

Abstract
Background
The cardiac isoform of the sarco/endoplasmic reticulum Ca2+ATPase (SERCA2a) plays a major role in controlling excitation/contraction coupling. In both experimental and clinical heart failure (HF), SERCA2a expression is significantly reduced which leads to abnormal Ca2+ handling and deficient contractility. A large number of studies in isolated cardiac myocytes, in small and large animal models of HF showed that restoring SERCA2a expression by gene transfer corrects the contractile abnormalities and improves energetics and electrical remodeling. Following a long line of investigation, a clinical trial is underway to restore SERCA2a expression in patients with HF using adeno-associated virus type 1.
Objective
This review addresses the following issues regarding HF gene therapy: 1) new insights on calcium regulation by SERCA2a; 2) SERCA2a as a gene therapy target in animal models of HF; 3) advances in the development of viral vectors and gene delivery; 4) clinical trials of HF using SERCA2a
Methods
This review focuses on the new advances in SERCA2a- targeted gene therapy made in the last three years.
Conclusion
SERCA2a is an important therapeutic target in various cardiovascular disorders. Ongoing clinical gene therapy trials will provide answers on its safety and applicability.
Keywords: Sacroplasmic reticulum Ca2+ ATPase, heart failure, gene therapy
Introduction
Heart failure (HF) remains a major cause of morbidity and mortality in the developed world. In the population under the age of 65 HF prevalence approaches 1%1. Despite the fact that pharmacologic and device-based treatments have improved survival, the 5-year mortality rate remains at an alarming 50% in this population2. Faced with these challenges, researchers have vigorously explored novel therapeutic options, including gene-based treatments. Over the last decade, important progress has been made in understanding the various intracellular and molecular mechanisms that become abnormal during HF3–5. One of the key abnormalities in both human HF and experimental models of HF is abnormal intracellular calcium ion (Ca2+) handling caused by a defect in sacoplasmic reticulum (SR) function4, 6. Deficient SR Ca2+ uptake in myocytes is associated with a decrease in the expression and activity of the sarco/ endoplasmic reticulum calcium ATPase cardiac isoform 2a, (SERCA2a)3–5. Unloaded ([Ca2+])SR in turn regulates the effectors of SR Ca2+ release and Ca2+ influx channels, further worsening the abnormal Ca2+ distribution. SERCAs play a critical role in the control of spatio-temporal patterns of intracellular calcium signaling – a pattern, that controls virtually every cellular function, including contraction, proliferation/hyperthrophic growth and apoptosis6, 7. Normalization of SERCA2a function has been shown to increase contractility in failing human cardiomyocytes and to improve hemodynamics along with survival in rodent and large animal models of HF3–5. The overexpression of SERCA2a also has been found to restore energetic supply and to decrease ventricular arrhythmias in a model of ishemia/reperfusion8–10. Furthermore, a number of novel beneficial mechanisms have been uncovered in vascular smooth muscle cells (VSMCs) and endothelial cells (EC)11, 12. Therefore, SERCA2a is one of the most promising targets for the treatment of HF.
Advances in the understanding of the molecular basis of myocardial dysfunction together with the evolution of gene transfer technology, has finally placed CHF within reach of gene-based therapy5, 13, 14. Myocardial dysfunction results in myocardium that has a mixture of diseased, dysfunctional and healthy myocytes. The goal of gene therapy is to treat dysfunctional myocytes and to prevent disease or loss of healthy myocytes.
This review focuses on gene therapy using SERCA2a or small molecules regulating SERCA2a activity to treat HF. We present new insight in the mechanism of Ca2+ regulation by SERCA2a, examine the complex beneficial effects as well as the risk associated with SERCA2a overexpression. We describe the new advances in the development of viral vectors and gene delivery and discuss the potential of SERCA2a as a therapeutic target in cardiovascular disease.
1. Calcium cycling abnormalities in heart failure
Calcium is crucial to the very process that enables the chambers of the heart to contract and relax, a process called exitation-contraction coupling4–6. During systole, the action potential induces a minor Ca2+-influx through sarcolemmal L-type calcium channel (LTCC) (Figure 1). This triggers further calcium release from the SR via the major SR Ca2+-release channel ryanodine receptor (RyR2, the cardiac isoform of RyR). The combination of Ca2+ influx and release raises free intracellular Ca2+ concentration ([Ca2+]) from 0.1–0.2 μM to 2.0 – 10.0 μM, thereby allowing Ca2+ to bind to the myofilament protein troponin C resulting in sarcomere shortening and muscle contraction. Muscle relaxation is initiated by RyR2 closure accompanied by Ca2+ dissociation from the troponin complex and its reuptake into the SR by SERCA2a (75% of Ca2+ removal in human) its removal by the sarcolemmal Na2+/ Ca2+ exchanger (NCX) (25% of Ca2+ removal in human). SERCA pump serves a dual function: 1) to cause muscle relaxation by lowering cytosolic [Ca2+], and 2) to restore SR Ca2+ load necessary for muscle contraction. SERCA2a enzymatic activity is controlled by the inhibitory peptide phospholamban (PLN). In its dephosphorylated form, PLN inhibits SERCA2a activity by lowering its affinity for Ca2+.
Figure 1. Excitation-contraction coupling in cardiomyocytes.
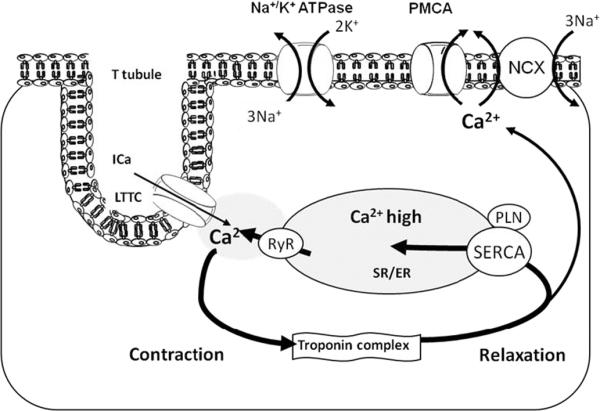
A small amount of Ca2+ uptake via LTCC causes a large amount of Ca2+ release from the SR via RyR – Ca2+-induced Ca2+ release. Rise of intracellular Ca2+ causes contraction of myofilaments. Ca2+ uptake into the SR by SERCA or extrusion of Ca2+ trough NCX channels results in relaxation of myofilaments. Abbreveations: Na-K ATPase: plasma membrane sodium-potassium ATPase; Ca2+: calcium ion; Ica: L-type calcium current; LTCC: L-type calcium channel; K+: potassium ion; PLN: Phospholamban; PMCA: Plasma Membrane Calcium ATPase; RyR: Ryanodine Receptor calcium channel; Na+: sodium ion; NCX: - sodium-calcium exchanger; SR/ER: Sarco/Endoplasmic Reticulum; SERCA: Sarco/Endoplasmic Reticulum Ca2+ ATPase 2a.
The system of Ca2+ cycling is finely regulated by the β-adrenergic signaling system through phosphorylation and dephosphorylation (Figure 2). In response to stress, binding of agonists to β-Adrenergic receptors (βARs) results in adenylyl cyclase (AC) stimulation, cAMP production and activation of protein kinase A (PKA). PKA phosphorylates LTCC and RyR, thus increasing cytosolic Ca2+ influx, whereas phosphorylation of troponin-I increases the overall contractility, mediating the inotropic and chronotropic effect of βAR. Phosphorylation of PLN on serine-16 by PKA relieves its inhibition of SERCA2a and contributes to a frequency-dependent acceleration of SR calcium reuptake, mediating the lusitropic effect of βARs. An additional critical target in calcium cycling is inhibitor-1 (I-1). I-1 becomes active upon PKA phosphorylation and inhibits the type 1 serine/threonine protein phosphatase (PP1) resulting in amplification of βAR responses in the heart15 (Figure 2).
Figure 2. The regulation of calcium cycling via adrenergic pathway and its alterations in failing heart.

Schematic showing the positive control of cardiac function through classic β-adrenergic pathway and a negative control of cardiac function through the α-adrenergic pathway. In response to stress, binding of agonist to the βARs results in inotrope (increased contractility), chronotrope (increased heart rate) and lusiotrope (enhanced relaxation) effects. These effects are mediated by PKA phosphorylation of several targets, resulting in enhanced Ca2+ cycling. Negative control of heart function through αARs is mediated by PP1 dephosphorylation of several PKA targets. Abbreveations: αAR: α-adrenergic receptor; AC: Adenylyl cyclase; βAR: β-adrenergic receptor; βARK: β-adrenergic receptor kinase; CIRC: Calcium induced calcium release; Gαq: Gq protein α-subunit; Gαs: Stimulatory G-protein α-subunit; I-1: Inhibitor 1; Ica: L-type calcium current; LTCC: L-type calcium channel; P: Phosphorylation; PKA: Protein kinase A; PKC: Protein kinase C; PLN: Phospholamban; PLC: Phospholipase C; PP1: Serine/threonine phosphatase 1; RyR: Ryanodine receptor calcium channel; SERCA: Sarco/endoplasmic reticulum Ca2+ ATPase.
Chronic HF is associated with increased sympathetic outflow, which may be compensatory early in the disease state; however long-term neurohormonal activation induces significant damage to cardiomyocytes. Long-term chronic stimulation of βARs results in multiple alterations in the βAR signaling cascade including βAR down regulation, upregulation of βAR kinase (βARK) and increased inhibitory G-protein α-subunit function3, 13. In failing cardiomyocytes the intracellular [Ca2+] is elevated, the amplitude of the [Ca2+] transient is decreased, and its duration is prolonged. These significant alterations in Ca2+ handling along with SR Ca2+ leak trough the RyR, decreased SR Ca2+ uptake and decreased SR Ca2+ content, result in abnormal excitation-contraction coupling3, 5.
The importance of SERCA2a in HF has been studied extensively in both animal models and in humans. These studies have shown that SERCA expression and activity is reduced in failing myocardium3, 5. Increased inhibitory function of PLN16 and increased activity of PP1 resulting in dephosphorylation and inactivation of PKA targets15 also contribute to impaired SR Ca2+ uptake. The common finding is a substantial decrease in SR Ca2+ uptake which is a crucial component of the impaired contractile performance of the failing heart.
In many cases, altered Ca2+ cycling precedes the observed depression of mechanical performance; consequently, improving of Ca2+ cycling has potential as an attractive strategy against HF3, 5.
2. Effect of SERCA2a gene transfer on calcium regulation and cell functions
Calcium is an ubiquitous and pleiotropic second messenger, thus, modulation of calcium signaling by SERCA2a gene transfer can affect multiple functions in different cardiovascular cell types. At least 11 isoforms of SERCA encoded by three separate homologous genes (ATP2A1, ATP2A2 and ATP2A3) have been discovered today7, 17, 18. Given the functional differences between isoforms, only cardiac specific isoform SERCA2a is therapeutically useful, others are or may be harmful or ineffective19–21.
Contractility
Adenovirus-mediated gene transfer of SERCA2a to failing human cardyomyocytes restored the calcium transient and improved contraction and relaxation velocity to the level of non-failing myocytes22. The enhanced contractility induced by SERCA2a overexpression was due to an enhanced SR Ca2+ uptake during diastole, increased SR Ca2+ content, and more effective Ca2+ efflux during systole (Figure 3).
Figure 3. Effect of SERCA2a overexpression on contractile function in diseased heart.

The overexpession of SERCA2a in failing hearts has been shown to result in the recovery of contractility. SERCA2a overexpression improves calcium cycling and myocyte shortening. Abbreveations: CIRC: Calcium induced calcium release; L-type calcium current; LTCC: L-type calcium channel; PLN: Phospholamban; RyR: Ryanodine receptor calcium channel; SERCA: Sarco/endoplasmic reticulum Ca2+ ATPase.
Mechanoenergetics
Calcium overload leading to increased calcium uptake by mitochondria, resulted in mitochondrial dysfunction and decreased ATP production23, 24. Normalization of intracellular calcium by SERCA2a overexpression should prevent mitochondrial dysfunction and decrease energy cost for Ca2+ regulation. Sakata et al. investigated the oxygen cost of left ventricular (LV) contractility, obtained by plotting myocardial oxygen consumption per beat against equivalent maximal elastance8, 9. In diseased hearts, the oxygen cost of LV contractility was increased; SERCA2a gene transfer restored the increased oxygen cost to the normal level. In addition, restoring SERCA2a was shown to correct the creatine kinase activity by normalizing the creatine to ATP ratio in animal model of HF25. Therefore SERCA2a gene transfer restores cardiac energetics both by improving mechanoenergetic efficiency and by enhancing energy supply.
Arrhythmias
Diastolic Ca2+ overload occurring in ischemia and chronic HF is responsible for cellular electric instability resulting in tachyarrythmias5. The increase in Ca2+ ions in the cell and SR leads to generation of asynchronous Ca2+ waves during diastole via abnormal activity of RyRs. These waves activate the electrogenic NCX and thereby produce arrhythmogenic spontaneous aftercontractions and afterdepolarizations26. Recent data demonstrated that SR Ca2+ load via SERCA2a is responsible to sensitization of RyR, thereby creating a traveling wave of high Ca2+ sensitivity initiating Ca2+ release27. SERCA2a overexpression in rabbit cardiomyocytes, favoring sequestration of Ca2+ in the SR, result in rapid inactivation of subsequent Ca2+ currents and reduced Ca2+ entry through LTCC28. In this model, reduction of trans-sarcolemmal Ca2+ flux by SERCA2a overexpression without changes of normal systolic transient also reduced the occurrence of aftercontractions and afterdepolarization28. As such, reduction of cytosolic Ca2+ overload might constitute a viable strategy for combating arrhythmia (Figure 4). In support of this hypothesis, Del Monte et al. showed that SERCA2a gene transfer in a model of ischemia/reperfusion injury effectively suppressed arrhythmias and reduced myocardial infarct size10.
Figure 4. Model showing the effect of SERCA2a overexpression on arrhythmias.

The development of an unstable equilibrium is the mechanistic basis of increased arrhythmogenic risk. At the basal level (left) Ca2+ homeostasis is maintained by the balanced activities of LTCC and NCX, and between RyR2 and SERCA2a. Normal cardiac activation (middle) proportionally increases LTCC, NCX, RyR2 and SERCA function to maintain synchronized Ca2+ fluxes resulting in a stable state. Acute disruption of a single component relative to others (e.g. RyR) results in the ablation of synchronized Ca2+ fluxes and directly increases the arrhythmogenic propensity of the system (right). This state, illustrated using the experimentally observed findings of increased RyR2 leak and NCX activity and diminished SERCA activity, is inherently more prone to destabilization. SERCA2a gene transfer removes Ca2+ overload, stabilizes RyR2 and prevents arrhythmias. Abbreveations: Ca2+: calcium ion; LTCC: L-type calcium channel; PLN: Phospholamban; RyR: Ryanodine Receptor calcium channel; NCX: - sodium-calcium exchanger; SR: Sarcoplasmic Reticulum; SERCA: Sarco/Endoplasmic Reticulum Ca2+ ATPase 2a.
Transcription
Calcium also acts on Ca2+ -dependent transcription pathways via the activation of various kinases and phosphatases6 (Figure 5). The Ca2+-dependent serine/threonine phosphatase calcineurin was identified as a central pro-hypertrophic signaling molecule in the myocardium29. Its target, nuclear factor of activated T lymphocytes (NFAT), was shown to be necessary and sufficient for mediating pathological hypertrophic remodeling30 apoptosis31 and inflammation32 in cardiomyocytes and proliferation of VSMC12. During hypertrophic /proliferative remodeling, reduced SR Ca2+ store and increased expression of transient receptor potential channels (TRPC)7 favor store-operated Ca2+ entry and sustained activation of calcineurin. In VSMC overexpression of SERCA2a inhibits calcineurin activity and NFAT-driven gene transcription12.
Figure 5. Model showing Ca2+ dependent hyperthrophy/proliferation transcription pathways.

Excitation/contraction calcium cycling is not involved in the control of hyperthrophic/proliferation pathways. Cardiac myocytes also contain lipid rafts rich in caveolin in the sarcolemma and T-tubules that are thought to generate local microdomains of Ca2+. Activation of phosphoinositol-3-phosphate-coupled membrane receptors results in calcium release from the SR via IP3R. Depletion of SR calcium close to the sarcolemma induces the formation of Store Operated Calcium complex resulted in calcium influx in local submembrane area. This calcium activates calcineurin (PP2B), which dephosphorylates NFAT, inducing its nuclear translocation and transcriptional activation.
Hypertrophy Proliferation
Myocardial hypertrophy is an adaptive response to hormonal and mechanical stimuli. Several reports suggest that SERCA2a overexpression rescues or prevents cardiac hypertrophy33, 34. Although the mechanism of this benefit was not completely elucidated in the heart, it is speculated that they may result from inhibition of calcineurin, as shown in VSMC12. In VSMC SERCA2b (ubiquitous isoform) and SERCA2a are co-expressed in adulthood. During proliferation VSMC undergo de-differentiation and acquisition of a synthetic phenotype, associated with loss of SERCA2a. Restoring SERCA2a expression in VSMC by gene transfer inhibits VSMC proliferation of and post-injury restenosis in a rat carotid injury model12.
NO synthesis
NO synthesis is controlled by intracellular Ca2+ oscillations, and increase in SERCA activity by overexpression of S100A1 (see below) also increases NO production in endothelial cells (EC)35. Sakata et al showed that transcoronary gene transfer of SERCA2a increase coronary blood flow34. One of the reasons for this improvement is the increased production of NO caused by SERCA2a overexpression in EC11. This additional effect of SERCA2a overexpression might have a synergic benefit on cardiovascular disease.
3) SERCA2a as a target for heart failure gene therapy in animal models
Several studies have been designed to improve cardiac function using SERCA as a target. The SERCA isoform used is an important consideration. Data from transgenic animals demonstrated functional differences between the isoforms SERCA2a and SERCA2b in the heart. Long-term expression of SERCA2a in transgenic animals improves contractility without adverse effects36, but replacement of SERCA2a by SERCA2b isoform resulted in cardiac dysfunction and hypertrophy19, 20. Adenovirus-mediated gene transfer of SERCA2a improved cardiac hemodynamics and increased survival in a rat model of HF25, whereas gene transfer of the faster skeletal muscle isoform SERCA1 had limited functional and metabolic improvement in the same model and compromised function in hypertrophic hearts21.
SERCA2a has been the subject of most gene therapy approaches targeting altered calcium physiology5. The increase in contractility concomitant with an increase in SERCA2a expression has been shown by gene transfer and transgenic methods in healthy, HF or diabetes mellitus animals and by gene transfer in a large animal model of HF5, 37. Furthermore, SERCA2a gene transfer restored the oxygen cost of LV contractility to the normal level, indicating an improvement in energy utilization8, 9, 25.
LV dysfunction is a major predictor of electrical instability and arrhythmias. Hence, improvement of myocardial contractility by gene transfer could have a major beneficial electrophysiological effect. Adenovirus-directed gene transfer of SERCA2a in a rat model of ischemia/reperfusion injury effectively suppressed arrhythmias and reduced myocardial infarct size10. In contrast with this finding, overexpression of SERCA2a in transgenic rats increased the occurrence of fatal ventricular tachyarrhythmia following infarct38. These latter finding could result from adverse electrical remodeling caused by reduced NCX expression and function triggered by enhanced SR Ca2+ content. Overall, the overexpression of SERCA2a by gene transfer in the setting of ischemia reperfusion both in small and large animals has been shown to reduce ventricular arrhythmias10, 39.
Recently several preclinical HF studies were conducted on large animal models. A porcine model of HF was created by mitral regurgitation40. AAV1/SERCA2a was administrated using single intracoronary perfusion. Two months after treatment SERCA2a-injected animals demonstrated significant improvements in contractile function and restoration of ventricular volumes compared with untreated animals. There was an absolute increase of 16% in median ejection fraction in AAV1/SERCA2a treated animals40. A sheep model of HF was created by rapid pacing37. Delivery of AAV1/SERCA2a by cardiac recirculating elicited a dose-dependent improvement in LV function 1–2 month after virus administration 37. In both studies, safety-related parameters including histopathologic analysis, hematology, and clinical chemistry indicate that the treatment did not cause any organ damage or inflammatory response37, 40. Furthermore, in addition to favorable haemodynamic effects, brain natriuretic peptide (BNP) expression was reduced consistent with reversal of the HF molecular phenotype37, 40. To evaluate the potential toxicological effect of AAV1/SERCA2 gene therapy the virus was administrated using transcoronary perfusion in normal Göttingen minipigs41. The animals received the dose at least 3-fold higher that the highest dose intended for administration in clinical trials. AAV1/SERCA2a DNA was found in the majority of all the tissues sampled 5 days after injection. The highest concentrations were found in the lung and liver, low to non detectable levels of AAV1/SERCA2a were found in blood and brain tissue. Given the wide spread level of “junk” DNA in the human genome, the safety consequence of the presence of vector DNA in non cardiac cells such as liver is expected to be minimal or nonexistent41. On Days 30 to 90, the tissue concentrations of AAV1/SERCA2a decreased significantly, with many tissues having low to undetectable concentrations of AAV1/SERCA2a. On 90 days after gene transfer, there was an increase in SERCA2a protein expression in the left and right ventricular wall; in addition, coronary arteries, pulmonary arteries, and the diaphragm and lung tissues showed increased expression of SERCA2a, without apparent toxicologic effect41. However, there was no change in SERCA2 expression in liver, kidney, spleen, retina, biceps, sartorius, and gastrocnemius muscles41. AAV1/SERCA2a was well tolerated in minipigs; no signs of toxicity, clinical pathology or histology were observed, and values for echocardiography and electrocardiography parameters were found to be within normal range41.
The overall conclusion from animal studies is that AAV-directed SERCA2a gene transfer appears to be a safe, well-tolerated, and effective treatment for HF.
4. Small molecules targeting SERCA activity
Phospholamban
Inhibition of PLN is another therapeutic approach to target the Ca2+ handling pathway in the failing heart3, 13. Gene transfer of a dominant-negative PLN mutant42, PLN antisense RNAs43 and intracellular inhibitory PLN antibodies44 have been used to increase SERCA2a activity and to rescue HF in animal models. Furthermore, normalization of the calcium transient and restoration of cell contractility has been reported in cardiomyocytes isolated from failing human hearts after adenovirus-mediated PLN antisense gene transfer45, 46. Recently, successful treatment of HF was demonstrated in a rat model of aortic banding by RNAi targeting of PLN47. Simple intravenous injection of PLN RNAi carried by rAAV9 provided long-term stable cardiac shPLN expression and restores cardiac function, reduces dilatation, hyperthrophy and fibrosis after 3 months47. The excitement generated by these studies has been tempered by the discovery in human two mutations on PLN, leading to PLN-null genotype in homozygous individuals with a phenotype of dilated cardiomyopathy48–50. These latter developments raise doubts about using PLN ablation as a universal approach to treat all forms of HF.
Inhibitor 1
Targeting PP1 by overexpression of its inhibitory peptide (I-1) to increase SERCA2a activity may constitute a promising therapeutic strategy in HF13, 15. Indeed, silencing PP1 activity in transgenic mice by the expression of a constitutively active I-1 protein (I-1T35D) results in enhanced PLN phosphorylation, augmented cardiac contractility, rescues from HF development upon aortic constriction and improves mechanical recovery and survival in an ischemia/reperfusion model51, 52. Similarly, adenovirus gene transfer of I-1T35D results in marked restoration of contractility in animal models and failing human myocytes51.
S100A1
Another important protein that has been recently targeted is S100A1, a low-molecular weight Ca2+ binding protein that stabilizes RyR and increases SERCA ATPase activity, resulting in a balanced enhancement of SR Ca2+ release and uptake13, 53. In HF the expression of S100A1 is reduced. Gene transfer of S100A1 in rats following cryo-infarction induced an increase in SERCA2a and RyR activities, resulting in improved intracellular calcium handling and overall enhanced systolic and diastolic ventricular function54, 55.
Pharmacotherapy
Recently, a novel inotropic agent that enhances SERCA2a activity was discovered and tested in HF models. Istaroxime, (E, Z)-3-((2-aminoethoxy)imino) androstane-6,17-dione hydrochloride) enhances SR Ca2+ content and Ca2+ uptake in failing cardiomyocytes and improves heart function in a guinea pig aortic binding model56, 57. This finding opens new perspective for the pharmacotherapy of HF.
5. Advances in the development of viral vectors
The molecular vectors for cardiac gene therapy generally employ expressing cassettes containing strong virus promoters that are constitutively active in a wide spectrum of cells (e.g. cytomegalovirus promoter [CMV]), the gene of interest (SERCA2a) and an appropriate mRNA stabilizing polyadenylation signal. The most commonly used virus vectors are recombinant adeno- and adeno-associated viruses (Table 1).
Table 1.
Comparison of major viral vector systems for cardiovascular gene transfer.
| Vectors | Adenovirus | Adeno-Associated Virus |
|---|---|---|
| Functional titer (per ml) | Up to 1012 | Up to 1010 |
| Genome | dsDNA | ssDNA (can also be dsDNA) |
| Insert Capacity | 7–30 kb | 4.8 kb |
| Integration | No | Yes:chromosome 19 for wild-type AAV2 Low for recombinant vectors |
| Pattern of gene expression | Transient | Long-term |
| Cell cycle-dependent transduction | No | No |
| Immunoreactions | Cytotoxic and immunogenic | Minimally immunogenic |
| Clinical trial approved | Yes | Yes |
Adenoviruses
Recombinant human adenovirus (Ad) vectors are used essentially in preclinical small animal models. Adenoviruses are non-enveloped vectors that are very efficient at transducing target cells in vitro and in vivo, and can be prepared at high titers. The wild type Ad genome is approximately 35 kb of which up to 30 kb can be replaced with foreign DNA. These vectors have demonstrated efficient transduction of adult myocardium in vivo resulting in alteration of global heart function. In animal models, Ads mediate high-level expression for only short term (1 week). However, Ad cause intense local inflammation and stimulate potent cellular and humoral immune responses14.
Adeno-associated viruses
Recombinant adeno-associated viruses vectors (rAAV), derived from nonpathogenic parvoviruses, have many characteristics that make them particularly well suited for cardiac gene transfer58. First, AAV effectively transfect dividing and non-dividing cells. Second, AAV provide stable long term (up to a year) expression in most systems59. Third, AAV evoke minimal immune response in humans and are not known to cause human disease. AAV vectors have been studied in hundreds of patients, and have demonstrated an excellent safety profile14, 60.
Major limitation of rAAV vector systems is the limited packaging capacity of 4.8 kb. AAV vectors package single- stranded genomes and require host cell synthesis of complementary strand for transduction. Recently, a new AAV vector (self-complementary [sc]AAV) which can package either two copies of DNA or dimeric-inverted repeat DNA molecules, thereby alleviating the requirement for host-cell DNA synthesis, was developed61, 62. However, the cassette sizes have to be half the size of a normal sized single-stranded vector (i.e., 2.3 kb), which further limits the number of genes that can be used with scAAV.
Today, 12 different AAV serotypes are known, each with different tissue tropisms63. AAV1, 2, 6, 8 and 9 have higher tropism for myocardium64–66. AAV8 and 9 efficiently cross the endothelial barrier and can be injected intravenously or intraperitoneously65, 67. New mutant AAV pseudotypes have been constructed recently, to improve tissue specificity and safety of gene transfer58, 68. The pseudotype used in human Phase I clinical trial, AAV2/1, contains the capsid sequence of AAV1 and the inverted terminal repeats (ITR) sequences of AAV2. The AAV2 ITRs are incorporated because they have been used in many previous clinical trials and their safety profile is established68. The AAV2/1 vector was selected because AAV1 has been shown to be superior to the other AAV serotypes in transducing cardiomyocytes64. Newly constructed pseudotypes rAAV2/8 and rAAV2/9 are able to transduce myocardium of neonatal mice at approximately 20- and 200-fold (respectively) higher levels than rAAV2/147, 65.
Thus, recombinant rAAV vectors hold a great promise for delivering genes to treat heart diseases.
6. Techniques of Gene Delivery
A number of mechanical approaches have been developed for cardiac gene delivery including direct intramyocardial injection, endocardial delivery, ventricular cavity infusion during aortic cross-clamping, pericardial delivery and percutaneous coronary catheterization3, 5.
Direct intramyocardial injection let to efficient local gene delivery, but was often limited by a patchy pattern of vector transfer and procedure related risks69, 70. Intravenous delivery requires injection of at least 1011 AAV genomes to initiate transduction in small animals and is not applicable to human diseases. The best method developed for the rat model involves injecting the virus into the aortic root just above the aortic valve, while the aorta and pulmonary artery are transiently cross-clamped, achieving homogeneous transduction of the myocardium71.
Endocardial delivery is limited to focal gene transfer to the myocardium. This method of vector delivery for therapeutic angiogenesis has been using in Phase I clinical trials of patients with medically refractory severe angina72.
Intracoronary delivery or retrograde infusion of the virus via the coronary veins results are the attractive solutions since all of the myocardial territory supplied by a coronary artery is accessible. Percutaneous catheter-based gene delivery to the myocardium in vivo can be achieved by the endocardial or the intracoronary veins delivery by several approaches:
Antegrade Epicardial Coronary Artery Infusion (AECAI). The vector is slowly infused into the coronary arteries42.
Modified percutaneous antegrade myocardial gene transfer (PAMGT). Catheter-based antegrade intracoronary viral gene delivery with coronary venous blockade73.
V-Focus Cardiac Delivery System. The V-Focus system is an investigational device that isolates the coronary circulation from the remainder of the systemic circulation while maintaining perfusion of the myocardium74. The V-Focus system enables a closed circuit to be percutaneously established between the coronary arteries and coronary sinus in which the virus vector circulates through the myocardium for approximately 10 min.
The catheter-based techniques were developed for cardiac gene delivery in large animals and may be amenable to translation into human.
7. Clinical trials of heart failure using SERCA2a
The central role of SERCA2a in human HF has been established in patients treated with heart transplant, drugs and devices. Diastolic dysfunction in heart transplant recipients has been shown associated with decreased SERCA2a expression75. Improved LV ejection fraction in DCM (dilated cardiomyopathy) patients treated with β-blockers was correlated with increases in SERCA2a mRNA concentrations in myocardial biopsies76. Patients with DCM and end-stage HF in whom combined LV assist device and pharmacological therapy induced sufficient clinical recovery to allow removal of the device without recurrence of HF, had increased SR Ca2+ content compared with patients who did not recover77. In patients (n=11) with advanced CHF, functional improvement related to CRT (cardiac resynchronization therapy) was associated with favorable changes in molecular markers of HF, including increase in SERCA2a mRNA; no significant changes were observed in non responders patients78. Improved symptoms and exercise tolerance in HF patients after 3 months treatment with Cardiac Contractility Modulator (CCM) has been associated with increase in SERCA2a mRNA79, 80.
Following a long line of investigation on animal models, gene transfer of SERCA2a has shown tremendous promise and two clinical trials using SERCA2a as a target have been initiated: a Phase I, randomized, double-blinded, placebo-controlled, dose escalation trial of intracoronary administration of AAV1. SERCA2a (MYDICAR™) in patients with CHF (Celladon Corp., CA, USA) and a Phase I study evaluating the safety and biological effects of AAV6. SERCA2a in non-ischemic patients undergoing left ventricular assist device placement.
The investigatioal agent (MYDICAR™) used in the first clinical trial41 is a rAAV1 vector and consists of a single-stranded 4486 nucleotide DNA containing the human SERCA2a expression cassette (CMV promoter- SERCA2a-bovine growth hormone pA signal) flanked by ITRs derived from AAV2. The MEDRAD Mark V ProVis angiographic injection system is used to produce a slow, diffuse, and homogenous myocardial exposure to AAV1/SERCA2a via AECAI over 10 min74.
The objectives of phase I clinical trial are the following: 1) to evaluate the safety and feasibility of a 1 time AECAI administration of MYDICAR™ to subjects with ischemic or nonishemic DCM and Class III/IV symptoms of HF; 2) to explore the activity/efficacy of MYDICAR™ and identify appropriate doses for future studies41.
The method of cardio-specific vector administration chosen for human testing is AECAI42. Vector is slowly infused into the coronary arteries after percutaneous access obtained using a femoral approach. Laboratory assessment is monitored for 12 months after administration of AAV1/SERCA2a and includes following parameters: assessment of cardiac enzymes (creatine kinase-MB and cardiac troponin; BNP and N-terminal prohormone brain natriuretic peptide (NT-proBNP); ELISPOT assay to assess possible anti-AAV1 capsid T-cell response; anti-AAV1 neutralizing antibody assay. Sequential echocardiograms are used to assess response to therapy. Global systolic and diastolic functions are analyzed. Parameters measured include left ventricular sizes and volumes. The primary safety end-point is based on the Stage 1 12-months follow-up data, the primary activity/efficacy analysis is based on the Stage 2 6 –month postdose data.
In the phase 1 trial a total of 12 patients with advanced HF have received intracoronary infusion of AAV1.SERCA2a81. Of the 9 reported patients treated, several demonstrated improvements from baseline to month 6 of a number of HF parameters81. Of note, 2 patients who failed to improve had preexisting anti-AAV-1 antibodies.
Conclusion
Advances in the understanding of the molecular basis of HF together with the evolution of increasingly efficient gene transfer technology, has finally placed congestive HF within reach of gene-based therapy. The intracellular handling of calcium is central to the process of excitation-contraction coupling and a number of calcium cycling proteins have been involved in the pathogenesis of HF. Of these, increasing SERCA2a function have been the subject of a large number of gene therapy approaches targeting calcium physiology. Beyond its beneficial effects on contractile function, SERCA2a had profound effect on hypertrophic remodeling, mechanoenergetics, arrhythmogenesis, vascular smooth muscle and endothelial function. These additional effects have heightened the interest in this versatile Ca2+ transporter. However the majority of SERCA2a studies have used animal models, which do not precisely mirror human HF. The patients with end-stage HF may have a greater degree of fibrosis or chronic oxidative stress, reducing efficacy of gene therapy. Early results in the Phase I of a first in-human study of AAV1/SERCA2a in advanced HF showed improvement of a number of HF parameters. Further human studies are greatly needed. Phase 2 of this trial which is placebo controlled randomized and blinded with three therapeutic arms and one control arm is currently under way with more than 30 patients having been enrolled. The results of this important phase 2 trial will be in available in 2010.
Expert opinion
The clinical cardiovascular gene therapy experience is very limited. The available data, however, emphasize the importance of randomized placebo-controlled trials in assessing the efficacy of novel treatments. Larger trials of this type are in progress and the results of these are eagerly awaited. Meanwhile, the large volume of preclinical data is likely to continue to expand as knowledge of the molecular basis of cardiovascular disease increases. Importantly, the accumulation of this data provides the scientific basis from which the human evaluation of novel gene-based therapies can be pursued.
With regard to gene transfer technology, recent serious adverse reactions in young human subjects have been attributed to the vector employed. As a result, efforts to improve vector biosafety have been revitalized; vectors will undergo modifications to further improve biosafety and gene transfer efficiency. Conventional treatment modalities have made significant advances over the last decade in the areas of pharmacotherapy, vascular intervention, surgery and implantable devices. The modest success of gene therapy in cardiovascular disease to date has been in filling a therapeutic need. Future success will depend on either improving conventional therapies of finding effective gene-based therapies. In the next few years, a number of Phase I clinical trials using AAV.SERCA2a based gene therapy for heart failure will provide much needed new information in this field.
Essential to the success of the clinical gene therapy trials in heart failure is the ability to safely use AAV vectors targeting the heart. Even though AAV vectors have been proven to be safe in our trials in patients with heart failure, in our recent experience with the clinical gene therapy trials, we have found that their use for gene delivery has the following limitations: 1) they are not specific for the heart and 2) pre-existing neutralizing antibodies to any individual serotype result in 40% of exclusion of these patients from clinical trials. To identify hybrid viruses that can selectively transduce cardiomyocytes in vivo, and which can also escape the antecedent netralizing antibodies, a number of groups have been employing a combinatorial approach to generate a library of diverse AAV variants, obtained by DNA shuffling, with an enrichment of cardiotropic AAV variants, by directed evolution82, 83
This has allowed the generation of cardiotropic chimerics of AAVs that more specifically targets the heart and escapes the inherent immunity in patients. In the next ten years, this approach will be used to generate novel cardiotropic AAV derived vectors that can also escape patients neutralizing antibodies.
Acknowledgments
This work is supported by AHA SDG 0930116N (LL), by NIH R01 HL078691, HL057263, HL071763, HL080498, & HL083156 (RJH) and by Leducq Foundation through the Caerus network (05 CVD 03, AML and RJH).
Abbreveations
- αAR
α-adrenergic receptor
- Ca2+
calcium ion
- CaM
calmodulin
- CaMK
Ca2+/CaM – dependent protein kinase
- DAG
Diacylglycerol
- ET-1
Endothelin-1 receptor
- Gαq
Gq protein α-subunit
- HDAC
Histone deacetylase
- IP3
Inositol-1,4,5-triphosphate
- IP3R
IP3 receptor
- Ica
L-type calcium current
- LTCC
L-type calcium channel
- MEF
Myocyte-enhancing factor
- NFAT
Nuclear Factor of Activated T lymphocytes
- Orai
SOC sub-unite
- P
Phosphorylation
- PLC
Phospholipase C
- PLN
Phospholamban
- PP2B
serine/threonin phosphatase 2B, calcineurin
- RyR
Ryanodine Receptor calcium channel
- SR
Sarcoplasmic Reticulum
- SERCA
Sarco/Endoplasmic Reticulum Ca2+ ATPase 2a
- SRF
Serum Response Factor
- TRPC
Transient Receptor Potential Ca2+ channel, SOC sub-unite
- STIM 1
stromal interacting molecule 1, SR calcium sensor., SOC sub-unite
Bibliography
Papers of special note have been highlighted as either of interest (*) or of considerable interest (**).
- 1.Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, et al. Heart disease and stroke statistics--2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008 Jan 29;117(4):e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 2.Jessup M, Brozena S. Heart failure. N Engl J Med. 2003 May 15;348(20):2007–18. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- 3.Lipskaia L, Ly H, Kawase Y, Hajjar R, Lompre AM. Treatment of heart failure by calcium cycling gene therapy. Future Cardiology. 2007;3(4):413–23. doi: 10.2217/14796678.3.4.413. [DOI] [PubMed] [Google Scholar]
- 4.Del Monte F, Hajjar RJ. Intracellular devastation in heart failure. Heart Fail Rev. 2008 Jun;13(2):151–62. doi: 10.1007/s10741-007-9071-9. [DOI] [PubMed] [Google Scholar]
- 5.Kawase Y, Hajjar RJ. The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: a potent target for cardiovascular diseases. Nat Clin Pract Cardiovasc Med. 2008 Sep;5(9):554–65. doi: 10.1038/ncpcardio1301. [DOI] [PubMed] [Google Scholar]; **Review on calcum cycling gene therapy
- 6.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]; **Well -documented review on calcium cycling.
- 7.Lipskaia L, Hulot JS, Lompre AM. Role of sarco/endoplasmic reticulum calcium content and calcium ATPase activity in the control of cell growth and proliferation. Pflugers Arch. 2009 Jan;457(3):673–85. doi: 10.1007/s00424-007-0428-7. [DOI] [PubMed] [Google Scholar]; *Calcium-regulated transcription pathways
- 8.Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, et al. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007 Apr;42(4):852–61. doi: 10.1016/j.yjmcc.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang LF, et al. Mechanical and metabolic rescue in a type II diabetes model of cardiomyopathy by targeted gene transfer. Mol Ther. 2006 May;13(5):987–96. doi: 10.1016/j.ymthe.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 10.del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci U S A. 2004 Apr 13;101(15):5622–7. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hadri L, Lipskaia L, Kawase Y, Clement N, Plenge T, Lebeche D, et al. Transcoronary gene transfer of SERCA2a increases coronary blood flow trought an increase of eNOS activity in endothelial cells. Circ Res. 2007;101:E65–E. [Google Scholar]
- 12.Lipskaia L, del Monte F, Capiod T, Yacoubi S, Hadri L, Hours M, et al. Sarco/endoplasmic reticulum Ca2+-ATPase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ Res. 2005 Sep 2;97(5):488–95. doi: 10.1161/01.RES.0000180663.42594.aa. [DOI] [PubMed] [Google Scholar]
- 13.Vinge LE, Raake PW, Koch WJ. Gene therapy in heart failure. Circ Res. 2008 Jun 20;102(12):1458–70. doi: 10.1161/CIRCRESAHA.108.173195. [DOI] [PMC free article] [PubMed] [Google Scholar]; **Well documented review on HF gene therapy
- 14.Lyon AR, Sato M, Hajjar RJ, Samulski RJ, Harding SE. Gene therapy: targeting the myocardium. Heart. 2008 Jan;94(1):89–99. doi: 10.1136/hrt.2007.116483. [DOI] [PubMed] [Google Scholar]; **Well documented review on HF gene therapy
- 15.Nicolaou P, Hajjar RJ, Kranias EG. Role of protein phosphatase-1 inhibitor-1 in cardiac physiology and pathophysiology. J Mol Cell Cardiol. 2009 May 27; doi: 10.1016/j.yjmcc.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Exhaustive review of inhibitor-1.
- 16.MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003 Jul;4(7):566–77. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 17.Bobe R, Bredoux R, Corvazier E, Lacabaratz-Porret C, Martin V, Kovacs T, et al. How many Ca(2)+ATPase isoforms are expressed in a cell type? A growing family of membrane proteins illustrated by studies in platelets. Platelets. 2005 May–Jun;16(3–4):133–50. doi: 10.1080/09537100400016847. [DOI] [PubMed] [Google Scholar]
- 18.Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007 Apr;35(4):430–42. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 19.Vangheluwe P, Tjwa M, Van Den Bergh A, Louch WE, Beullens M, Dode L, et al. A SERCA2 pump with an increased Ca2+ affinity can lead to severe cardiac hypertrophy, stress intolerance and reduced life span. J Mol Cell Cardiol. 2006 Aug;41(2):308–17. doi: 10.1016/j.yjmcc.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 20.Ver Heyen M, Heymans S, Antoons G, Reed T, Periasamy M, Awede B, et al. Replacement of the muscle-specific sarcoplasmic reticulum Ca(2+)-ATPase isoform SERCA2a by the nonmuscle SERCA2b homologue causes mild concentric hypertrophy and impairs contraction-relaxation of the heart. Circ Res. 2001 Oct 26;89(9):838–46. doi: 10.1161/hh2101.098466. [DOI] [PubMed] [Google Scholar]
- 21.O'Donnell JM, Fields A, Xu X, Chowdhury SA, Geenen DL, Bi J. Limited functional and metabolic improvements in hypertrophic and healthy rat heart overexpressing the skeletal muscle isoform of SERCA1 by adenoviral gene transfer in vivo. Am J Physiol Heart Circ Physiol. 2008 Dec;295(6):H2483–94. doi: 10.1152/ajpheart.01023.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.del Monte F, Harding SE, Schmidt U, Matsui T, Kang ZB, Dec GW, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999 Dec 7;100(23):2308–11. doi: 10.1161/01.cir.100.23.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipskaia L, Pinet C, Fromes Y, Hatem S, Cantaloube I, Coulombe A, et al. Mutation of delta-sarcoglycan is associated with Ca(2+) -dependent vascular remodeling in the Syrian hamster. Am J Pathol. 2007 Jul;171(1):162–71. doi: 10.2353/ajpath.2007.070054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandebrouck A, Ducret T, Basset O, Sebille S, Raymond G, Ruegg U, et al. Regulation of store-operated calcium entries and mitochondrial uptake by minidystrophin expression in cultured myotubes. FASEB J. 2006 Jan;20(1):136–8. doi: 10.1096/fj.04-3633fje. [DOI] [PubMed] [Google Scholar]
- 25.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation. 2001 Sep 18;104(12):1424–9. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venetucci LA, Trafford AW, O'Neill SC, Eisner DA. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc Res. 2008 Jan 15;77(2):285–92. doi: 10.1093/cvr/cvm009. [DOI] [PubMed] [Google Scholar]
- 27.Keller M, Kao JP, Egger M, Niggli E. Calcium waves driven by “sensitization” wave-fronts. Cardiovasc Res. 2007 Apr 1;74(1):39–45. doi: 10.1016/j.cardiores.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Davia K, Bernobich E, Ranu HK, del Monte F, Terracciano CM, MacLeod KT, et al. SERCA2A overexpression decreases the incidence of aftercontractions in adult rabbit ventricular myocytes. J Mol Cell Cardiol. 2001 May;33(5):1005–15. doi: 10.1006/jmcc.2001.1368. [DOI] [PubMed] [Google Scholar]
- 29.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006 Aug;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]; *Transcription pathways in cardiac hypertrophy.
- 30.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, et al. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol. 2002 Nov;22(21):7603–13. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q, Wilkins BJ, Lee YJ, Ichijo H, Molkentin JD. Direct interaction and reciprocal regulation between ASK1 and calcineurin-NFAT control cardiomyocyte death and growth. Mol Cell Biol. 2006 May;26(10):3785–97. doi: 10.1128/MCB.26.10.3785-3797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki J, Bayna E, Li HL, Molle ED, Lew WY. Lipopolysaccharide activates calcineurin in ventricular myocytes. J Am Coll Cardiol. 2007 Jan 30;49(4):491–9. doi: 10.1016/j.jacc.2006.10.043. [DOI] [PubMed] [Google Scholar]
- 33.Nakayama H, Otsu K, Yamaguchi O, Nishida K, Date MO, Hongo K, et al. Cardiac-specific overexpression of a high Ca2+ affinity mutant of SERCA2a attenuates in vivo pressure overload cardiac hypertrophy. FASEB J. 2003 Jan;17(1):61–3. doi: 10.1096/fj.02-0474fje. [DOI] [PubMed] [Google Scholar]
- 34.Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang L, et al. Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am J Physiol Heart Circ Physiol. 2007 Feb;292(2):H1204–7. doi: 10.1152/ajpheart.00892.2006. [DOI] [PubMed] [Google Scholar]
- 35.Pleger ST, Harris DM, Shan C, Vinge LE, Chuprun JK, Berzins B, et al. Endothelial S100A1 modulates vascular function via nitric oxide. Circ Res. 2008 Apr 11;102(7):786–94. doi: 10.1161/CIRCRESAHA.108.172031. [DOI] [PubMed] [Google Scholar]
- 36.He H, Giordano FJ, Hilal-Dandan R, Choi DJ, Rockman HA, McDonough PM, et al. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997 Jul 15;100(2):380–9. doi: 10.1172/JCI119544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Byrne MJ, Power JM, Preovolos A, Mariani JA, Hajjar RJ, Kaye DM. Recirculating cardiac delivery of AAV2/1SERCA2a improves myocardial function in an experimental model of heart failure in large animals. Gene Ther. 2008 Dec;15(23):1550–7. doi: 10.1038/gt.2008.120. [DOI] [PubMed] [Google Scholar]
- 38.Chen Y, Escoubet B, Prunier F, Amour J, Simonides WS, Vivien B, et al. Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation. 2004 Apr 20;109(15):1898–903. doi: 10.1161/01.CIR.0000124230.60028.42. [DOI] [PubMed] [Google Scholar]
- 39.Prunier F, Kawase Y, Gianni D, Scapin C, Danik SB, Ellinor PT, et al. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation. 2008 Aug 5;118(6):614–24. doi: 10.1161/CIRCULATIONAHA.108.770883. [DOI] [PubMed] [Google Scholar]
- 40.Kawase Y, Ly HQ, Prunier F, Lebeche D, Shi Y, Jin H, et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol. 2008 Mar 18;51(11):1112–9. doi: 10.1016/j.jacc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 41.Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, et al. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail. 2008 Jun;14(5):355–67. doi: 10.1016/j.cardfail.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 42.Kaye DM, Preovolos A, Marshall T, Byrne M, Hoshijima M, Hajjar R, et al. Percutaneous cardiac recirculation-mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J Am Coll Cardiol. 2007 Jul 17;50(3):253–60. doi: 10.1016/j.jacc.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 43.Tsuji T, Del Monte F, Yoshikawa Y, Abe T, Shimizu J, Nakajima-Takenaka C, et al. Rescue of Ca2+ overload-induced left ventricular dysfunction by targeted ablation of phospholamban. Am J Physiol Heart Circ Physiol. 2009 Feb;296(2):H310–7. doi: 10.1152/ajpheart.00975.2008. [DOI] [PubMed] [Google Scholar]
- 44.Dieterle T, Meyer M, Gu Y, Belke DD, Swanson E, Iwatate M, et al. Gene transfer of a phospholamban-targeted antibody improves calcium handling and cardiac function in heart failure. Cardiovasc Res. 2005 Sep 1;67(4):678–88. doi: 10.1016/j.cardiores.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 45.del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002 Feb 26;105(8):904–7. doi: 10.1161/hc0802.105564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziolo MT, Martin JL, Bossuyt J, Bers DM, Pogwizd SM. Adenoviral gene transfer of mutant phospholamban rescues contractile dysfunction in failing rabbit myocytes with relatively preserved SERCA function. Circ Res. 2005 Apr 29;96(8):815–7. doi: 10.1161/01.RES.0000163981.97262.3b. [DOI] [PubMed] [Google Scholar]
- 47.Suckau L, Fechner H, Chemaly E, Krohn S, Hadri L, Kockskamper J, et al. Long-term cardiac-targeted RNA interference for the treatment of heart failure restores cardiac function and reduces pathological hypertrophy. Circulation. 2009 Mar 10;119(9):1241–52. doi: 10.1161/CIRCULATIONAHA.108.783852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003 Feb 28;299(5611):1410–3. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 49.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A. 2006 Jan 31;103(5):1388–93. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003 Mar;111(6):869–76. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005 Apr 15;96(7):756–66. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 52.Nicolaou P, Rodriguez P, Ren X, Zhou X, Qian J, Sadayappan S, et al. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ Res. 2009 Apr 24;104(8):1012–20. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Most P, Remppis A, Pleger ST, Katus HA, Koch WJ. S100A1: a novel inotropic regulator of cardiac performance. Transition from molecular physiology to pathophysiological relevance. Am J Physiol Regul Integr Comp Physiol. 2007 Aug;293(2):R568–77. doi: 10.1152/ajpregu.00075.2007. [DOI] [PubMed] [Google Scholar]
- 54.Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, et al. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004 Dec;114(11):1550–63. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pleger ST, Most P, Boucher M, Soltys S, Chuprun JK, Pleger W, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007 May 15;115(19):2506–15. doi: 10.1161/CIRCULATIONAHA.106.671701. [DOI] [PubMed] [Google Scholar]
- 56.Rocchetti M, Alemanni M, Mostacciuolo G, Barassi P, Altomare C, Chisci R, et al. Modulation of sarcoplasmic reticulum function by PST2744 [istaroxime; (E,Z)-3-((2-aminoethoxy)imino) androstane-6,17-dione hydrochloride)] in a pressure-overload heart failure model. J Pharmacol Exp Ther. 2008 Sep;326(3):957–65. doi: 10.1124/jpet.108.138701. [DOI] [PubMed] [Google Scholar]
- 57.Micheletti R, Palazzo F, Barassi P, Giacalone G, Ferrandi M, Schiavone A, et al. Istaroxime, a stimulator of sarcoplasmic reticulum calcium adenosine triphosphatase isoform 2a activity, as a novel therapeutic approach to heart failure. Am J Cardiol. 2007 Jan 22;99(2A):24A–32A. doi: 10.1016/j.amjcard.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 58.Gray SJ, Samulski RJ. Optimizing gene delivery vectors for the treatment of heart disease. Expert Opin Biol Ther. 2008 Jul;8(7):911–22. doi: 10.1517/14712598.8.7.911. [DOI] [PubMed] [Google Scholar]
- 59.Samulski RJ. AAV vectors, the future workhorse of human gene therapy. Ernst Schering Res Found Workshop. 2003;(43):25–40. doi: 10.1007/978-3-662-05352-2_3. [DOI] [PubMed] [Google Scholar]
- 60.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006 Mar;12(3):342–7. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 61.Choi VW, Samulski RJ, McCarty DM. Effects of adeno-associated virus DNA hairpin structure on recombination. J Virol. 2005 Jun;79(11):6801–7. doi: 10.1128/JVI.79.11.6801-6807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003 Dec;10(26):2112–8. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 63.Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, et al. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008 Jul;16(7):1252–60. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 64.Palomeque J, Chemaly ER, Colosi P, Wellman JA, Zhou S, Del Monte F, et al. Efficiency of eight different AAV serotypes in transducing rat myocardium in vivo. Gene Ther. 2007 Jul;14(13):989–97. doi: 10.1038/sj.gt.3302895. [DOI] [PubMed] [Google Scholar]
- 65.Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006 Aug 18;99(4):e3–9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- 66.Raake PW, Hinkel R, Muller S, Delker S, Kreuzpointner R, Kupatt C, et al. Cardio-specific long-term gene expression in a porcine model after selective pressure-regulated retroinfusion of adeno-associated viral (AAV) vectors. Gene Ther. 2008 Jan;15(1):12–7. doi: 10.1038/sj.gt.3303035. [DOI] [PubMed] [Google Scholar]
- 67.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006 Jul;14(1):45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schultz BR, Chamberlain JS. Recombinant adeno-associated virus transduction and integration. Mol Ther. 2008 Jul;16(7):1189–99. doi: 10.1038/mt.2008.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wright MJ, Wightman LM, Lilley C, de Alwis M, Hart SL, Miller A, et al. In vivo myocardial gene transfer: optimization, evaluation and direct comparison of gene transfer vectors. Basic Res Cardiol. 2001 May–Jun;96(3):227–36. doi: 10.1007/s003950170053. [DOI] [PubMed] [Google Scholar]
- 70.Muller OJ, Katus HA, Bekeredjian R. Targeting the heart with gene therapy-optimized gene delivery methods. Cardiovasc Res. 2007 Feb 1;73(3):453–62. doi: 10.1016/j.cardiores.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 71.Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK, et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci U S A. 1998 Apr 28;95(9):5251–6. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yla-Herttuala S. An update on angiogenic gene therapy: vascular endothelial growth factor and other directions. Curr Opin Mol Ther. 2006 Aug;8(4):295–300. [PubMed] [Google Scholar]
- 73.Hayase M, Del Monte F, Kawase Y, Macneill BD, McGregor J, Yoneyama R, et al. Catheter-based antegrade intracoronary viral gene delivery with coronary venous blockade. Am J Physiol Heart Circ Physiol. 2005 Jun;288(6):H2995–3000. doi: 10.1152/ajpheart.00703.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Preovolos A, Mennen M, Bilney A, Mariani J, Kaye D, Power J. Developpement of a novel perfusion technique to allow targeted delivery of Gene Therapyapy– the V-Focus system. Journal of Extra-corporeal Technology. 2006;38:51–2. [PMC free article] [PubMed] [Google Scholar]; *Gene delivery techniques for the heart
- 75.Studeli R, Jung S, Mohacsi P, Perruchoud S, Castiglioni P, Wenaweser P, et al. Diastolic dysfunction in human cardiac allografts is related with reduced SERCA2a gene expression. Am J Transplant. 2006 Apr;6(4):775–82. doi: 10.1111/j.1600-6143.2006.01241.x. [DOI] [PubMed] [Google Scholar]
- 76.Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002 May 2;346(18):1357–65. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- 77.Terracciano CM, Hardy J, Birks EJ, Khaghani A, Banner NR, Yacoub MH. Clinical recovery from end-stage heart failure using left-ventricular assist device and pharmacological therapy correlates with increased sarcoplasmic reticulum calcium content but not with regression of cellular hypertrophy. Circulation. 2004 May 18;109(19):2263–5. doi: 10.1161/01.CIR.0000129233.51320.92. [DOI] [PubMed] [Google Scholar]
- 78.Vanderheyden M, Mullens W, Delrue L, Goethals M, de Bruyne B, Wijns W, et al. Myocardial gene expression in heart failure patients treated with cardiac resynchronization therapy responders versus nonresponders. J Am Coll Cardiol. 2008 Jan 15;51(2):129–36. doi: 10.1016/j.jacc.2007.07.087. [DOI] [PubMed] [Google Scholar]
- 79.Butter C, Rastogi S, Minden HH, Meyhofer J, Burkhoff D, Sabbah HN. Cardiac contractility modulation electrical signals improve myocardial gene expression in patients with heart failure. J Am Coll Cardiol. 2008 May 6;51(18):1784–9. doi: 10.1016/j.jacc.2008.01.036. [DOI] [PubMed] [Google Scholar]
- 80.Mullens W, Bartunek J, Wilson Tang WH, Delrue L, Herbots L, Willems R, et al. Early and late effects of cardiac resynchronization therapy on force-frequency relation and contractility regulating gene expression in heart failure patients. Heart Rhythm. 2008 Jan;5(1):52–9. doi: 10.1016/j.hrthm.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 81.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009 Apr;15(3):171–81. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, Qiao C, et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci U S A. 2009 Mar 10;106(10):3946–51. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koerber JT, Jang JH, Schaffer DV. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol Ther. 2008 Oct;16(10):1703–9. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]