultimately impact the nature of their respective signaling mechanisms. Furthermore, based on sequence considerations, ICOS will certainly exhibit similarities to CD28 and CTLA-4. However, given the important role of ICOS in costimulation, and the precedence in this family for small functionally important structural differences, the structures of ICOS and that of the complex with its cognate ligand (ICOSL) are highly desirable, so as to reveal the details unique to its overall organization and function.
Unique biochemical properties
The presence of unique biochemical properties also provides a strong rationale for structure determination. An interesting example is provided by leukocyte-associated Ig-like receptor 1 (LAIR-1), a type-I transmembrane glycoprotein that functions as an inhibitory receptor on both NK and T cells (6, 7). Recently, collagen, a major component of the extracellular matrix (ECM), was identified as an important ligand for leukocyte-associated Ig-like receptor-1 (LAIR-1), as engagement of LAIR-1 by collagen directly inhibits immune cell activation both in cell lines and primary cells (8-10). The ectodomain of LAIR-1 contains a single Ig domain, and the gene encoding human LAIR-1 is localized to the leukocyte receptor complex on human chromosome 19, which spans 1 Mb and contains approximately thirty Ig-like receptors, including two additional collagen-binders, LAIR-2 and glycoprotein VI (GPVI) (11). LAIR-2, a soluble homolog of LAIR-1 (84% sequence identity), efficiently blocks the LAIR-1-collagen interaction, providing a mechanism for the regulation of ECM-associated LAIR-1 through direct competition for collagen binding sites (12). The third member, GPVI, is more distantly related to the other two (<40% sequence identity in each case) and acts as an activating platelet collagen receptor (11). Thus, a structure of any of these molecules bound to collagen could reveal a new paradigm by which a costimulatory pathway is regulated by engagement with the ECM, and may provide new opportunities for the development of protein-based and small molecule modulators of these interactions and the associated signaling pathways.
Systems considerations
In addition to specific biological function and unique biochemical properties, the consideration of genome-scale mapping of protein-protein interaction networks (13-15) provides another guide for the selection of high value candidates for structure determination. Analysis of protein interaction networks show that they are often organized around a small number of proteins, with multiple connections, that serve as ‘hubs’ for the network (16). These hub proteins are frequently associated with unique structural features that may be associated with critical biological functions (17, 18). DcR3, a secreted decoy receptor belonging to the TNFR superfamily represents a relevant example, as it modulates multiple signaling pathways as the consequence of neutralizing three different ligands: TNF superfamily, member 6 [Fas ligand (FasL)], TNF (ligand) superfamily, member 14 (LIGHT), and TNF (ligand) superfamily, member 15 (TL1A) (Fig. 2) (19-21). The sequences of these three ligands do not distinguish them from other TNF family members and provide no immediate insights into their ability to participate in high affinity associations with the same DcR3 receptor. Moreover, each of these ligands binds different functional receptors and subsequently direct distinct immune responses. The pivotal position of DcR3 in this interaction network and its binding promiscuity immediately suggest a unique mode of receptor:ligand recognition. Structural characterization of hub proteins like DcR3 and the complexes formed with their binding partners are likely to reveal new paradigms in immune regulation.
Fig. 2. Biological and genomic strategies for identifying protein targets for structure determination.
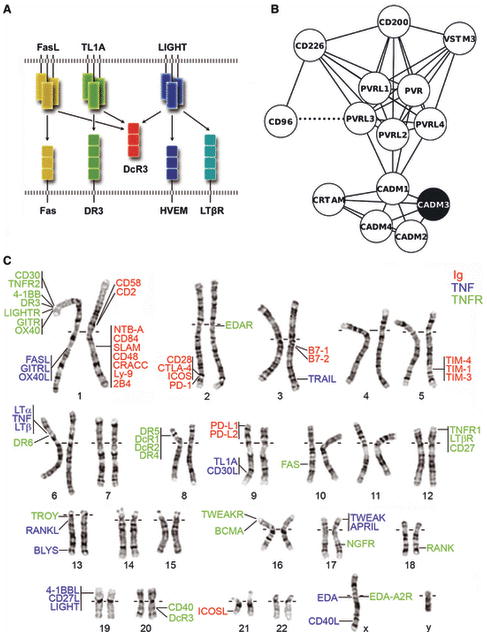
(A) Systems considerations. DcR3 (red), a soluble TNFR superfamily member, represents a hub protein, as it neutralizes three trimeric TNF ligands, FasL (yellow), TL1A (green) and LIGHT (blue). Arrows indicate functional interactions between the ligands and their cognate receptors. Cysteine rich domains of the receptors are represented as squares. (B) Sequence-based network graph highlighting the sequence similarities between genes in the Nectin family. Genes are illustrated as nodes (circles) and related sequences are connected by edges (i.e., lines). A dashed line depicts adjacent genes in the genome. The only member of this diverse family for which a structure has been determined (CADM3; PDB 1Z9M) is highlighted in black. Cytoscape (131) was used to layout the network. (C) Physically Linked Gene Families. The mapping of selected Ig superfamily (red) and all known TNF (blue) and TNFR (green) genes on the human karyotype highlights clusters of evolutionary related adjacent genes. Genes sharing a vertical line have immediately adjacent chromosomal positions.
Genomic considerations
The power of biological and biochemical rationales to drive structure determination is indisputable; however, function has not been assigned to a large fraction of all existing sequences. Thus, approaches that are independent of functional knowledge are required to aid in the identification of those proteins for which a structure is likely to be most informative in terms of unique function and mechanism. In this regard, the genome sequencing projects provide immediate opportunities, as they are a powerful resource for the identification of unique primary amino acid sequence signatures that suggest the existence of unique structural features, which are in turn responsible for unique biological function. This paradigm for identifying candidates for structure determination requires the dissection of superfamilies (e.g., Ig superfamily) into smaller evolutionarily related families, which readily highlight distinguishing sequence features responsible for the unique structural characteristics that ultimately underlie function (Fig 3). Genome sequencing efforts provide two pieces of information that are useful for the initial identification of such families: 1) Physical proximity in the genome and 2) Primary sequence similarity. Furthermore, this approach readily identifies families that are under-represented in terms of structural characterization. In addition, primary sequence analysis can also reveal subtle sequence differences between members of the same family, which can indicate specialized structural and functional features within a family. Thus the structures of multiple members of a given family are likely to be highly informative in terms of the origins of overlapping, as well as unique biological functions.
Fig. 3. Unique sequence features reveal structural and functional specialization.
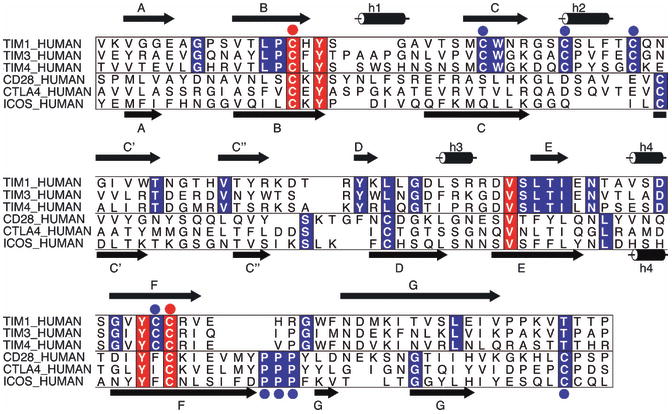
As an example, multiple sequence alignments of the IgV domains of three members of the human TIM family (TIM1, TIM3, and TIM4) and three members of the human CD28 family (CD28, ICOS, and CTLA4) are displayed. Secondary structure calculated from representatives of each family is illustrated at the top and bottom of the alignment for the TIM and CD28 families. Highlighted in red are positions that are invariant in both families; for example the two cysteines that form the canonical disulfide bond between the B and the F strands (red circles). In blue are sequence positions that are conserved in one family but are absent in the other; these positions define family-specific sequence signatures, and suggest unique structural features that are directly relevant to functional specialization. ALSCRIPT (132) was used to format the multiple sequence alignment.
Physical proximity
Gene duplication represents one of the major mechanisms for the generation of new function. The second copy of a gene is often subjected to reduced selective pressure and can evolve at enhanced rates relative to the original gene, allowing for the acquisition of new function. Frequently, the duplicated genes, or paralogs, are immediately adjacent to one another and sizeable clusters of duplicated genes are found in the genome. Because these genes are evolutionarily related, they share primary sequence signatures that are responsible for similar structural features supporting related biological functions. Detailed sequence differences within these clusters of physically proximal genes point to determinant(s) responsible for specialized structure and function. Fig. 2 highlights several examples of proximal gene clusters in the Ig superfamily, including the CD28, T cell Ig and Mucin domain (TIM), and signaling lymphocyte activation molecule (SLAM) families, as well as various families from the TNF and TNFR superfamilies. The identification of proximally related clusters of genes represents a direct, simple and intuitive strategy for defining protein families, which can be examined for unique primary sequence signatures to identify candidates for structure determination.
Primary sequence similarities
Not all related sequences are physically linked, but are more widely distributed across the genome. Thus, more general approaches to define related families of proteins based solely on primary amino acid sequence must be employed. We have implemented an algorithm that defines a ‘profile’ of features for each individual primary sequence. Sequences that share similar profiles are assigned to the same sequence family. Although this approach is conceptually straightforward, it can represent a significant challenge in large divergent superfamilies, such as the Ig superfamily, where sequence identities can be lower than 15%. We applied this approach to ∼550 genes in the human genome that are predicted with high confidence to encode secreted or cell surface proteins that contain Ig domains. A representative example is provided by the nectin and nectin-like family of Ig-containing proteins that mediate both homophilic and heterophilic cell-cell adhesion interactions. The family is composed of nectin-1 to 5 (PVRL1–PVRL4, and PVR) and nectin-like-1 to 4 (also called CADM1–CADM4) (Fig. 2). Our sequence-based clustering methods also identified five additional Ig-containing molecules, CD226, CRTAM (Class-I MHC-restricted T cell associated molecule), CD96, T-cell immunoreceptor with Ig and immunoreceptor tyrosine-based inhibition motif (ITIM) domains (TIGIT), and CD200, as members of this family. In this family, only two genes (CD96 and PVRL3) were observed to be adjacent on the genome. The genes in this cluster also demonstrate similarities in both domain organization (specifically between nectin and nectin-like) and a similarity in function. With the exception of CD200, the ligands for all these molecules reside within this same family (22, 23). Taken together, these observations support the hypothesis that these molecules all form a single sequence-based cluster or family within the Ig superfamily. Notably, the structure of only a single family member, nectin-like-1, has been determined to date (24). Because the sequence identity of nectin-1-like is less than 30% in comparison to most members of the family, structures of additional family members are likely to reveal shared structural features common to the entire family, as well as unique features related to functional diversity. Thus, comprehensive sequence considerations not only allow for an expanded description of protein families, but also highlight those families where additional structure determination efforts are warranted.
Summary of approaches
As described above, simple informatics considerations can be exploited to identify proteins with unique structural features that directly contribute to mechanism and function. Importantly, the constructions of families on the basis of proximity and explicit sequence considerations represent testable hypotheses for the structural and functional relatedness of the individual members, which provides an opportunity to enhance existing informatics approaches. These sequence-based approaches complement more traditional approaches that rely on biochemical and functional annotations to identify candidates for structure determination. In the remainder of this review we provide a number of examples from the Ig and TNF superfamilies that highlight the power of primary sequence considerations for the formulation of specific structural hypotheses regarding receptor:ligand specificity and affinity, oligomeric state, valency, the formation and localization of higher-order assemblies, and the importance of these features for the adaptive and innate immune responses. Most importantly, these structures provide the foundation for the generation of mutant molecules with unique biochemical properties for cell-based studies and for the generation of in vivo structure-function correlations in mammalian model systems that directly bear on costimulation. The examples and approaches described here begin to define a strategy for the comprehensive structural and mechanistic analysis of costimulation.
The Ig superfamily
The Ig superfamily makes the largest contribution to costimulatory receptors and ligands and adhesion molecules. All the Ig-like domains are composed of ∼100 residues that share a common two-layered fold composed of two anti-parallel β-sheets (Fig. 4). Several basic variants of this architecture are found in nature and are classified according to β-strand topology and conserved sequence signatures. The Ig variable (IgV) domains are generally composed of front sheets formed by the A′, G, F, C, C′, and C″ strands and back sheets formed by the A, B, E, and D strands. Of particular importance are the loops connecting the BC, CC′ and FG strands, which correspond to complementarity-determining region 1 (CDR1), CDR2 and CDR3 in the antigen receptors, respectively. Notably, the majority of dimers involving IgV domains utilize an interface formed by the front sheets. The Ig constant 1 (IgC1) domain contains a front sheet formed by G, F, and C strands and a back sheet formed by A, B, E and D strands, while the truncated IgC2 domain possesses the C′ strand in the front sheet, but lacks the D strand in the back sheet. Nearly all Ig domains contain a conserved disulfide bond that links the B and F strands in the back and front sheets, respectively. Despite the common overall architecture, Ig domains can share as low as 15% sequence identity, and this variability results in a wide range of structural and organizational variations that underlie the enormous diversity of function exhibited by members of the Ig superfamily (Fig. 4).
Fig. 4. Structure of the IgV domain and its diversification in costimulation.
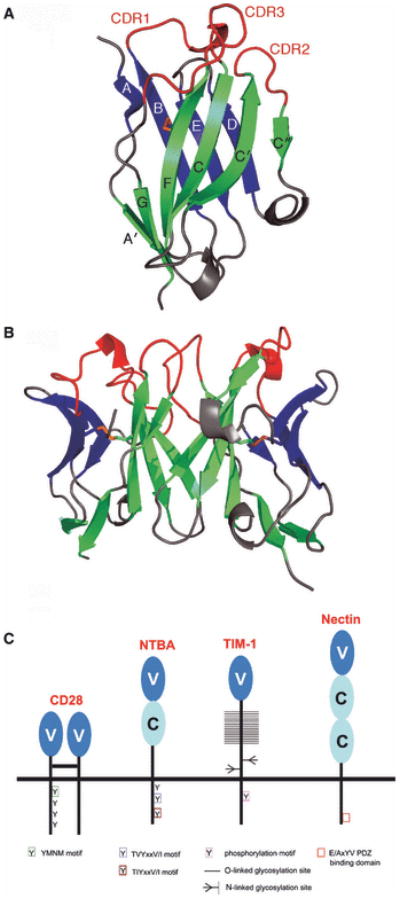
(A) Structure of an IgV domain. Strands of the front (green) and back (blue) sheets of the heavy-chain IgV domain from an Fv molecule (PDB Code 1A6W) are labeled according to convention and the conserved disulfide bond connecting strands B and F is colored in orange. The CDR loops connecting strands B and C (CDR1), strands C′ and C″ (CDR2) and strands F and G (CDR3) are highlighted in red. (B) Typical IgV interface formed by the front sheets. IgV dimer formed by heavy- and light-chain IgV domains of an Fv molecule (PDB Code 1A6W). (C) Structural and organizational variations of the Ig superfamily. Costimulatory receptors of the CD28 superfamily are predominantly disulfide-linked dimers of single IgV domains; SLAM family members contain both IgV and IgC domains; TIM family molecules contain one IgV domain attached to a highly glycosylated stalk region; nectin and nectin-like molecules consist of three Ig domains (one IgV and two IgC)s.
CD28/CTLA-4/ICOS family
The members of the CD28/B7 receptor/ligand families are among the best characterized of all costimulatory molecules and play crucial roles in T-cell activation and tolerance (25-27). These families include the CD28, CTLA-4, ICOS cell-surface receptors expressed on T cells and the B7 ligands expressed on APCs. CD28 and CTLA-4 share the ligands B7-1 and B7-2, with CTLA-4 exhibiting 10-100-fold higher affinities (28). The constitutively expressed CD28 delivers positive signals upon binding to B7-1 or B7-2, which synergizes with the TCR signal to promote T-cell activation. The main effects of CD28 signaling are to augment and sustain T-cell responses, promote survival of T cells and direct cytokine production for the induction of clonal expansion and differentiation (29). In contrast, CTLA-4 is expressed subsequent to T-cell activation, and its interactions with B7-1 and B7-2 have profound inhibitory effects on T-cell activation, including inhibition of IL-2 synthesis and impaired cell cycle progression, and results in the termination of a T-cell response (28). CTLA-4-deficient mice develop fatal lymphoproliferative diseases due to unopposed costimulation through the CD28:B7 pathways, demonstrating a crucial role for CTLA-4 in regulating peripheral tolerance (30). ICOS is another positive costimulatory receptor expressed on activated T cells, which binds to the B7-homolog ICOS-Ligand (ICOSL) expressed on APCs (31). In contrast to CD28, ICOS is not expressed on naive T cells but is rapidly upregulated after TCR engagement, suggesting that ICOS might primarily provide costimulatory signals to previously activated T cells.
In humans, the genes encoding CD28, CTLA-4 and ICOS are immediately adjacent to one another on chromosome 2; B7-1 and B7-2 are located on chromosome 3, and ICOSL on chromosome 21 (Fig. 2) (27). CD28, CTLA-4 and ICOS are type-I transmembrane proteins composed of a single extracellular IgV domain linked to a stalk region and a transmembrane segment followed by a relatively short cytoplasmic tail, which contains at least one tyrosine-based signaling motif (Fig. 5). The ectodomains of the B7 ligands also share a similar architecture that includes a membrane distal IgV domain and membrane proximal IgC domain (Fig. 5).
Fig. 5. Organizational and sequence features of the CD28:B7 families.
(A) Domain organization of the CD28:B7 families. CD28, CTLA-4 and ICOS are immediately adjacent on the chromosome and form disulfide-linked homodimers; PD-1 is monomeric and not part of this cluster. CD28-type receptors consist of a single IgV domain, linked to stalk region and cytoplasmic tail containing tyrosine-based signaling motifs. (B) Organization of B7 ligands. The B7 ligands possess ectodomains composed of a membrane proximal IgC and a membrane distal IgV domain, linked to a stalk region and cytoplasmic tail. (C) Sequence alignment of the ectodomains of CD28 family receptors. Secondary structure is denoted on the basis of the CTLA-4 crystal structure; conserved residues are white with red background; residues with similar properties are labeled red; residues that contribute to the dimer interface of CTLA-4 are highlighted with black circles, those contributing to the CD28 dimer interface with red circles, grey circles annotate those positions that contribute to the dimer interface in both molecules. The conserved cysteine mediating covalent dimerization in CD28, CTLA-4 and ICOS is highlighted with a red asterisk; PD-1 has a Ser at this position. Residues that contribute to the proline-rich ligand-binding motif in CD28, CTLA-4 and ICOS are highlighted with green asterisks; PD-1 lacks this motif. Numbering corresponds to the human CTLA-4.
CD28, CTLA-4 and ICOS share primary amino acid sequence features that have proven important for their molecular organization and function (Fig. 5). In particular, the stalk regions of CD28, CTLA-4 and ICOS contain a unique cysteine residue that is responsible for the formation of biologically functional disulfide-linked homodimers. This cysteine, the equivalent of residue 122 in human CTLA-4, is conserved in all known sequences, with the exception of chicken CD28, suggesting that covalent dimerization contributes to effective signaling and costimulation (32). A second distinguishing feature of CD28, CTLA-4 and ICOS is a consensus motif that includes three consecutive proline residues in the FG loops. In CD28 and CTLA-4, these prolines are embedded within the MYPPPY sequence, while in ICOS they are embedded in the FDPPPF sequence.
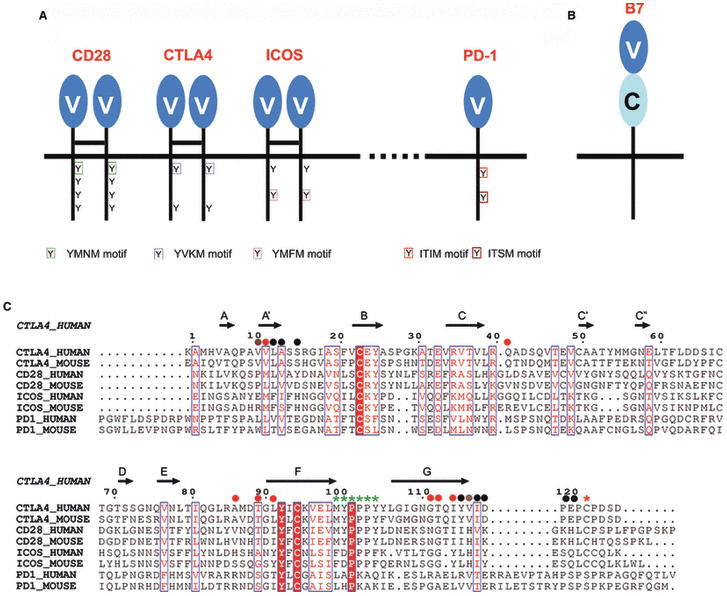
The crystal structures of murine CTLA-4 bound to B7-1 or B7-2 (33, 34) reveal that the binding interface is formed predominately by contacts between the MYPPPY sequence of the CTLA-4 FG loop and a concave surface on the front sheet of the B7 ligands (Fig. 6). The three proline residues adopt a unique high-energy cis-trans-cis main chain configuration that provides the geometric complementarity required for specific recognition of the B7-1 and B7-2 surfaces. This same cis-trans-cis main chain conformation is present in both unliganded CTLA-4 and CD28 (35, 36). Importantly, mutagenesis experiments indicate that the FG loop is essential for the binding of CTLA-4, CD28 and ICOS to their respective ligands (37-39). The extreme sequence, structural and functional similarity of the FG loops suggests that all of these receptors share a common mode of recognition for their B7 ligands.
Fig. 6. CD28 and CTLA-4 share a similar binding mode for the B7 molecules.
(A) Superposition of the CD28 monomer (PDB Code 1YJD) and the CTLA-4:B7-1 (PDB Code 1I8L) complex. The tip of the FG loop contains the MYPPPY motif that is crucial for binding. (B) Detailed view of the MYPPPY motif. The consensus ligand-binding sequence shared in CD28 and CTLA-4 makes contacts with residues on the front face (C, F, and G strands) of B7-1. Residues in the MYPPPY motif are numbered according to human CTLA-4.
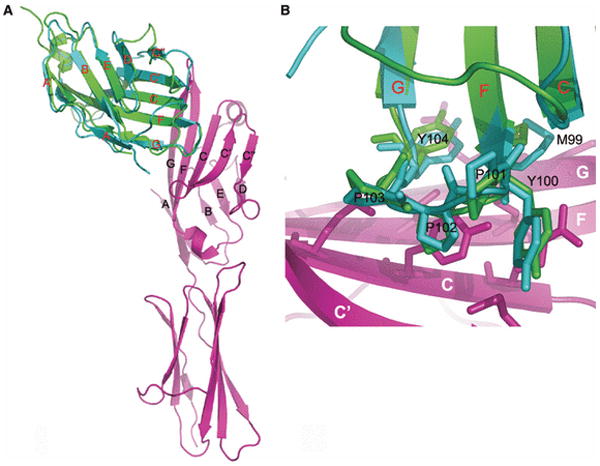
The liganded and unliganded structures of CTLA-4 reveal an unusual side-to-side dimerization interface that involves invariant residues in the base of the A and G strands, including the beginning of the stalk and Cys122 (Fig. 7) (33-35). This interface buries only ∼700 Å2 of accessible surface area, which is considerably lower than that associated with the front sheet-to-front sheet interactions typical of IgV domain associations. The unusual organization of this dimer places the FG ligand recognition loops distal from the dimer interface suggesting the possibility of bivalent ligand binding. The crystal structures of the CTLA-4 complexes with B7-1 and B7-2 confirm this bivalent interaction. B7-1 and B7-2 lack a cysteine in the stalk region and thus, unlike the receptors, cannot form covalent oligomers. However, B7-1 was shown to exist predominantly as non-covalent dimer on the cell surface, while B7-2 is predominantly monomeric (40). ICOSL was also shown to form non-covalent dimers on the cell surface (41). The B7-1 structure reveals another unusual dimer interface, in this case formed by association of the back sheets, which places the receptor binding surface distal to the dimer interface and again suggests the potential for bivalent interactions (42).
Fig. 7. CTLA-4 and CD28 have different valencies for the B7 ligands.
(A) Structure of the CTLA-4 dimer. This structure (PDB Code 1I8L) reveals a side-to-side dimer in which the interface is formed by the bases of the A and G strands, placing the FG ligand-binding loop distal from the dimer interface. This relative orientation of the monomers allows for a bivalent ligand binding. (B) Structure of the CD28 dimer. This structure (PDB Code 1YJD) shows a different dimer interface, which results in a more compact dimer and a different placement of the FG ligand-binding loops. (C) Crystal structure of bivalent CTLA-4 dimers binding to bivalent B7-1 dimers. These properties result in a periodic network that may have important functional consequences (PDB Code 1I8L). (D) The CD28 dimer is monovalent. A model of the putative CD28:B7-1 complex was constructed by superimposing a single CTLA-4:B7-1 complex on each of the two molecules in the CD28 dimer. This model is incompatible with the simultaneous binding of two distinct B7 molecules to the CD28 dimer due to unfavorable steric interactions and supports monovalency.
The bivalent binding potential of both the CTLA-4 receptor and the B7-1 ligand dimers may have important functional consequences. In the crystal lattice, the CTLA-4 and B7 homodimers form a periodic array of receptor and ligand homodimers with a characteristic spacing of ∼100 Å (Fig. 7). This periodic network provides a model for the assembly of these molecules at the T cell-APC interface and offers a mechanism for the localized enrichment of signaling molecules within the immunological synapse. While the organization of these assemblies are driven by interactions involving the ectodomains, these same constraints are imposed on the non-covalently associated cytoplasmic signaling and scaffolding proteins that are responsible for propagating and amplifying extracellular signals. This unique arrangement of the alternating CTLA-4-B7-1 homodimers may provide a structural basis for the potent immune inhibitory function of CTLA-4. Due to the apparent monomeric nature of B7-2 it has the potential to form signaling complexes composed of a single CTLA-4 dimer and two B7-2 monomers. The existence and the extent of higher-order assemblies involving CTLA-4 and the B7 ligands will likely depend on a number of factors, including the local concentrations of receptor and ligand on the cell surface, and the mechanistic importance of these putative interactions remains to be established, as does the generality of this behavior for CD28 and ICOS.
The structures of CD28 and CTLA-4 exhibit striking overall similarity, but there exist significant differences in the relative organization of monomers within their respective homodimers, resulting in a more ‘compact’ CD28 assembly compared to CTLA-4 (Fig. 7) (36). Whereas the CTLA-4 interface is formed predominately by residues at the bases of the A and G strands, the dimer interface in CD28 is formed by residues located at the top of these strands (Fig. 5). The distinct dimeric organization of CD28 has profound functional impact as it alters the relative orientation of the ligand binding FG loops as compared to those in CTLA-4. On the basis of this model, unfavorable steric interactions would be predicted to preclude the simultaneous binding of two B7 ligands molecules, rendering CD28 monovalent (Fig. 7). This model is consistent with solution binding studies suggesting monovalent behavior of CD28 (43) and predicts a significant reduction in the complexity of the CD28:B7 assemblies that can be formed relative to CTLA-4. Specifically, one would predict that a dimer of B7-1 could bind two independent CD28 dimers, while the monomeric B7-2 could only bind a single CD28 dimer.
These differences in receptor and ligand valency are expected to result in receptor:ligand complexes with a range of stoichiometries, apparent affinities and half-lives (e.g., CTLA-4:B7-1 versus CTLA-4:B7-2), all of which can be exploited to fine tune responses to different antigens and at different points during a response. These considerations are not restricted to signals directly mediating T-cell responses, but could potentially effect ‘reverse signaling’ into the ligand-bearing cells. For example, CTLA-4 can deliver signals via B7-1 and B7-2 into dendritic cells (DCs), which result in the induction of indoleamine 2,3-dioxygenase (IDO), an enzyme that degrades tryptophan to by-products that inhibit T-cell proliferation (44). These bidirectional CTLA-4:B7 interactions may participate in downregulation of T-cell responses and induction of T-cell tolerance. It will be of considerable interest to determine the structures of ICOS and its complex with ICOSL to define the effective valency of these molecules and the overall organization of this complex.
The CTLA-4:B7 structures provide important physical constraints that must be accommodated for the proper localization within the immunological synapse. Among the challenges facing T cells are the natural low abundance of most peptide antigens and the characteristic low affinity between TCRs and their antigenic peptide-MHC ligands (45). In addition, an abundance of large, negatively charged glycoproteins (i.e., the glycocalix) on the cell surface hinders the intimate intercellular contacts required for the engagement of the smaller cell surface receptors, including TCR, CD28, and CD2. To overcome these barriers, T cells have developed mechanisms to ensure that signaling and adhesion molecules are differentially compartmentalized at the T cell:APC interface. The central zone of the immunological synapse is characterized by the receptor:ligand pairs that control T-cell activation including the TCR:peptide-MHC, CD28:B7, CTLA-4:B7, and CD2:CD58 complexes (Fig. 8), and associated intracellular signaling molecules such as the leukocyte-specific protein tyrosine kinase (Lck), Fyn, and protein kinase C θ (PKC-θ) kinases and the protein phosphatase 2 regulatory subunit A (PP2A), protein tyrosine phosphatase non-receptor type 6 (SHP1), and Src homology 2 (SH2) domain-containing protein tyrosine phosphatase-2 (SHP2) phosphatases (46). Surrounding the central zone is the peripheral zone, which is composed of large cellular adhesion molecules and components of the actin cytoskeleton. It has been proposed that localization within the immunological synapse is controlled by a mechanism that discriminates on the basis of molecular dimension, with similar sized molecules partitioning to the same compartment. Notably, most of the ‘signaling’ molecular pairs within the central zone have maximal linear dimensions of ∼100-140 Å, while the adhesive molecules that reside in the peripheral zone have linear dimensions that exceed several hundred Å (5). This compartmentalization affords for significant cell-cell adhesion interactions, while at the same time concentrating TCRs and their rare antigenic peptide-MHC ligands and a wide range of costimulatory molecules to a small contact area that facilitates signaling. Recent studies support the biological significance of this compartmentalization, as TCR:MHC assemblies with increased linear dimensions exhibited greatly reduced TCR signaling (47).
Fig. 8. ‘Crystallographic view’ of the immunological synapse.
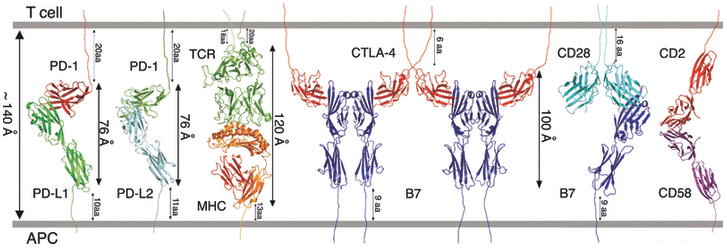
Composite model of the MHC:TCR complex and costimulatory receptor:ligand complexes in the central region of the immunological synapse. The MHC:TCR (PDB Code 1G6R), PD-1:PD-L1 (PDB Code 3BIK), PD-1:PD-L2 (PDB Code 3BP5) and CTLA-4:B7-1 (PDB Code 1I8L) complexes are based on existing crystal structures; the model of the CD28:B7-1 complex was generated as described in Fig. 7; the generation of the model of the full length CD2:CD58 complex was described previously (97). The approximate dimensions (i.e., lengths) of the complexes are shown, as well as the number of residues connecting the structured Ig domains to the membrane. Also noted is the ∼140 Å distance that characterizes the separation between the plasma membranes in the immunological synapse.
PD-1 (programmed death-1)
PD-1 is a unique inhibitory receptor that is often considered part of the CD28 family. Unlike other members of the family, PD-1 expression is rapidly upregulated after activation not only on T cells, but also on B cells and myeloid cells, suggesting a broad role for PD-1 in regulation of immune responses (31). The cytoplasmic tail of PD-1 contains two tyrosine residues, one that belongs to an ITIM, and the other an immunoreceptor tyrosine-based switch motif (ITSM) (Fig. 5). Engagement of PD-1 by either of its two ligands, PD-L1 (B7-H1, CD274) or PD-L2 (B7-DC), induces negative signals through the recruitment of phosphatases that dephosphorylate effector molecules involved in downstream TCR or BCR signaling. PD-1 signaling plays an important role in inducing and maintaining peripheral tolerance as PD-Ligands (PD-Ls) on APCs inhibit autoreactive T cells and induce peripheral tolerance, and those on parenchymal cells prevent tissue destruction by suppressing effector T cells to maintain tolerance (48). The inhibitory role of PD-1 is highlighted by the phenotypes of PD-1-deficient mice, which develop various background-dependent autoimmune diseases (49, 50). It is of considerable interest that recent studies have shown PD-1 to be an important regulator of pathogen-induced immune anergy (51). Functional impairment or exhaustion of virus-specific CD8+ T cells in chronic viral infections in mice [e.g. lymphocytic choriomeningitis virus (LCMV) infection] and in humans [e.g. human immunodeficiency virus (HIV) infection] are associated with elevated PD-1 expression on the exhausted cells (52, 53). Importantly, blockade of the PD-1:PD-L interactions reverses the exhaustion of the virus-specific T cells and restores effector functions, cytokine production and proliferation. Upregulation of the PD-1:PD-L pathway also plays a central role in the immune evasion mechanisms exploited by a variety of other pathogens, including, bacteria, parasites (51) and fungi (54).
Human and mouse PD-1 share ∼60% amino acid identity, while the PD-1 extracellular IgV domain shares only very modest sequence identity with CD28 (21%), CTLA-4 (16%) and ICOS (14%). Consistent with this modest sequence identity, PD-1 exhibits several features that set it apart from members of the CD28 costimulatory family. Although the gene encoding PD-1 is located on the same chromosome (chromosome 2 in humans and chromosome 1 in mice) as the CD28/CTLA4/ICOS cluster, it is separated by a distance of ∼25 cM (Fig. 2) (55). Another unique feature of PD-1 is its oligomeric state. The nearly invariant extracellular cysteine in the stalk region of CD28, CTLA4 and ICOS is absent from PD-1 (Fig. 5). Therefore, PD-1 is incapable of forming covalent dimers and it has been shown to exist as a monomer on the cell surface (56). Furthermore, PD-1 lacks the proline-rich ligand binding motif present in all CD28, CTLA4, and ICOS receptors. These differences in primary sequence and oligomeric behavior suggest differences in the mode of ligand recognition and a distinct signaling mechanism.
The ligands, PD-L1 and PD-L2 (human chromosome 9; Fig. 2), both have a domain organization similar to the B7 ligands, but differ in their expression patterns and exhibit different affinities for PD-1. PD-L2 expression is restricted to activated macrophages and dendritic cells, while PD-L1 has a three-fold lower affinity and is widely expressed not only on APCs, but also on non-hematopoietic cells and T cells. Recent findings have shown that PD-L1 expressed on T cells delivers negative signals upon binding to B7-1 expressed on APCs (57). In turn, PD-L1 expressed on APCs specifically binds B7-1 on T cells, inhibiting T-cell responses. This bi-directional inhibitory interaction between B7-1 and PD-L1 increases the complexity of T cell costimulation, as competitive binding interactions between multiple molecules provide mechanisms for linking the PD-1, CTLA-4 and CD28 pathways.
The recent structure of the PD-1:PD-L1 complex by Garboczi and colleagues (58) and our structure of the PD-1:PD-L2 complex (59) highlight the overall similarity of these two inhibitory complexes (Fig. 9). Most strikingly, despite the low sequence identity of the receptors, the PD-1:PD-L and CTLA-4:B7 complexes exhibit gross similarities in overall organization, albeit with significant detailed differences (Fig. 9). Like the CTLA-4:B7 complexes, the PD-1:PD-L interface is formed by the front ®-sheets of both PD-1 and the PD-L IgV domains; however, the CTLA-4 and the B-7 IgV domains cross at ∼90°, as opposed to 60° in the PD-1:PD-L complexes. The PD-1:PD-L interfaces are formed by residues distributed over the front sheets and associated loops of both molecules, in contrast to the proline-rich FG loops in CD28, CTLA-4, and ICOS that make the majority of the contacts with their B7 ligands. Furthermore, consistent with the primary sequence differences (e.g., lack of proline-rich motif), the FG loop in PD-1 makes few and no contacts with PD-L1 and PD-L2, respectively.
Fig. 9. Comparison of the PD-1:PD-L and CTLA-4:B7 complexes.
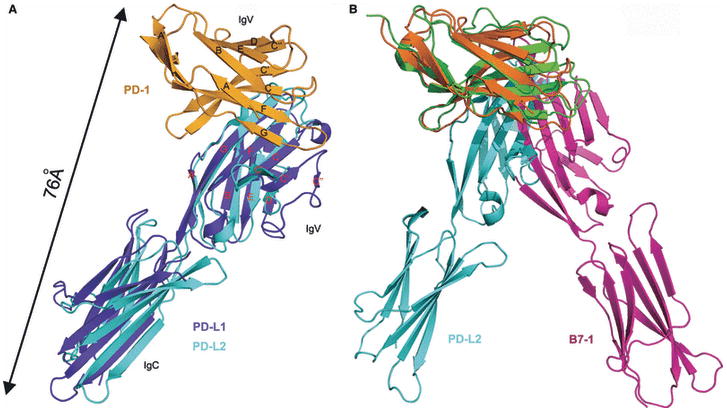
(A) PD-L1 and PD-L2 form similar complexes with PD-1. Superposition of the PD-1:PD-L1 and PD-1:PD-L2 complexes shows very similar overall structure features. (B) Different organization of the PD-1:PD-L and CTLA-4:B7 complexes. Superposition of the PD-1:PD-L2 and CTLA-4:B7-1 complexes reveal different overall organizations. The PD-1:PD-L complexes are more compact, spanning an end-to-end distance of ∼76 Å, compared to ∼100Å in the CTLA-4:B7 complexes. Longer stalk regions in PD-1 and PD-Ls presumably compensate for this difference, allowing for all of these complexes to be recruited to the central zone of the immunological synapse.
Of particular note, the more acute angle between IgV domains in the PD-1:PD-L complexes results in more compact assemblies with end-to-end distances that span ∼76 Å, as compared to the CTLA-4:B7 complexes that span ∼100 Å. These differences in dimension must be accommodated in order to support the localization of both types of inhibitory complex to the central zone of the immunological synapse. Notably, the linker regions connecting the ectodomains and transmembrane segments are longer for PD-1 (20 residues) and PD-L1 (10 residues) and PD-L2 (11 residues) than those present in CTLA-4 (6 residues) and B7-1 (9 residues), and could easily allow the PD-1:PD-L complexes to span an end-to-end distance comparable to the linear dimensions of the pMHC:TCR complex and other pairs of signaling molecules (i.e., ∼100-140 Å) within the immunological synapse (Fig. 8).
Mutagenesis studies have validated the crystal structures of the PD-1:PD-L complexes, as alteration of residues at the crystallographically observed interface result in significant changes in receptor:ligand affinity. The importance of such confirmatory studies cannot be over-emphasized, as by definition, crystallization absolutely requires the formation of intermolecular contacts, and it can sometimes be a considerable challenge to identify those intermolecular interactions that are biologically meaningful. Beyond their value in validation, these structure-based mutagenesis experiments can provide unanticipated and highly useful results. For example, we have identified two single point mutants of the PD-1 receptor that exhibit novel biochemical properties. The first mutant A99L, exhibited 2 and 3-fold higher affinity for PD-L1 and PD-L2, respectively. Although a relatively modest enhancement in affinity, the therapeutic potential of such a reagent can be appreciated by the consideration of belatacept, a modified CTLA-4-Ig fusion protein in clinical trials for renal transplantation, which exhibits only a two-fold increase in avidity for the B7 ligands but a ten-fold enhancement in biological activity (60, 61). The second mutant, L95R, exhibited wild type affinity for PD-L2, but essentially no binding to PD-L1. This remarkable uncoupling of ligand recognition provides a novel opportunity to dissect the distinct roles of the two PD-1 ligands. Soluble forms of these mutant proteins (e.g., Ig fusions) are supporting mechanistic studies by allowing for the manipulation of the PD-1:PD-L pathways in cell-based and animal model systems, and may afford new targeted therapeutic approaches. Furthermore, we are now generating knock-in mouse models expressing these two biochemically defined PD-1 mutants to begin defining in vivo structure-function relationships for costimulation and adaptive immunity. This integrated atoms-to-animals approach maximally leverages high resolution structural information by supporting the generation of functionally altered mutants that ultimately support in vivo mechanistic analyses.
TIM family receptors
TIM family receptors regulate T-cell activation and tolerance and appear to play important roles in the etiology of asthma, autoimmune, and allergic diseases (62). In mice, the TIM family genes are tightly clustered on chromosome 11 and encode four expressed genes (TIM-1 through TIM-4) and four additional predicted genes (TIM-5 through TIM-8). Human orthologs of TIM-1, TIM-3 and TIM-4 are tightly clustered on chromosome 5 and are all expressed on the cell surface (63) (Fig. 2). All TIM family receptors are type-I transmembrane proteins, containing an ectodomain composed of a single membrane distal IgV domain and a membrane proximal mucin domain of varying length that can be heavily modified with O-linked carbohydrate (Fig. 10) (64). The N-terminal IgV domains of TIM receptors share ∼40% sequence identity and, with the exception of TIM-4, all family members contain a tyrosine-kinase phosphorylation motif in the cytoplasmic domain (65). These molecules exhibit distinct expression patterns and interact with a wide range of ligands, suggesting they play important roles in integrating multiple signaling pathways and processes.
Fig. 10. Overall organization of TIM family receptors.
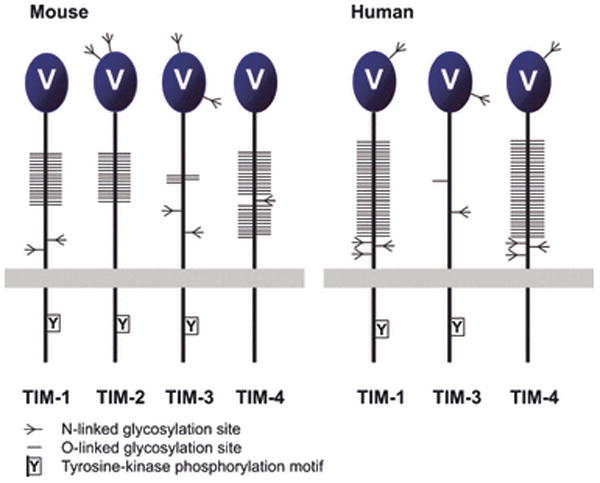
The TIM family receptors are encoded by tightly clustered genes (see Fig. 2). All TIM receptors possess an IgV domain and a variable length mucin domain that can be highly highly O-glycosylated, followed by stalk region, transmembrane region, and cytoplasmic tail with tyrosine-based signaling motif, except for TIM-4.
A wide range of approaches, including agonist and blocking antibodies and knockout mouse models, have demonstrated that the TIM family members play important roles in T helper (Th) cell type 1 (Th1) Th1/Th2 polarization (66-68). Recent in vivo studies have also implicated TIM-1 in the regulation of Th17 and regulatory T cell (Treg) functions (69, 70). It is notable that TIM-4, which is expressed on APCs, has been reported as a ligand for TIM-1 and signaling through this pathway appears to positively regulate CD4+ T-cell activity (71). It has also been reported that the IgV domains of TIM-1 and TIM-4 can bind to a wide variety of cell lines in a calcium-dependent manner, suggesting the existence of an evolutionarily conserved ligand(s) for TIM-1 and TIM-4 (72, 73). Semphorin4A (Sema4A) and the ferritin heavy chain (H-ferrtin) have been reported as putative ligands of TIM-2 (74, 75) and blockade of TIM-2:Sema4A interaction inhibits the development of experimental autoimmune encephalomyelitis, a Th1-dependent disease (75). Ferritin is a large multi-subunit assembly that serves as an intracellular iron storage particle and can also bind extracellularly to subsets of lymphocytes and myeloid cells (76). Ectopic expression of TIM-2 facilitates the uptake of H-ferritin, but the exact immunoregulatory roles of the TIM-2:H-ferritin interaction remain to be elucidated (74). Galectin-9 specifically recognizes N-linked carbohydrate moieties on the IgV domain of TIM-3, and results in the induction of apoptosis of Th1 cells but not Th2 cells (77). Most interestingly, it was recently reported that TIM-3 up-regulation resulted in exhaustion of HIV-1-specific CD8+ T cells from HIV infected patients and that blockade of the TIM-3 signaling pathway restored the function of these HIV-1-specific T cells (78).
Given the involvement of the TIM family in a wide range of normal, pathological and therapeutically important processes, these molecules, and their complexes, were clear candidates for high resolution structural analysis. However, an equally compelling reason for pursuing structural analysis of this family is the remarkable and unique primary sequence signature present in all family members. As illustrated in Fig. 3, in addition to the two cysteines that form the canonical disulfide linkage between the B and F strands, every TIM family IgV domain identified to date also possesses four additional cysteine residues that are not present in any other Ig superfamily members. The invariance of these four cysteines within the TIM family, and their absence in all other Ig superfamily members, immediately suggests that TIM family members possess unique structural features that are likely to be important for function. Most simply, one would predict that these four cysteines form either four intermolecular disulfide bonds at a dimer interface, or two intramolecular disulfide bonds.
Our recent crystal structure of murine TIM-3 and the contemporaneous structures of TIM-1 and TIM-2 by Santiago et al. (65, 79) reveal that these four invariant cysteines form two intramolecular disulfide bonds that are responsible for the novel restructuring of the classic IgV fold. This reorganization is characterized by the close apposition of the CC′ and FG loops, resulting in the formation of a continuous surface not previously observed in any other members of the Ig superfamily (Fig. 11) (79). The subsequent structure determination of TIM-4 confirmed the presence of this unique cleft structure in all TIM family receptors (80). Directly relevant to the functional role of this unique cleft, we and Wilker et al., reported that TIM-3 exhibited galectin-9-independent binding to a very wide range of primary mammalian immune cells, established cancer cell lines and insect cells (72, 79). This behavior suggested the recognition of a ubiquitous and evolutionarily conserved ligand such as a highly conserved protein or protein-associated glycan. Despite the unknown identity of this ligand(s), through a series of mutagenesis experiments we demonstrated that the unique cleft formed by the CC′ and FG loops was directly responsible for this binding activity (79).
Fig. 11. The TIM family IgV Domains are unique in the Ig superfamily.
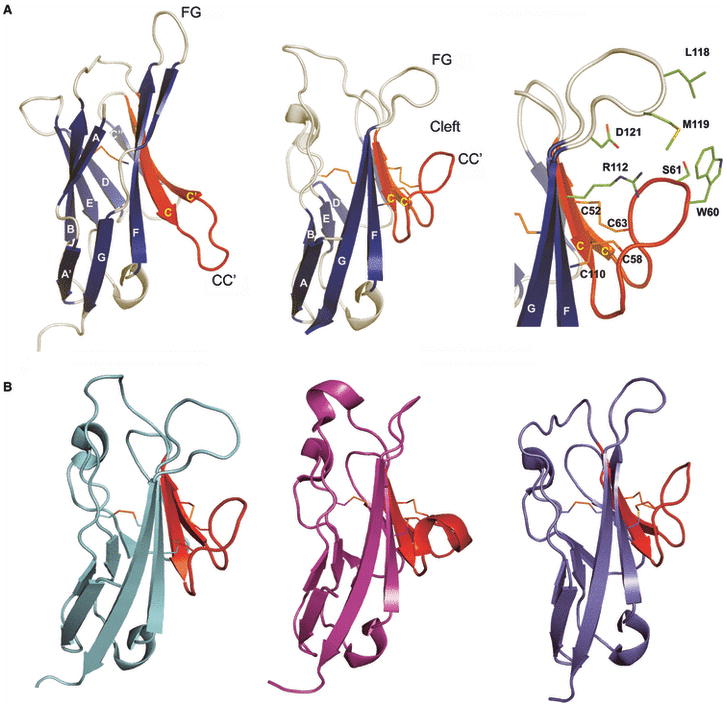
(A) Comparison of a typical IgV domain (PD-1; PDB Code 1NPU) and the TIM-3 (PDB Code 2OYP) IgV domain. The strands are labeled, as are the CC′ and FG loops; the C and C′ strands and the CC′ loop are displayed in red. Residues in TIM-3 CC′-FG cleft are highlighted in green stick representation. (B) Structures of TIM-1 (PDB Code 2OR8), TIM-2 (PDB Code 2OR7), and TIM-4 (PDB Code 3BIB). The C and C′ strands and the CC′ loop are colored as red. These structures highlight the unique structural variation present in all TIM family.
New functional roles within the TIM family were recently brought to light with the demonstration that TIM-1 and TIM-4 expressed on macrophages act as phosphatidylserine (PS) receptors and play important roles in the clearance of apoptotic cells (81-83). In vitro biochemical approaches demonstrated that both TIM-1 and TIM-4 bound PS with a Kd in the nanomolar range, while TIM-2 and TIM-3 did not appear to exhibit detectable binding to PS (82, 83). Consistent with these in vitro activities, transfection of fibroblasts with TIM-1 and TIM-4, but not TIM-2 and TIM-3, resulted in enhanced phagocytosis of apoptotic cells. This activity could be inhibited by TIM-1 or TIM-4 specific mAb. TIM-4 blocking mAb also inhibited the phagocytosis of apoptotic cells by macrophages in vitro (82, 83).
Notably, the crystal structure of TIM-4 bound to PS demonstrated that the unique CC′-FG cleft is a metal-dependent ligand site for PS (80). The PS molecule binds to TIM-4 with its polar head group penetrating into the cleft formed by the CC′ and FG loops, while the hydrophobic alkyl chains point away from the cleft, as would be expected for the recognition of membrane associated PS (Fig. 12). The metal ion is coordinated by the side chains of N121 and D122, main chain carbonyl oxygens from the FG loop, a phosphoryl oxygen from PS and one water molecule. N121 and D122 and the main chain atoms are invariant in mouse and human TIM-1, TIM-3 and TIM-4, suggesting that all of these family members are metalloproteins. In contrast, the equivalent of D122 in TIM-4 is phenylalanine (F119) in TIM-2, which cannot participate in metal ion coordination, suggesting functional specialization for TIM-2. In addition to its role in metal coordination, the PS headgroup also forms several hydrogen bonds with the side chains of S62 and D122 and the main chain nitrogen of K63. The two hydrophobic fatty acid chains of PS interact with the side chains of W119 and F120 in the FG loop. Interestingly, in mouse TIM-1, TIM-3 and TIM-4, S62 and D122 are invariant and the equivalents of residues 119 and 120 are all hydrophobic. Although recent reports indicate that PS and other phospholipids are not ligands for TIM-3, the apparent sequence and structural conservation at the ligand binding site suggest that this point merits re-examination.
Fig. 12. Structure of TIM-4:phosphatidylserine (PS) complex.
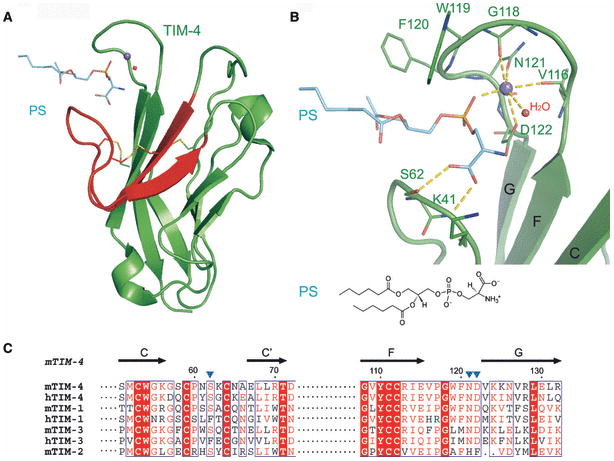
(A) Overall arrangement of the TIM-4:PS structure. TIM-4 exploits the CC′-FG cleft to bind PS in a metal-dependent fashion (purple sphere) involving one water molecule (red sphere) (PDB Code 3BIB). The C and C′ strands and the CC′ loop are highlighted in red. (B) Detailed view of the PS binding site. The TIM-4:PS binding site, including the CC′-FG cleft, metal atom, PS and water, in the same orientation as panel A. The metal atom (purple sphere) is coordinated to side chains of N121, D122, main chain of V116 and G118, PS and water. The PS head group forms hydrogen bonds with side chains of S62 and K63. (C) The multiple sequence alignment of CC′ and FG loop in human and mouse TIM family members. Residues with greater than 50% conservation are colored red; invariant residues are in bold white with red background. The secondary structure of the TIM-4 IgV domain is shown above the alignment.
It has also been reported that TIM-1, TIM-3 and TIM-4 can recognize cell surface carbohydrates in a calcium-dependent manner (72). This behavior is consistent with the suggestion that all three proteins possess similar metal-dependent ligand binding sites. In vitro screening identified a number of potential carbohydrate ligands for TIM-3, of which the top candidates are complex N-glycans, suggesting a potential epitope for TIM-3 recognition (72). In addition, the interaction between TIM-1 and TIM-4 has been proposed to be the consequence of interactions between the IgV domains of each molecule and the opposing mucin domain, suggesting the recognition of O-linked glycans (72). It is of particular note that glycan, PS and cell surface binding are metal-dependent processes, suggesting that these ligands compete for the same or overlapping binding sites associated with the unique CC′-FG surface.
The TIM family represents a remarkable example of the relationship between sequence, structure and biological function. The TIM family also highlights the power of hypothesis-driven structural biology, where unique primary sequence signatures are used to identify targets for structure determination. These structures are directly driving functional studies, as they revealed a novel surface on the TIM family molecules that is responsible for the recognition of diverse glycan and PS ligands. These studies provide the foundation for future in vitro and in vivo efforts focusing on the roles of the TIM family members in coordinating and integrating multiple intersecting signaling pathways.
CD2/SLAM family heterophilic and homophilic interaction
The SLAM family, a subset of the CD2 family, modulates the activity of a wide range of immune cell types, including T cells, B cells, macrophages, dendritic cells, and NK cells (84). Interactions involving SLAM family members on these immune cells control a variety of responses including, T-cell activation, memory-B-cell generation, antibody production, and NK-cell activation (84). The SLAM family is composed of nine members: SLAM, CD84, natural killer-, T-, and B-cell antigen (NTB-A), 2B4, CD48, lymphocyte antigen 9 (Ly9), CD2-like receptor activating cytotoxic cells (CRACC), B lymphocyte activator macrophage expressed (BLAME), and SLAM family member 9 (SF2001) (85). In humans, seven of these proteins are encoded by a cluster of immediately adjacent genes on chromosome 1, with the other two (BLAME and SF2001) in close proximity (Fig. 2). The genes encoding CD2 and CD58 are close to one another (i.e., separated by two intervening genes) at a second locus on chromosome 1 (Fig. 2). With the exception of CD58, orthologs of each these CD2/SLAM family genes are present, and similarly arranged, in the mouse genome (86).
Although the extracellular domains of these receptors share less than 15% identity, the CD2/SLAM family members exhibit a common organization, with an N-terminal membrane-distal IgV and a membrane-proximal IgC2 domain; Ly-9 is the sole exception with a tandem repeat of this IgV-IgC2 motif (Fig. 13) (87). It is notable that all CD2/SLAM family IgV domains lack the disulfide bond that typically links the B and F strands, highlighting their evolutionary relatedness. With the exception of CD48, all SLAM family receptors contain ITSMs in their cytoplasmic tails, which serve as docking sites for the SLAM-associated protein (SAP), as well as the related Ewing's sarcoma-associated transcript (EAT)-2 and EAT-2-related transducer (ERT) proteins (88). SAP recruits the Fyn tyrosine kinase, which triggers a signaling cascade that ultimately results in modulation of interferon interferon-γ (IFN-γ) secretion in T cells and the stimulation of Th2 responses (89). The SAP:Fyn pathway also operates in NK cells and enhances cytotoxicity against tumor cells (90). In contrast to SAP, EAT-2 and ERT both repress NK cell cytotoxicity and IFN-γ secretion.
Fig. 13. Domain organization of the CD2/SLAM family receptors.
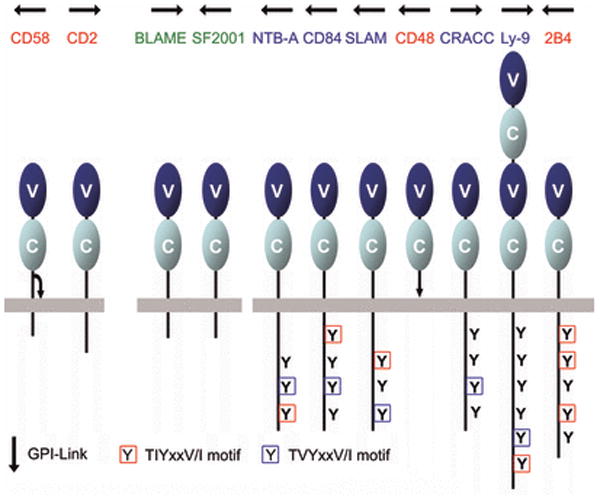
Seven of these genes are immediately proximal to one another; two others are nearby; CD2 and CD58 form a second cluster on the same chromosome (see Fig. 2). All CD2/SLAM family members are composed of a membrane distal IgV domain and a membrane proximal IgC2 domain; Ly-9 is the sole exception with tandem repeats of IgV-IgC2 motif. Tyrosine-based signaling motifs are highlighted. The GPI-linkages in CD48 and CD58 are denoted as arrows. The homophilic receptors and the heterophilic receptors are labeled in red and blue, respectively. Receptors with unknown ligand are denoted in green.
Notably the binding partners of all characterized SLAM/CD2 family members reside within the CD2/SLAM family itself (Fig. 13). Both homophilic (e.g. CD150, CD84, NTB-A and Ly-9) and heterophilic (e.g. 2B4 with CD48; CD2 with CD58) receptor:ligand interactions occur via interactions involving their extracellular IgV domains (91-96). Gene proximity, overall architectural similarities, receptor/ligand binding specificity and their related biological functions support the hypothesis that the CD2/SLAM family arose via successive gene duplication events. Several high resolution structures have revealed the biologically important structural similarities shared by these molecules and have highlighted the functionally relevant differences associated with individual family members.
The first structural description of a CD2/SLAM family interaction was of the heterophilic complex formed by the CD2 and CD58 IgV domains (97). The CD2:CD58 complex is characterized by an equilibrium dissociation constant (Kd) of ∼10 μM (98), and exhibits an asymmetric assembly formed by interactions involving the front sheets of each IgV domain (Fig. 14). Notably, the interface is almost entirely ionic in nature, with nine basic residues from CD2 and eleven acidic residues from CD58 participating in ten salt bridges. Recently, the crystal structure of a second heterophilic complex, formed by the IgV domains of 2B4 and CD48 (Kd∼ 4 μM), was reported (99). This structure also revealed an overall architecture grossly similar to the CD2:CD58 complex (99), with the front faces of the 2B4 and CD48 IgV forming the heterophilic dimer interface composed of predominantly hydrophilic residues (99) (Fig. 14). While both CD2:CD58 and 2B4:CD48 complex interfaces are dominated by polar interactions, the distribution and the number of salt bridges (ten versus six) and hydrogen bonds (five versus 18) are significantly different between these two complexes.
Fig. 14. Specificity of homophilic and heterophilic interactions within CD2/SLAM family.
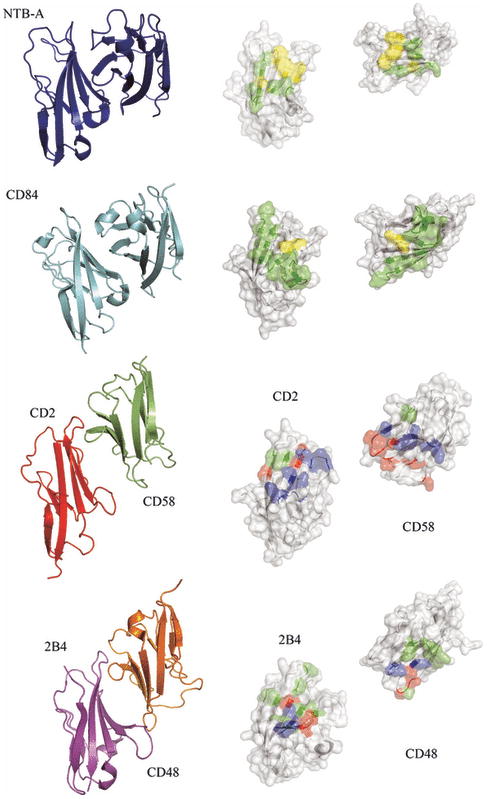
The left panels show ribbon representations of the structures of the interacting IgV domains of NTB-A (PDB Code 2IF7) and CD84 (PDB Code 2PKD) homophilic dimers and the CD2:CD58 (PDB Code 1QA9) and 2B4:CD48 heterophilic dimers (2PTT). The right panels show surface representations of the homophilic or heterophilic dimer interfaces; the two molecules are each rotated 90° in opposite directions about a vertical axis to expose the dimer interface. The residues involved forming hydrogen bonds and hydrophobic interactions across the dimer interface are colored as green and yellow, respectively. Positively and negatively charged residues involved in ionic interactions across the dimer interface are colored as blue and red, respectively. Each pair of molecules presents a distinct set of surface features that underlie binding specificity.
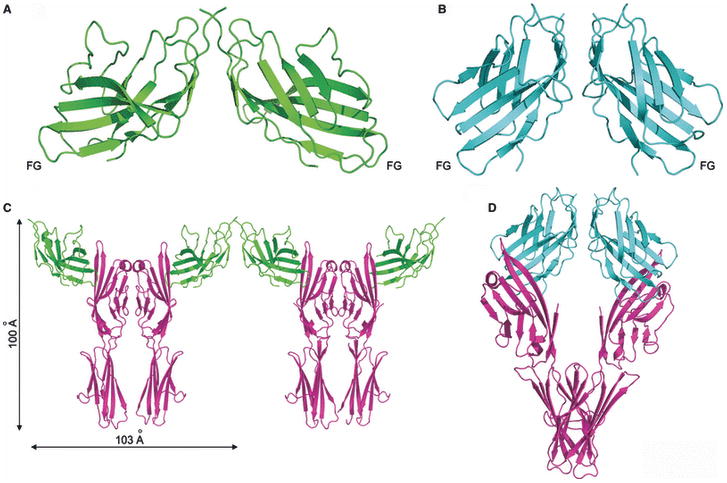
The chemical and physical features responsible for homophilic receptor recognition and signaling in the SLAM family were first defined by our crystal structure of the full length human NTB-A ectodomain (Ly108 in mice) (100) (Fig. 15). The N-terminal IgV domain and the C-terminal IgC2 domains are organized as an extended rod-like structure with dimensions of ∼20 × 25 × 85Å. A similar organization is observed in the structure of the full length CD2 ectodomain, suggesting that this architecture is common to all members of the CD2/SLAM family. In the NTB-A crystal structure, the front sheets of the IgV domains interact to form a two-fold symmetric dimer in which the interface is formed by the nearly orthogonal association of the C, C′, C″, and F strands from each monomer (100). This mode of association results in a highly kinked NTB-A dimer with an end-to-end linear distance of ∼100Å, and provides a model for all homophilic and heterophilic interactions within the CD2/SLAM family. Six residues from each monomer are involved in eleven potential hydrogen bonds, while a tightly packed cluster of aromatic residues forms the center of the interface (Fig. 14-15). Thus, both hydrogen bonding and hydrophobic packing interactions, but not ionic interactions, contribute to the NTB-A homophilic association that is characterized by a Kd of ∼2 μM.
Fig. 15. Structure and organization of the NTB-A homophilic dimer.
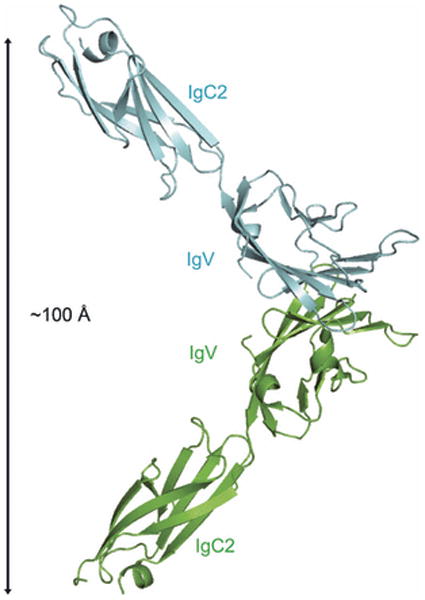
The crystal structure of full length NTB-A shows a homophilic dimer formed by the interaction of the two front sheets of the IgV domains (cyan and green); the IgV and IgC2 domains are labeled (PDB Code 2IF7). The end-to-end distance of NTB-A homophilic dimer is ∼100 Å and this organization is consistent with the engagement of monomers from two interacting cell surfaces.
The structure of the CD84 IgV domain provides a second example of a SLAM family homophilic interaction (87). Similar to NTB-A, the CD84 IgV domain forms a symmetric dimer with the front sheets packing in a nearly orthogonal fashion. The similar association of the IgV domains in NTB-A and CD84 indicates that the dimer of full length CD84 is also highly kinked and spans an end-to-end distance of ∼100 Å. In contrast to NTB-A, the CD84 interface is composed predominantly of neutral polar residues, with few hydrophobic residues (Fig. 14). Solution studies demonstrate that the CD84 dimer is at least an order of magnitude more stable than the NTB-A dimer and is characterized by a Kd with an upper limit in the hundred nM range.
The structures of the CD2:CD58 and 2B4:CD48 complexes, and those of the CD84 and NTB-A homodimers provide the basis for defining the determinants responsible for the overlapping and unique functions of the members within the CD2/SLAM family. Both homophilic and heterophilic dimers show overall similarities, with their interfaces formed by the front sheets of the interacting IgV domains. However, these interfaces all possess distinct chemical and physical properties with different geometric complementarities and unique distributions of hydrophobic, polar, and ionic groups. These differences likely play a primary role in promoting the formation of physiologically important homophilic and heterophilic associations, while at the same time preventing inappropriate interactions among the CD2/SLAM family receptors.
In addition to contributing to receptor:ligand specificity, differences in these interfaces directly control the strength of the homophilic and heterophilic interactions. The NTB-A and SLAM (CD150) homophilic interactions are characterized by Kds of 2 μM and 200 μM, respectively, with the Kd for CD84 dimerization residing in the hundred-nM range (87, 100), while the CD2:CD58 and 2B4:CD48 heterophilic interactions are characterized by Kds of 10 μM and 4 μM, respectively. This range of three orders of magnitude in affinities provides a mechanistic basis for the overlapping, but distinct, biological functions exhibited by the SLAM family members. As the engagement of most SLAM family members direct the recruitment of similar cytoplasmic signaling proteins (e.g., SAP, EAT, and ERT), the wide range of homophilic and hetrophilic affinities may allow for the signalling pathways operating through these proteins to be optimally tuned to appropriately respond to the wide range of challenges encountered in vivo.
Of particular importance, our existing structures provide a rational basis for the generation of mutants with altered biochemical properties and we have already produced NTB-A and CD84 mutants that span at least a 100-fold range in Kd. These reagents now provide a unique opportunity to carefully dissect the contribution of homophilic and heterophilic affinity and association/dissociation kinetics to immunity in cell-based and whole animal models (i.e., knock-in models). These capabilities are relevant to recent reports that extensive polymorphisms within SLAM family genes, including four polymorphisms located in the first IgV domain of Ly-9 (CD229), contribute to susceptibility to systemic lupus erythematosis (SLE). Two of these polymorphisms are predicted to reside at the Ly-9 homophilic interface, suggesting these polymorphisms may contribute to the disease by affecting the affinity and/or kinetics of the Ly-9 homophilic interaction (101). In addition to differences in affinities and association/dissociation rates, the SLAM family receptors are also distinguished by unique sequences in the cytoplasmic tails responsible for binding adaptor and signalling molecules. The importance of these differences is highlighted by the recent report that NTB-A (Ly108) splice variants possessing different numbers of ITSMs (i.e., two or three) exhibit distinct signalling capabilities in B cells. Specifically, the isoform with three ITSMs, and presumably an enhanced capability to recruit intracellular signalling and scaffolding proteins, sensitizes immature B cells to undergo apoptosis, suggesting that Ly108 may censor self-reactive B cells and protect against autoimmune responses (102). These results indicate that SLAM family function may rely on a complex interplay between affinity and recruitment activity.
While the structures of homophilic and heterophilic complexes have identified differences that may contribute to the specific functions of the individual CD2/SLAM family members, they also highlight common features that are essential for these diverse family members to function co-ordinately and synergistically within the context of the numerous cell-cell interactions that are required for innate and adaptive immunity. Of particular importance are the overall architectural features that are likely shared by all homophilic and heterophilic pairs in the CD2/SLAM family. As described, the rigid rod-like design of the CD2/SLAM family monomers in combination with the nearly orthogonal association of the IgV domains results in a highly kinked structure that spans an end-to-end distance of ∼100 Å. This distance is comparable to the linear dimensions of other pairs of signaling molecules that localize within the T cell and NK cell immunological synapses (e.g., TCR:pMHC, killer cell Ig-like receptor (KIR):human leukocyte antigen-C (HLA-C), and CTLA-4:B7). These similarities in molecular dimension may allow for all two-domain SLAM/CD2 family receptors to co-localize, along with their cytoplasmic signaling molecules, within the immunological synapse and bridge the T cells and NK cells with APCs.
The multiple structures of the homophilic and heterophilic pairs within the SLAM/CD2 family have highlighted the importance of systematically examining the structural features of evolutionarily related proteins encoded by physically proximal clusters of genes. These structures have revealed the chemical and physical features that control specificity, affinity and likely the half-lives of these interactions, all of which make important contributions to function. These structures have also revealed general architectural features needed to integrate the function of all family members into the overall immune response. Perhaps most exciting, these structures are supporting the development of biochemically defined mutants that can be exploited in cell-based and knock-in mouse models to generate in vivo structure-function correlations that allow for the mechanistic dissection of SLAM/CD2 family function.
The TNF:TNFR superfamilies
High resolution structures of TNF family ligands and their receptors TNFRs have given unique insights into aspects of receptor:ligand specificity, oligomeric state, and overall molecular architecture that are important for function. The TNF/TNFR superfamilies modulate activation, proliferation and survival signals important for a wide array of T cell-mediated immune responses. At present, 19 TNF ligands and 29 TNFRs have been described, and a considerable number of cognate TNF:TNFR interactions among these family members are known to play direct roles in immunity and T-cell regulation. Some prominent examples include, TNFα/TNFβ and their receptors, OX40L:OX40, glucocorticoid-induced tumor necrosis factor receptor (GITR) ligand (GITRL):GITR, CD70:CD27, 4-1BBL:4-1BB, CD30L:CD30, LIGHT: herpes virus entry mediator (HVEM), and TL1A:DR3 (21, 103-105). TNF family members are typically type-II transmembrane proteins, a number of which can be proteolytically released from the plasma membrane as soluble forms. Central to TNF superfamily function is the C-terminal extracellular TNF homology domain (THD), characterized by a conserved signature of aromatic and hydrophobic residues, which forms non-covalently associated trimers (Fig. 16). The individual protomers adopt a classic β-sandwich ‘jelly-roll’ fold, with inner and outer sheets composed of A′AHCF and B′BGDE strands, respectively. TNFR family molecules are, in general, type-I transmembrane proteins characterized by pseudorepeats of one to four extracellular cysteine-rich domains (CRDs). Existing structures of TNF ligands in complex with their receptors display a common organization in which a TNF trimer engages three elongated receptor molecules at the grooves formed by the interfaces between each pair of adjacent TNF protomers (103, 104) (Fig. 16). These interactions result in a three-fold symmetric complex, with 3:3 receptor:ligand stoichiometry and a separation of ∼35 Å between individual receptor molecules. Importantly, these geometric constraints are also imposed on the cytoplasmic tails of the receptors and are believed to direct their clustering so as to facilitate recruitment of the signal adapter proteins like TNF receptor-associated factors (TRAFs) and/or death domain proteins, resulting in the activation of downstream signaling pathways (104). The generalization of this model to the entire superfamily, however, requires additional structural data because the members of the TNF/TNFR superfamilies share relatively low sequence identity in the range of ∼15-20% (103). In fact, recent studies have revealed unique structural features within these superfamilies, suggesting significant deviations from this common paradigm for signal transduction mechanism. Here we discuss some of these recent structural insights and their possible impact on the immune function of TNF/TNFR family costimulatory molecules.
Fig. 16. Structure and assembly of a classical TNF trimer.
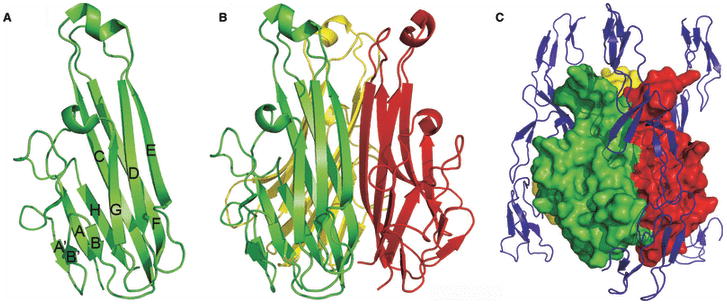
(A) TNF-β monomer. Ribbon diagram of human TNF-β monomer (PDB Code 1TNR). The ten anti-parallel β-strands are labeled. (B) Conventional TNF trimer. Ribbon diagram of human TNF-β trimer shows typical compact architecture of conventional THDs. (C) Conventional TNF:TNFR complex (PDB Code 1TNR). Human TNF-β trimer (shown in surface representation) engages its receptor (blue ribbon) to yield a complex with 3:3 receptor:ligand stoichiometry and results in a separation of ∼35 Å between individual receptor molecules.
On the basis of sequence and structure, the TNF superfamily can be subdivided into the conventional, the EF-disulfide and the divergent families (103). The conventional family includes TNF, FasL, receptor activator of nuclear factor κB ligand (RANKL), CD40L, LIGHT, lymphotoxin α (LTα), LTβ, TNF-related apoptosis inducing ligand (TRAIL), and TL1A, of which six have been structurally characterized. These structures, and additional sequence considerations, indicate that all members of this family will exhibit the characteristic tightly packed truncated pyramidal or bell-shaped trimeric assembly as the consequence of relatively long connecting loops between the CD, DE and EF strands and possess a disulfide bond linking the CD and EF loops. The EF-disulfide family includes tumor necrosis factor ligand superfamily member 13 (APRIL), TNF (ligand) superfamily member 13b (BAFF), EDA, and TNF (ligand) superfamily, member 12 (TWEAK), of which three have been structurally characterized. Members of this family all contain a disulfide bond linking the E and F strands and are more globular in appearance than the conventional family due to the presence of shorter CD and EF loops. In contrast to the conventional and EF-disulfide families, until very recently, no member of the divergent family had been structurally characterized. The members that compose the divergent family, OX40L, GITRL, CD27L, CD30L, and 4-1BBL, exhibit extensive sequence divergence with each other and with the members of the conventional and EF-disulfide families. Most notably, several members of the divergent family, OX40L, GITRL, and CD30L, appeared to possess THD that were significantly shorter (∼120-125 residues) than those found in most other TNF superfamily members (∼150 residues). Thus structural studies on members of this family might be particularly informative in terms of structure, function and mechanism. Indeed, this prediction has been borne out by the recent reports of the structures of OX40L and its complex with OX40 (106), and the subsequent structures of human and murine GITRL (107-110). The GITRL studies are particularly revealing and are highlighted below.
GITRL
GITRL is a recently discovered member of the TNF superfamily that binds its receptor GITR present on both effector and Tregs. Engagement of these molecules induces a positive costimulatory signal leading to increased T-cell proliferation and cytokine production (105, 111, 112). Interestingly, GITR activation on effector T cells has also been shown to inhibit the suppressive effects of Tregs in mice (113-115). The GITRL:GITR-mediated costimulatory pathway therefore appears to be a potentially important therapeutic target for manipulating T cell immune responses in various diseases including autoimmunity, viral infections and tumors (112).
Atypical human GITRL structure
We and Zhou et al. (107, 109), recently reported the crystal structure of human GITRL, which displayed the classic THD jelly-roll topology. However, as predicted from amino acid sequence analysis, the human GITRL THD is only ∼119 residues long, and is thus considerably shorter than conventional THDs (∼150 aa) (103). OX40L (a THD of ∼126 residues) and CD30L (with a predicted THD of ∼117 residues) are the only other known examples of TNF family members to possess such a similar short THD (103, 106). The reduced size of the THD in human GITRL is the consequence of two pairs of shortened β-strand pairs, the C and F strands in the inner face, and the D and E strands in the outer face of the molecule.
The most striking feature of the human GITRL crystal structure is that the human GITRL protomers self-assemble to form a homotrimer with an atypical expanded organization that significantly contrasts the more compact truncated pyramidal assembly of the classical TNF trimers (103, 107, 109) (Fig. 17). As a consequence, the protomers in the human GITRL trimer are oriented at an angle of ∼45° with respect to the trimer axis, while in most other TNF ligands the subunits are displaced by only ∼20-30°. This difference in orientation leads to a strikingly reduced height of the human GITRL trimer (∼40 Å) compared to most other TNF trimers (∼60 Å). Furthermore, as a consequence of the greater displacement of human GITRL subunits from the trimer axis, the human GITRL trimer shows substantially smaller intersubunit interfaces with correspondingly fewer contacts between interacting protomers (i.e, ∼40 residues contributing to the inter-protomer interface of human TNF-α versus ten residues in human GITRL). The conventional TNF trimers assemble such that one edge of each protomer is tightly packed against the inner sheet of its neighbor, forming a large and mostly hydrophobic inter-subunit interface that runs the entire extent of the molecule. In contrast, the inter-protomer interfaces in the human GITRL are confined to the lower half of the trimer interior. The lack of inter-protomer contacts in the upper half of the human GITRL trimer is the consequence of the strikingly short length of the EF loop.
Fig. 17. Atypical TNF trimers.
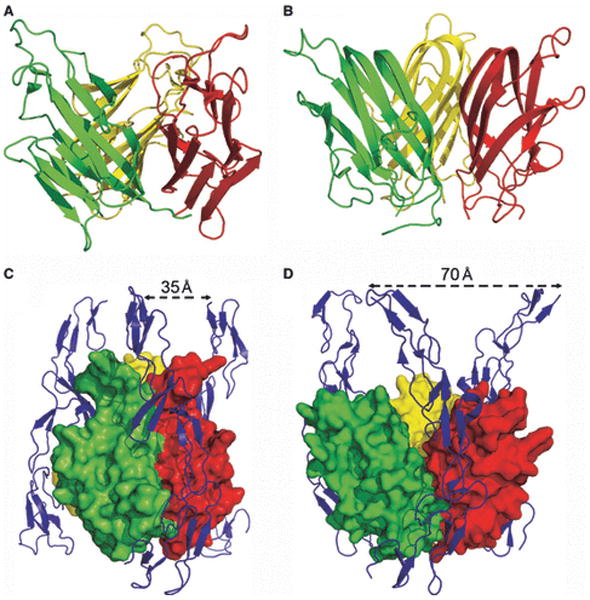
(A) and (B) Ribbon diagrams of human GITRL (A: PDB Code 2Q1M) and human OX40L (B: PDB Code 2HEV) from the divergent family of the TNF superfamily exhibiting an atypical expanded trimeric assemblies. (C) and (D) Comparison of a conventional TNF:TNFR complex (C: PDB Code 1TNR) and the atypical human OX40L:OX40 complex (D: PDB Code 2HEV), highlighting the differences in overall organization, including the distinct placement of the receptor C-termini.
It is notable that OX40L, another member of the divergent family, is the only other structurally characterized TNF ligand to possess a roughly comparable expanded organization with sparse inter-protomer interfaces (106) (Fig. 17). Despite differences in overall organization, the structure of the OX40L:OX40 complex demonstrates that this divergent TNF ligand binds its cognate receptor with 3:3 stoichiometry at the interfaces between each pair of adjacent TNF protomers in a fashion similar to other TNF:TNFR complexes (Fig. 17). Importantly, mutagenesis experiments also support a similar mode of receptor recognition for human GITRL (107). Though similar to the conventional ligands, the specific differences in the organization of these divergent ligands may impact downstream signaling. For example, recruitment of the trimeric signal adaptor molecules, TRAFs, depends on the presence of a specific recruitment motif on the TNFRs cytoplasmic domains, as well as on the particular geometry of the receptor cytoplasmic tails (116, 117). Structures of TRAFs bound to peptides corresponding to the TRAF-recruiting segments of TNFR cytoplasmic tails are consistent with an assembly with 3:3 stoichiometry, in which the peptides are separated from each other by a distance of ∼55 Å. Most TNF trimers place the membrane-proximal C-terminal ends of three TNFRs at the vertices of an equilateral triangle with an edge length of ∼35 Å (Fig. 17). In contrast, the atypical expanded structures of OX40L and human GITRL would be predicted to support the binding of receptors such that their cytoplasmic tails are positioned at a considerably greater distance. Indeed the structure of the human OX40L:OX40 complex shows that the C-termini of the receptors are separated by a distance of ∼70 Å (106) (Fig. 17). Thus, the overall architecture of each ligand imposes specific constraints on the spatial organization of the associated receptor, which in combination with the length and composition of the receptor cytoplasmic tail may contribute to the energetics of the different receptor–TRAF interactions. These structural and energetic characteristics would promote the biologically appropriate/optimal recruitment of adaptor and signaling molecules.
The unusual expanded organization of human GITRL has considerable impact on its self-association behavior in solution. The sparse inter-protomer interfaces in the human GITRL trimer result in a highly reversible monomer-trimer equilibrium, a phenomenon that has not been reported for other members of the TNF superfamily (103, 104, 107, 118, 119). Of particular importance, this dynamic self association behavior has a profound effect on the biochemical, and hence the biological activity of human GITRL, as soluble human GITRL exhibits a considerably weaker apparent affinity (Kd ∼ 560 nM) for its receptor than other TNF ligands (Kd ∼ 0.1–10 nM) (107). As only intact trimers of TNF ligands are competent to bind receptor, on the basis of thermodynamic considerations, the existence of the monomer–trimer equilibrium reduces the effective affinity between receptor and ligand because an energetic penalty must be paid to drive ligand trimerization (Fig. 18). Consequently, not all of the interaction energy goes to support the human GITRL:GITR binding interaction, but some of it must be used for the formation of the intermolecular interfaces in human GITRL trimer. This finding is supported by the observation that the forced trimerization of human GITRL with a triple helical coiled coil sequence results in a ∼100-fold increase in affinity for receptor (Fig. 18). The in vivo impact of this dynamic equilibrium will depend on the cell surface expression level of human GITRL, as well as any specific effects of the plasma membrane environment. The dynamic behavior of human GITRL may represent a mechanism to ensure that the receptor:ligand interaction is not too tight, but is carefully tuned to give the biologically optimal signal. Indeed, under conditions typically used to study GITR costimulation, the enforced trimeric version of human GITRL resulted in a significant enhancement of the T-cell proliferation response as compared to the WT human GITRL (107). In addition, a number of reports have suggested that GITRL can support ‘reverse signaling’ in a variety of circumstances (120), including the production of proinflamatory cytokines by macrophages, immune evasion by cancer cells and the activation of IDO in plasmacytoid DCs (121-124). These activities that operate through human GITRL would also be expected to be sensitive to putative reversible self-association processes on the cell surface.
Fig. 18. Dynamic self-assembly of human GITRL.
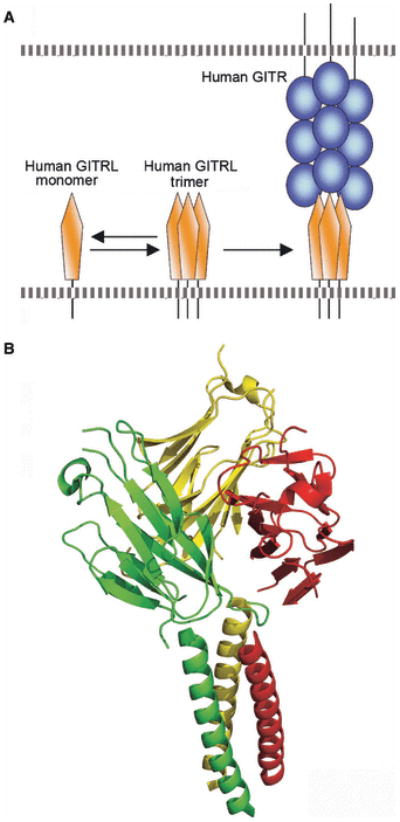
(A) Schematic of the reversible monomer-trimer equilibrium of human GITRL. As only the trimeric GITRL is competent bind receptor, this self-association behavior imposes an energetic penalty that results in modest apparent receptor binding affinity. (B) Ribbon diagram of the high affinity coiled-coil construct of human GITRL trimer that does not exhibit measurable dissociation (PDB Code 2R32).
The TNF superfamily provides a powerful example of the application of genome-wide sequence considerations to define families that aid in the identification of candidate molecules for structure determination. The targeting of GITRL and OX40L on the basis of sequence resulted in the discovery of a new trimeric organization within the TNF superfamily. The GITRL structure was particularly informative as it revealed a novel self-association processes that may be biologically relevant and most importantly allowed for the generation of GITRL constructs with altered dissociation properties that directly impacted function. It will be of particular interest to examine the structural and biochemical properties of CD30L, which also possesses a notably short THD.
An unexpected surprise: mouse GITRL adopts a novel dimeric assembly
Human and mouse orthologs of GITRL/GITR display a sequence identity of ∼50-60%, which is comparable to human and mouse orthologs of other TNF/TNFR family members (125, 126). Surprisingly, human GITRL does not recognize the mouse receptor, and mouse GITRL does not bind the human receptor (127), suggesting that these putative orthologs do not share a common mechanisms for the recognition of their cognate binding partners. Furthermore, recent reports suggest that GITRL:GITR signaling may play different roles in mice and humans (128), as, in contrast to mice, in humans the Treg-mediated suppression of effector T-cell function is not inhibited by GITR stimulation (129). The mechanistic basis for these distinct costimulatory effects remains to be defined.
We and Zhou et al. (108, 110) recently reported the crystal structure of mouse GITRL, which as expected exhibits the typical TNF superfamily jelly-roll topology. Most remarkably however, in the crystalline state mouse GITRL is organized as a novel two-fold symmetric dimer (Fig. 19), a quaternary structure completely novel in the TNF family. In the mouse GITRL dimer, the two subunits are oriented at an angle of ∼40° with respect to each other, with significant association of the inner β-strands from the two engaging protomers. The most notable aspect of murine GITRL dimer is the conformation of the C-terminus of each protomer, which is markedly different from the equivalent segment in the human ortholog. The C-terminus of each murine subunit adopts a conformation such that it contacts the A strand and the AB loop of the adjacent subunit, resulting in a classic example of ‘domain swapping’ (130) (Fig. 19). This strand exchange is stabilized by a set of specific interactions, with Phe171 and Ile172 (at the C-terminus) from one subunit interacting with His64 (A strand) and Pro68 (AB loop) from the other subunit, respectively. Although Phe171 and Ile172 are conserved in both mouse and human proteins, in human GITRL His64 and Pro68 are replaced by Gln and Ser, respectively (Fig. 19). Thus, the hydrophobic and packing interactions observed in the mouse GITRL dimer are not available in the human ortholog. Furthermore, the four residue linker sequence (167Pro-Asp-Leu-Pro170) that connects the immediate C-terminus to the core of the ectodomain in mouse GITRL is distinct in the human protein (Ala-Asn-Pro-Gln). This specific sequence, including the presence of prolines at positions 167 and 170, may bias the conformation of the mouse GITRL C-terminal segment and thus favor the observed dimeric assembly.
Fig. 19. Novel dimeric organization of mouse GITRL.
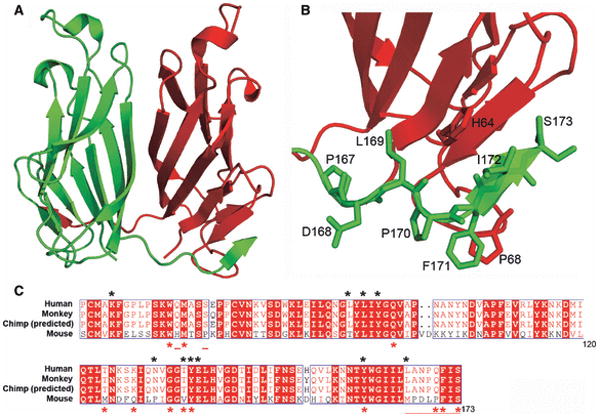
(A) Mouse GITRL dimmer. This structure exhibits a novel dimer that involves domain swapping of the C-termini (PDB Code 2QDN). (B) Detailed view of the C-terminal domain swap. (C) GITRL amino acid sequence alignment. Residues involved in hydrogen bond interactions at the murine dimer interface are denoted with red asterisks. Residues involved in the domain-swap interactions are underlined in red. Residues forming contacts at the subunit interfaces of human GITRL trimer are marked with black asterisks on the top of the alignment.
The unique dimeric structure of mouse GITRL immediately provides an explanation for the lack of cross reactivity between the human and mouse receptors and ligands. In the murine structure, the equivalents of the loops that form the receptor binding sites in the conventional TNF ligands are organized in a completely different fashion. Furthermore, the mutagenesis of these loops in mouse GITRL does not affect the interaction between the mouse receptor and ligand (108). These structural and biochemical observations clearly demonstrate that mouse GITRL exploits a mode of receptor engagement that is distinct from other members of the TNF superfamily.
The unique dimeric assembly of mouse GITRL also impacts the mechanisms of signaling and may begin to provide a basis for its distinct biological properties. Engagement of the mouse receptor and ligand would likely result in an assembly with a 2:2 receptor:ligand stoichiometry. While the cytoplasmic tail of mouse GITR contains the TRAF recruitment motifs typical of other TNFR family members, the unique dimeric assembly would seem to preclude a typical interaction with trimeric TRAFs. These properties suggest the formation of unique signaling assemblies that may ultimately be responsible for the distinct outcome of GITRL:GITR engagement in mouse and human.
Structure-function analysis with the GITRL costimulatory system represents a case where considerable unanticipated insights were revealed. Despite a high degree of sequence identity, very modest local changes in primary sequence resulted in dramatically altered structure and biochemical activity that may be directly related to biological function. Indeed it is possible that these studies have caught the evolution of new function in the GITRL/GITR subfamily. Based on the conservation of residues involved in the domain swap in mouse GITRL, known GITRL sequences appear to fall into two groups, human-like sequences that can be predicted to form trimers (e.g., monkey, chimpanzee) and the mouse GITRL sequence that forms dimer (Fig. 19). These studies provide a particularly important cautionary note for the development of animal models of human diseases, as it would appear that the murine GITRL:GITR system does not faithfully represent all aspects of human GITRL/GITR structure, function and mechanism. Finally, these studies highlight the need for improved informatics tools to predict further surprises, and underscore the value of the structural characterization for even closely related proteins, especially those with clinical and therapeutic potential.
Concluding remarks
We have presented a number of important examples where structural information has directly contributed to our mechanistic understanding of costimulation by defining the features responsible for receptor:ligand specificity, affinity, oligomeric state, and valency. Structures of the CD28:B7 families highlighted how alterations in oligomeric state and valency can influence the types of complexes that are available for signaling. Structures of the SLAM family revealed the strategies employed to direct the formation of multiple discrete homophilic and heterophilic complexes with overlapping and distinct functional properties. The TIM family provided a remarkable example of how unique primary sequence features can remodel a classic protein fold to support new biological function. The structure of human GITRL shows another direct correlation between unique sequence and structural features, which in this case result in unanticipated dynamic self-association properties that directly impact function. Finally, the structure of murine GITRL reminds us that many surprises remain to be discovered, as this molecule exhibits a completely unexpected organization (i.e., dimer rather than trimer) that directly impacts receptor recognition and function. In addition to these properties that rely on specific atomic interactions, the structures of these costimulatory receptor:ligand complexes have defined their overall geometries and dimensions. These architectural features are critical determinants for the localization of receptor:ligand pairs to the immunological synapse and for the integration of these interactions and associated signaling pathways into overall cellular physiology and immunity.
Importantly, the remarkable diversity of structural, biochemical, and functional features described above are direct consequences of unique primary sequence signatures within the individual costimulatory families. In the future we envision broad efforts that exploit this powerful, albeit simple, relationship as a strategy to identify those proteins for which structure determination would most readily reveal new biology. We further envision that insights obtained from these structures will serve as the basis to design specifically tailored mutant costimulatory molecules in order to define in vivo structure-function correlations in mammalian model systems. This integrated atoms-to-animals paradigm will provide a comprehensive approach for defining atomic and molecular mechanisms and may result in the identification of new therapeutic strategies that target costimulatory pathways.
Acknowledgments
The work and ideas described here were supported by National Institute of Health Grants RO1AI007289, U54 GM074945 (New York SGX Research Center for Structural Genomics; NYSGXRC), Immunooncology & Immunology Training Program (5T32CA009173), Cancer Center Support Grant (P3OCAO13330), and the Albert Einstein Macromolecular Therapeutics Development Facility.
References
- 1.Roughton FJW, Kendrew JC. A symposium based on a conference held at Cambridge in June 1948 in memory of Sir Joseph Barcroft. Presented at A symposium based on a conference held at Cambridge in June 1948 in memory of Sir Joseph Barcroft; Cambridge. 1948. [Google Scholar]
- 2.Perutz MF, Muirhead H, Cox JM, Goaman LC. Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: the atomic model. Nature. 1968;219:131–139. doi: 10.1038/219131a0. [DOI] [PubMed] [Google Scholar]
- 3.Almo SC, et al. Structural genomics of protein phosphatases. J Struct Funct Genomics. 2007;8:121–140. doi: 10.1007/s10969-007-9036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Page R, Deacon AM, Lesley SA, Stevens RC. Shotgun crystallization strategy for structural genomics II: crystallization conditions that produce high resolution structures for T. maritima proteins. J Struct Funct Genomics. 2005;6:209–217. doi: 10.1007/s10969-005-1916-7. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz JC, Zhang X, Nathenson SG, Almo SC. Structural mechanisms of costimulation. Nat Immunol. 2002;3:427–434. doi: 10.1038/ni0502-427. [DOI] [PubMed] [Google Scholar]
- 6.Meyaard L, Adema GJ, Chang C, Woollatt E, Sutherland GR, Lanier LL, Phillips JH. LAIR-1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity. 1997;7:283–290. doi: 10.1016/s1074-7613(00)80530-0. [DOI] [PubMed] [Google Scholar]
- 7.Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305) J Leukoc Biol. 2008;83:799–803. doi: 10.1189/jlb.0907609. [DOI] [PubMed] [Google Scholar]
- 8.Lebbink RJ, et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med. 2006;203:1419–1425. doi: 10.1084/jem.20052554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lebbink RJ, de Ruiter T, Kaptijn GJ, Bihan DG, Jansen CA, Lenting PJ, Meyaard L. Mouse leukocyte-associated Ig-like receptor-1 (mLAIR-1) functions as an inhibitory collagen-binding receptor on immune cells. Int Immunol. 2007;19:1011–1019. doi: 10.1093/intimm/dxm071. [DOI] [PubMed] [Google Scholar]
- 10.Jansen CA, et al. Regulated expression of the inhibitory receptor LAIR-1 on human peripheral T cells during T cell activation and differentiation. Eur J Immunol. 2007;37:914–924. doi: 10.1002/eji.200636678. [DOI] [PubMed] [Google Scholar]
- 11.Martin AM, Kulski JK, Witt C, Pontarotti P, Christiansen FT. Leukocyte Ig-like receptor complex (LRC) in mice and men. Trends Immunol. 2002;23:81–88. doi: 10.1016/s1471-4906(01)02155-x. [DOI] [PubMed] [Google Scholar]
- 12.Lebbink RJ, et al. The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J Immunol. 2008;180:1662–1669. doi: 10.4049/jimmunol.180.3.1662. [DOI] [PubMed] [Google Scholar]
- 13.Stelzl U, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 14.Rual JF, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 15.Venkatesan K, et al. An empirical framework for binary interactome mapping. Nat Methods. 2008;6:83–90. doi: 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 17.Higurashi M, Ishida T, Kinoshita K. Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 2008;17:72–78. doi: 10.1110/ps.073196308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 19.Pitti RM, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature. 1998;396:699–703. doi: 10.1038/25387. [DOI] [PubMed] [Google Scholar]
- 20.Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem. 1999;274:13733–13736. doi: 10.1074/jbc.274.20.13733. [DOI] [PubMed] [Google Scholar]
- 21.Migone TS, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 22.Takai Y, Miyoshi J, Ikeda W, Ogita H. Nectins and nectin-like molecules:roles in contact inhibition of cell movement and proliferation. Nat Rev Mol Cell Biol. 2008;9:603–615. doi: 10.1038/nrm2457. [DOI] [PubMed] [Google Scholar]
- 23.Yu X, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 24.Dong X, et al. Crystal structure of the V domain of human Nectin-like molecule-1/Syncam3/Tsll1/Igsf4b, a neural tissue-specific immunoglobulin-like cell-cell adhesion molecule. J Biol Chem. 2006;281:10610–10617. doi: 10.1074/jbc.M513459200. [DOI] [PubMed] [Google Scholar]
- 25.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 26.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 27.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 28.Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol. 2006;24:65–97. doi: 10.1146/annurev.immunol.24.021605.090535. [DOI] [PubMed] [Google Scholar]
- 29.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 30.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 31.Okazaki T, Iwai Y, Honjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Curr Opin Immunol. 2002;14:779–782. doi: 10.1016/s0952-7915(02)00398-9. [DOI] [PubMed] [Google Scholar]
- 32.Young JR, Davison TF, Tregaskes CA, Rennie MC, Vainio O. Monomeric homologue of mammalian CD28 is expressed on chicken T cells. J Immunol. 1994;152:3848–3851. [PubMed] [Google Scholar]
- 33.Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature. 2001;410:604–608. doi: 10.1038/35069112. [DOI] [PubMed] [Google Scholar]
- 34.Stamper CC, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. 2001;410:608–611. doi: 10.1038/35069118. [DOI] [PubMed] [Google Scholar]
- 35.Ostrov DA, Shi W, Schwartz JC, Almo SC, Nathenson SG. Structure of murine CTLA-4 and its role in modulating T cell responsiveness. Science. 2000;290:816–819. doi: 10.1126/science.290.5492.816. [DOI] [PubMed] [Google Scholar]
- 36.Evans EJ, et al. Crystal structure of a soluble CD28-Fab complex. Nat Immunol. 2005;6:271–279. doi: 10.1038/ni1170. [DOI] [PubMed] [Google Scholar]
- 37.Kariv I, Truneh A, Sweet RW. Analysis of the site of interaction of CD28 with its counter-receptors CD80 and CD86 and correlation with function. J Immunol. 1996;157:29–38. [PubMed] [Google Scholar]
- 38.Peach RJ, et al. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med. 1994;180:2049–2058. doi: 10.1084/jem.180.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Truneh A, et al. Differential recognition by CD28 of its cognate counter receptors CD80 (B7.1) and B70 (B7.2): analysis by site directed mutagenesis. Mol Immunol. 1996;33:321–334. doi: 10.1016/0161-5890(95)00077-1. [DOI] [PubMed] [Google Scholar]
- 40.Bhatia S, Edidin M, Almo SC, Nathenson SG. Different cell surface oligomeric states of B7-1 and B7-2: implications for signaling. Proc Natl Acad Sci U S A. 2005;102:15569–15574. doi: 10.1073/pnas.0507257102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chattopadhyay K, Bhatia S, Fiser A, Almo SC, Nathenson SG. Structural basis of inducible costimulator ligand costimulatory function:determination of the cell surface oligomeric state and functional mapping of the receptor binding site of the protein. J Immunol. 2006;177:3920–3929. doi: 10.4049/jimmunol.177.6.3920. [DOI] [PubMed] [Google Scholar]
- 42.Ikemizu S, et al. Structure and dimerization of a soluble form of B7-1. Immunity. 2000;12:51–60. doi: 10.1016/s1074-7613(00)80158-2. [DOI] [PubMed] [Google Scholar]
- 43.Collins AV, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 44.Grohmann U, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3:1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 45.Stone JD, Chervin AS, Kranz DM. T-cell receptor binding affinities and kinetics:impact on T-cell activity and specificity. Immunology. 2009;126:165–176. doi: 10.1111/j.1365-2567.2008.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bromley SK, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 47.Choudhuri K, Wiseman D, Brown MH, Gould K, van der Merwe PA. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature. 2005;436:578–582. doi: 10.1038/nature03843. [DOI] [PubMed] [Google Scholar]
- 48.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27:195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 50.Nishimura H, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 51.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 52.Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 53.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 54.Lazar-Molnar E, Gacser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus Histoplasma capsulatum. Proc Natl Acad Sci U S A. 2008;105:2658–2663. doi: 10.1073/pnas.0711918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finger LR, Pu J, Wasserman R, Vibhakar R, Louie E, Hardy RR, Burrows PD, Billips LG. The human PD-1 gene: complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitors. Gene. 1997;197:177–187. doi: 10.1016/s0378-1119(97)00260-6. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337–347. doi: 10.1016/s1074-7613(04)00051-2. [DOI] [PubMed] [Google Scholar]
- 57.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin DY, et al. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci U S A. 2008;105:3011–3016. doi: 10.1073/pnas.0712278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lazar-Molnar E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc Natl Acad Sci U S A. 2008;105:10483–10488. doi: 10.1073/pnas.0804453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larsen CP, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5:443–453. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 61.Vincenti F, et al. Costimulation blockade with belatacept in renal transplantation. N Engl J Med. 2005;353:770–781. doi: 10.1056/NEJMoa050085. [DOI] [PubMed] [Google Scholar]
- 62.Kuchroo VK, Dardalhon V, Xiao S, Anderson AC. New roles for TIM family members in immune regulation. Nat Rev Immunol. 2008;8:577–580. doi: 10.1038/nri2366. [DOI] [PubMed] [Google Scholar]
- 63.Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol Med. 2005;11:362–369. doi: 10.1016/j.molmed.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 64.Kuchroo VK, Umetsu DT, DeKruyff RH, Freeman GJ. The TIM gene family: emerging roles in immunity and disease. Nat Rev Immunol. 2003;3:454–462. doi: 10.1038/nri1111. [DOI] [PubMed] [Google Scholar]
- 65.Santiago C, Ballesteros A, Tami C, Martinez-Munoz L, Kaplan GG, Casasnovas JM. Structures of T Cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity. 2007;26:299–310. doi: 10.1016/j.immuni.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chakravarti S, et al. Tim-2 regulates T helper type 2 responses and autoimmunity. J Exp Med. 2005;202:437–444. doi: 10.1084/jem.20050308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Monney L, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–541. doi: 10.1038/415536a. [DOI] [PubMed] [Google Scholar]
- 68.Umetsu SE, et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. 2005;6:447–454. doi: 10.1038/ni1186. [DOI] [PubMed] [Google Scholar]
- 69.Degauque N, et al. Immunostimulatory Tim-1-specific antibody deprograms Tregs and prevents transplant tolerance in mice. J Clin Invest. 2008;118:735–741. doi: 10.1172/JCI32562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ueno T, et al. The emerging role of T cell Ig mucin 1 in alloimmune responses in an experimental mouse transplant model. J Clin Invest. 2008;118:742–751. doi: 10.1172/JCI32451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meyers JH, et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005;6:455–464. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- 72.Wilker PR, Sedy JR, Grigura V, Murphy TL, Murphy KM. Evidence for carbohydrate recognition and homotypic and heterotypic binding by the TIM family. Int Immunol. 2007;19:763–773. doi: 10.1093/intimm/dxm044. [DOI] [PubMed] [Google Scholar]
- 73.Sizing ID, et al. Epitope-dependent effect of anti-murine TIM-1 monoclonal antibodies on T cell activity and lung immune responses. J Immunol. 2007;178:2249–61. doi: 10.4049/jimmunol.178.4.2249. [DOI] [PubMed] [Google Scholar]
- 74.Chen TT, et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med. 2005;202:955–965. doi: 10.1084/jem.20042433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kumanogoh A, et al. Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature. 2002;419:629–633. doi: 10.1038/nature01037. [DOI] [PubMed] [Google Scholar]
- 76.Fargion S, Fracanzani AL, Brando B, Arosio P, Levi S, Fiorelli G. Specific binding sites for H-ferritin on human lymphocytes: modulation during cellular proliferation and potential implication in cell growth control. Blood. 1991;78:1056–1061. [PubMed] [Google Scholar]
- 77.Zhu C, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–1252. doi: 10.1038/ni1271. [DOI] [PubMed] [Google Scholar]
- 78.Jones RB, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cao E, et al. T cell immunoglobulin mucin-3 crystal structure reveals a galectin-9-independent ligand-binding surface. Immunity. 2007;26:311–321. doi: 10.1016/j.immuni.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 80.Santiago C, et al. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity. 2007;27:941–951. doi: 10.1016/j.immuni.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118:1657–1668. doi: 10.1172/JCI34487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kobayashi N, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 84.Veillette A. Immune regulation by SLAM family receptors and SAP-related adaptors. Nat Rev Immunol. 2006;6:56–66. doi: 10.1038/nri1761. [DOI] [PubMed] [Google Scholar]
- 85.Calpe S, et al. The SLAM and SAP gene families control innate and adaptive immune responses. Adv Immunol. 2008;97:177–250. doi: 10.1016/S0065-2776(08)00004-7. [DOI] [PubMed] [Google Scholar]
- 86.Sidorenko SP, Clark EA. The dual-function CD150 receptor subfamily: the viral attraction. Nat Immunol. 2003;4:19–24. doi: 10.1038/ni0103-19. [DOI] [PubMed] [Google Scholar]
- 87.Yan Q, et al. Structure of CD84 provides insight into SLAM family function. Proc Natl Acad Sci U S A. 2007;104:10583–10588. doi: 10.1073/pnas.0703893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Latour S, Veillette A. The SAP family of adaptors in immune regulation. Seminars in Immunology. 2004;16:409–419. doi: 10.1016/j.smim.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 89.Latour S, Gish G, Helgason CD, Humphries RK, Pawson T, Veillette A. Regulation of SLAM-mediated signal transduction by SAP, the X-linked lymphoproliferative gene product. Nat Immunol. 2001;2:681–690. doi: 10.1038/90615. [DOI] [PubMed] [Google Scholar]
- 90.Bloch-Queyrat C, et al. Regulation of natural cytotoxicity by the adaptor SAP and the Src-related kinase Fyn. J Exp Med. 2005;202:181–192. doi: 10.1084/jem.20050449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mavaddat N, et al. Signaling Lymphocytic Activation Molecule (CDw150) Is Homophilic but Self-associates with Very Low Affinity. J Biol Chem. 2000;275:28100–28109. doi: 10.1074/jbc.M004117200. [DOI] [PubMed] [Google Scholar]
- 92.Brown MH, Boles K, Anton van der Merwe P, Kumar V, Mathew PA, Neil Barclay A. 2B4, the Natural Killer and T Cell Immunoglobulin Superfamily Surface Protein, Is a Ligand for CD48. J Exp Med. 1998;188:2083–2090. doi: 10.1084/jem.188.11.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Flaig RM, Stark S, Watzl C. Cutting Edge: NTB-A Activates NK Cells via Homophilic Interaction. J Immunol. 2004;172:6524–6527. doi: 10.4049/jimmunol.172.11.6524. [DOI] [PubMed] [Google Scholar]
- 94.Martin M, et al. CD84 Functions as a Homophilic Adhesion Molecule and Enhances IFN-{gamma} Secretion: Adhesion Is Mediated by Ig-Like Domain 1. J Immunol. 2001;167:3668–3676. doi: 10.4049/jimmunol.167.7.3668. [DOI] [PubMed] [Google Scholar]
- 95.Romero X, et al. CD229 (Ly9) lymphocyte cell surface receptor interacts homophilically through its N-terminal domain and relocalizes to the immunological synapse. J Immunol. 2005;174:7033–7042. doi: 10.4049/jimmunol.174.11.7033. [DOI] [PubMed] [Google Scholar]
- 96.Kumaresan PR, Lai WC, Chuang SS, Bennett M, Mathew PA. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Molecular Immunology. 2002;39:1–8. doi: 10.1016/s0161-5890(02)00094-9. [DOI] [PubMed] [Google Scholar]
- 97.Wang Jh, Smolyar A, Tan K, Liu Jh, Kim M, Sun ZyJ, Wagner G, Reinherz EL. Structure of a Heterophilic Adhesion Complex between the Human CD2 and CD58 (LFA-3) Counterreceptors. Cell. 1999;97:791–803. doi: 10.1016/s0092-8674(00)80790-4. [DOI] [PubMed] [Google Scholar]
- 98.van der Merwe PA, Barclay AN, Mason DW, Davies EA, Morgan BP, Tone M, Krishnam AK, Ianelli C, Davis SJ. Human cell-adhesion molecule CD2 binds CD58 (LFA-3) with a very low affinity and an extremely fast dissociation rate but does not bind CD48 or CD59. Biochemistry. 1994;33:10149–10160. doi: 10.1021/bi00199a043. [DOI] [PubMed] [Google Scholar]
- 99.Velikovsky CA, Deng L, Chlewicki LK, Fernandez MM, Kumar V, Mariuzza RA. Structure of natural killer receptor 2B4 bound to CD48 reveals basis for heterophilic recognition in signaling lymphocyte activation molecule family. Immunity. 2007;27:572–584. doi: 10.1016/j.immuni.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cao E, et al. NTB-A Receptor Crystal Structure: Insights into Homophilic Interactions in the Signaling Lymphocytic Activation Molecule Receptor Family. Immunity. 2006;25:559–570. doi: 10.1016/j.immuni.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 101.Wandstrat AE, et al. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769–780. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 102.Kumar KR, et al. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006;312:1665–1669. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- 103.Bodmer JL, Schneider P, Tschopp J. The molecular architecture of the TNF superfamily. Trends Biochem Sci. 2002;27:19–26. doi: 10.1016/s0968-0004(01)01995-8. [DOI] [PubMed] [Google Scholar]
- 104.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 105.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 106.Compaan DM, Hymowitz SG. The crystal structure of the costimulatory OX40-OX40L complex. Structure. 2006;14:1321–1330. doi: 10.1016/j.str.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 107.Chattopadhyay K, et al. Assembly and structural properties of glucocorticoid-induced TNF receptor ligand: Implications for function. Proc Natl Acad Sci U S A. 2007;104:19452–19457. doi: 10.1073/pnas.0709264104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chattopadhyay K, Ramagopal UA, Brenowitz M, Nathenson SG, Almo SC. Evolution of GITRL immune function:murine GITRL exhibits unique structural and biochemical properties within the TNF superfamily. Proc Natl Acad Sci U S A. 2008;105:635–640. doi: 10.1073/pnas.0710529105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou Z, et al. Human glucocorticoid-induced TNF receptor ligand regulates its signaling activity through multiple oligomerization states. Proc Natl Acad Sci U S A. 2008;105:5465–5470. doi: 10.1073/pnas.0711350105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou Z, et al. Structural basis for ligand-mediated mouse GITR activation. Proc Natl Acad Sci U S A. 2008;105:641–645. doi: 10.1073/pnas.0711206105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nocentini G, Ronchetti S, Cuzzocrea S, Riccardi C. GITR/GITRL: More than an effector T cell co-stimulatory system. Eur J Immunol. 2007;37:1165–1169. doi: 10.1002/eji.200636933. [DOI] [PubMed] [Google Scholar]
- 112.Shevach EM, Stephens GL. The GITR-GITRL interaction: co-stimulation or contrasuppression of regulatory activity? Nat Rev Immunol. 2006;6:613–618. doi: 10.1038/nri1867. [DOI] [PubMed] [Google Scholar]
- 113.Ji HB, et al. Cutting edge: the natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J Immunol. 2004;172:5823–5827. doi: 10.4049/jimmunol.172.10.5823. [DOI] [PubMed] [Google Scholar]
- 114.McHugh RS, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–323. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 115.Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins M, Shevach EM. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173:5008–5020. doi: 10.4049/jimmunol.173.8.5008. [DOI] [PubMed] [Google Scholar]
- 116.Park YC, Burkitt V, Villa AR, Tong L, Wu H. Structural basis for self-association and receptor recognition of human TRAF2. Nature. 1999;398:533–538. doi: 10.1038/19110. [DOI] [PubMed] [Google Scholar]
- 117.Ye H, Park YC, Kreishman M, Kieff E, Wu H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol Cell. 1999;4:321–330. doi: 10.1016/s1097-2765(00)80334-2. [DOI] [PubMed] [Google Scholar]
- 118.Eck MJ, Sprang SR. The structure of tumor necrosis factor-alpha at 2.6 A resolution. Implications for receptor binding. J Biol Chem. 1989;264:17595–17605. doi: 10.2210/pdb1tnf/pdb. [DOI] [PubMed] [Google Scholar]
- 119.Wingfield P, Pain RH, Craig S. Tumour necrosis factor is a compact trimer. FEBS Lett. 1987;211:179–184. doi: 10.1016/0014-5793(87)81432-1. [DOI] [PubMed] [Google Scholar]
- 120.Eissner G, Kolch W, Scheurich P. Ligands working as receptors: reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004;15:353–366. doi: 10.1016/j.cytogfr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 121.Baltz KM, et al. Cancer immunoediting by GITR (glucocorticoid-induced TNF-related protein) ligand in humans: NK cell/tumor cell interactions. Faseb J. 2007 doi: 10.1096/fj.06-7724com. [DOI] [PubMed] [Google Scholar]
- 122.Grohmann U, et al. Reverse signaling through GITR ligand enables dexamethasone to activate IDO in allergy. Nat Med. 2007;13:579–586. doi: 10.1038/nm1563. [DOI] [PubMed] [Google Scholar]
- 123.Shin HH, Kim SJ, Lee HS, Choi HS. The soluble glucocorticoid-induced tumor necrosis factor receptor causes cell cycle arrest and apoptosis in murine macrophages. Biochem Biophys Res Commun. 2004;316:24–32. doi: 10.1016/j.bbrc.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 124.Shin HH, Lee MH, Kim SG, Lee YH, Kwon BS, Choi HS. Recombinant glucocorticoid induced tumor necrosis factor receptor (rGITR) induces NOS in murine macrophage. FEBS Lett. 2002;514:275–280. doi: 10.1016/s0014-5793(02)02379-7. [DOI] [PubMed] [Google Scholar]
- 125.Gurney AL, et al. Identification of a new member of the tumor necrosis factor family and its receptor, a human ortholog of mouse GITR. Curr Biol. 1999;9:215–218. doi: 10.1016/s0960-9822(99)80093-1. [DOI] [PubMed] [Google Scholar]
- 126.Tone M, et al. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc Natl Acad Sci U S A. 2003;100:15059–15064. doi: 10.1073/pnas.2334901100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bossen C, et al. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem. 2006;281:13964–13971. doi: 10.1074/jbc.M601553200. [DOI] [PubMed] [Google Scholar]
- 128.Nocentini G, Riccardi C. GITR: a multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur J Immunol. 2005;35:1016–1022. doi: 10.1002/eji.200425818. [DOI] [PubMed] [Google Scholar]
- 129.Levings MK, et al. Human CD25+CD4+ T suppressor cell clones produce transforming growth factor beta, but not interleukin 10, and are distinct from type 1 T regulatory cells. J Exp Med. 2002;196:1335–1346. doi: 10.1084/jem.20021139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Barton GJ. ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]