

Abstract
Purpose
Polyethylene glycol (PEG) functionalized magnetic nanoparticles (MNPs) were tested as a drug carrier system, magnetic resonance imaging (MRI) agent, and ability to conjugate to an antibody.
Methods
An iron oxide core coated with oleic acid (OA) and then with OA-PEG forms a water dispersible MNP formulation. Hydrophobic doxorubicin partitions into the OA layer for sustained drug delivery. The T1 and T2 MRI contrast properties were determined in vitro and the circulation of the MNPs measured in mouse carotid arteries. An N-hydroxysuccinimide group (NHS) on the OA-PEG-80 was used to conjugate the amine functional group on antibodies for active targeting in the human MCF-7 breast cancer cell line.
Results
The optimized formulation had a mean hydrodynamic diameter of 184 nm with an 8 nm iron-oxide core. The MNPs enhance the T2 MRI contrast, and have a long circulation time in vivo with 30% relative concentration 50 min post-injection. Doxorubicin-loaded MNPs showed sustained drug release and dose-dependent antiproliferative effects in vitro; the drug effect was enhanced with transferrin antibody conjugated MNPs.
Conclusion
PEG functionalized MNPs could be developed as a targeted drug delivery system and MRI contrast agent.
Keywords: Iron-oxide, Anticancer drugs, Anti-proliferative effect, Reticuloendothelial system, Transferrin antibody
INTRODUCTION
Magnetic nanoparticles (MNPs) have gained significant interest in recent years for various biomedical applications. Their intrinsic magnetic properties have been explored in cell labeling/cell separation, developing bioassays, in magnetofection, drug delivery, to induce local hyperthermia in response to an external alternating magnetic field, and to selectively destroy cancer cells (1). In addition, MNPs are used in clinical diagnosis as a magnetic resonance imaging (MRI) contrast agent (2).
Our interest is in developing MNPs with dual functional characteristics for drug delivery and as a MRI agent. These two applications are complimentary in many ways, as it provides the unique ability to assess the efficiency of drug delivery to the target site in real time, as well as to monitor the progression of the disease in response to the therapy using a non-invasive MRI technique (3). Drug-loaded MNPs can play roles in therapy and/or detection, and therefore are of high clinical importance.
Although several MNP formulations have been developed to meet the specific needs of a particular application, there have been limited efforts towards developing MNPs with combined properties of drug delivery and imaging (2). This could, in part, be due to the different sets of issues involved in achieving both the objectives with one formulation of MNPs. The general approaches are to incorporate the drug of interest and MNPs in liposomes/emulsion/polymers, or to conjugate the drug of interest to the coating, usually dextran, that is used for stabilizing the core material in an aqueous system. However, such approaches usually result in low efficiency of drug association with MNPs; the associated drug rapidly dissociates (4), the process of drug conjugation often times results in the loss of magnetization of the core material, and/or causes significant increase in particle size. These changes significantly affect the imaging property of MNPs. Further, the rapid clearance of MNPs from the circulation and their sequestering by the reticuloendothelial system (RES) limit the drug targeting efficiency and imaging applications of MNPs to targets other than the organs of the RES such as the liver or lymphatic systems.
Thus, critical to the successful development of MNPs with multifunctional properties would be to meet the needs of drug delivery without compromising their MRI characteristics. Ideally, such a formulation should have the following functional properties- i) surface that would prevent the rapid clearance of the MNPs by the RES following intravenous administration; ii) ability to load drugs without influencing the physical properties of the MNP formulation or magnetization property of the core material; iii) ability to conjugate MNPs to a targeting ligand or antibody. With the above intention, we have been developing a MNP formulation that contains an iron-oxide core which is first coated with oleic acid (OA) and then with OA-conjugated poly(ethylene) glycol (PEG) (OA-PEG). It was hypothesized that the OA of OA-PEG would anchor on to the OA coated on the iron-oxide core, and the PEG would form a corona that would impart water dispersity and steric stability to the MNPs. In addition, functionalized PEG would prevent the rapid clearance of MNPs by the RES, a known characteristic of PEG, and provide the functional groups for conjugation to a targeting ligand. The OA coat around the iron-oxide core could act as a reservoir for loading of hydrophobic drugs. Further, functionalized groups on PEG can be used for conjugation to antibody or targeting ligands.
In this study, we have optimized the various formulation parameters and characterized the PEG-functionalized MNPs for physical properties and chemical analysis of the surface. Further, we have determined protein binding to MNPs and their uptake by macrophages in vitro. To determine their drug delivery application, we have demonstrated loading of doxorubicin (base) as a model anticancer cancer drug and studied the antiproliferative effect of the drug-loaded MNPs in the MCF-7 breast cancer cell line. The imaging properties of MNPs in phantom agar gels and clearance of the MNPs in the mouse carotid artery were determined using MRI. Magnetic resonance properties and clearance of our MNPs were compared with Feridex IV, a dextran-coated iron-oxide formulation used in clinical diagnostic applications.
MATERIALS AND METHODS
Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O) 99% pure granulated (Fe(III)), Iron(II) chloride tetrahydrate (FeCl2·4H2O) 99+% (Fe(II)), ammonium hydroxide (5 M), and oleic acid (OA) were purchased from Fisher Scientic (Pittsburgh, PA). SUNBRIGHT OE-020CS (OA-PEG-20), OE-040CS (OA-PEG-40) and OE-80CS (OA-PEG-80), namely (α-succinimidyloxysuccinyl-ω-oleyloxy) poly(oxyethylene)s differing in molecular weight of PEG chain length (i.e. 2104, 4352, and 8525, respectively) were purchased from NOF Corporation (Tokyo, Japan). Feridex IV (11.2 mg Fe/mL, 5.6–9.1 mg dextran/mL) was purchased from Berlex Laboratories, Inc. (Montville, NJ). Bacto nutrient agar dehydrated was purchased from Difco Laboratories (Detroit, MI). Doxorubicin hydrochloride was a gift from Dabur Research Foundation (Ghaziabad, India). A primary transferrin antibody FITC (rabbit polyclonal to transferrin) was purchased from Abcam Inc. (Cambridge, MA). Transferrin was received from Sigma (St. Louis, MO). Sterile nitrogen purged water was used in all steps in the synthesis of magnetic nanoparticles (MNPs).
Synthesis of Magnetic Nanoparticles
Aqueous solutions of 0.1 M Fe(III) (30 mL) and 0.1 M Fe(II) (15 mL) were combined in a 100 mL beaker with magnetic stirring for 10 min. Ammonium hydroxide (5 M, 3 mL) was added drop wise over 5 min and stirring was continued for 30 min under nitrogen-gas atmosphere. The reaction was stirred for 24 h for bare magnetite particles.
For oleic acid coated MNPs (OA-MNPs), after 30 min of stirring under nitrogen-gas atmosphere, 100 mg OA was added to the above mixture, and the mixture was heated to 80 °C while stirring for 30 min. The resulting mixture was cooled down to room temperature and stirred for 24 h. Poly(ethylene) glycol (PEG) functionalized OA-MNPs (PEG-OA-MNPs) were prepared by cooling the OA-MNPs to room temperature, and adding an aqueous solution of OA-PEG-20, OA-PEG-40, or OA-PEG-80 polymers (25–300 mg) dissolved in 5 mL of water over 5 min with stirring. The stirring was continued for an additional 24 h at room temperature. The PEG-OA-MNPs prepared with 200 mg of OA-PEG-20, OA-PEG-40 and OA-PEG-80 are designated as OE-20, OE-40, and OE-80 MNPs throughout the manuscript (Figure 1).
Figure 1.

Magnetic nanoparticle synthesis (A) and layering of various OA-PEG polymers on the surface of the magnetic nanoparticles (B).
Bare MNPs, OA-MNPs, and PEG-OA-MNPs were separated by magnetic separation by placing a magnet (12,2000 G, Edmund Scientific, Tonawanda, NY) below the beaker and allowing the solution to clear. The excess iron salts, ammonium hydroxide, OA, and OA-PEG in the wash were discarded. The particles were washed with 40 mL nitrogen purged sterile water four times using magnetic separation, dispersed in 25 mL of sterile water, and centrifuged at 1000 rpm for 10 min to remove large and aggregated particles. Purified OE-80 MNPs to be used in antibody conjugation were washed with water and centrifuged as mentioned above but at 4°C.
Magnetic Nanoparticle Characterization
A 5–10 μL nanoparticle suspension (3–5 mg/mL) was diluted to 2.5 mL with distilled water and the mean hydrodynamic particle size and zeta potential determined by dynamic laser light scattering (DLS, NICOMP 380™ ZLS, Particle sizing systems, Santa Barbara, CA). The particle size was also studied at the same concentration in mannitol citrate buffer, saline (0.9 wt.% sodium chloride), PBS (154 mM, pH 7.4), and 1mM HEPES buffer adjusted with 0.1 N sodium hydroxide or hydrochloric acid solutions to a final pH of 3 to 10. Particle stability was determined by storing 4 mg of particles in 1 mL of water, saline, PBS or mannitol buffer and measuring the size at varying time intervals.
Transmission electron microcopy (TEM) was used to determine the particle size of the formulations in dry state (Philips/FEI Inc., Briarcliff, Manor, NY). The particle size in TEM images were measured using NIH ImageJ software (http://rsbweb.nih.gov/ij/). Samples of lyophilized MNPs were used for Fourier transform infrared (FTIR) spectroscopy (PerkinElmer spectrum 100 spectrophotometer, PerkinElmer, Shelton, CT), thermogravimetric analysis (TGA, Thermogravimetric analyzer pyres 1 TGA, PerkinElmer, PerkinElmer, Shelton, CT), X-ray photoelectron spectroscopy (XPS) (PerkinElmer PHI-5600, Physical Electronics, Eden Prairie, MN), and magnetization measurements (MicroMag 2900 alternating gradient field magnetometer, AGFM, Princeton Measurements Corp, USA).
Protein Binding
To determine the protein binding, 1 mL of Bovine Serum Albumin (BSA) at 330 μg/mL in mannitol buffer was titrated with 1 mg/mL of OE-20, OE-40, OE-80 MNPs, or Feridex IV in mannitol buffer. The BSA binding onto MNPs was determined using the intrinsic fluorescence quenching of the tryptophan residue on BSA (5). The decrease in the fluorescence intensity of the tryptophan residue due to interaction with MNPs was recorded between 300–500 nm at λex = 295 nm at varying MNP concentrations. MNPs alone did not change the spectral intensity. The Lehrer and Fashman (6) and Chipman (7) methods were used in equation (1) to determine the binding constant (Kb) and number of binding sites or binding stochiometry (n).
| (1) |
where F0 and Fs are the relative fluorescence intensities of the BSA solution and BSA solution saturated with MNPs, respectively. The relative fluorescence intensity (F) was obtained from the area under the fluorescence curve. The magnetic nanoparticle concentration [MNP] is in mg/mL. Number of binding sites (n) was obtained from the slope of log [(F0−F)/(F−Fs)] vs log [MNP]. Logarithm of dissociation constant (Kdiss) equals log [MNP] at log [(F0−F)/(F−Fs)] = 0. Binding constant (Kb) is 1/Kdiss.
Macrophage Uptake
Macrophage cells (RAW 264.7 Cell Line murine, Macrophage from blood) were purchased from Sigma (St. Louis, MO) and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 μg/mL streptomycin (Gibco BRL, Grand Island, NY) and 100 μg/mL penicillin G (Gibco BRL, Grand Island, NY). Cells were kept in a humidified, 5% CO2 atmosphere at 37 °C, seeded in 6 well plates and allowed to reach 80% confluence. The media was then replaced with 2 mL of 0.05 and 0.10 mg/mL OA-PEG-MNPs or Feridex IV suspensions in growth media, and incubated for 4 h. The cells were washed three times with PBS, collected by scraping, and centrifuged at 1000 rpm at 4 °C. The pellets obtained were analyzed for iron levels using 1,10-phenanthroline colorimetric method (8) and the levels were normalized with respect to cell protein content using Pierce BCA Protein Assay (Pierce, Rockford IL/Fisher Scientific).
Preparation and Magnetic Resonance Imaging of Phantom Gels
A 2.5% w/v agar solution was prepared by heating 250 mg agar in 10 mL PBS at 80 °C for about 20 min and was maintained at 80 °C. Phantom gels were prepared by mixing 160 μL of the above agar solution with 840 μL dispersion of PEG-OA-MNPs with different concentrations of Fe so that the final concentrations are in the range of 0–50 μg Fe/mL. Dispersions of PEG-OA-MNPs were preheated to 60 °C to prevent gelation while mixing with agar solution. A 250 μL aliquot of each mixture was transferred quickly to 300 μL microcentrifuge tubes and allowed to cool to room temperature. Similarly, agar gels with Feridex IV were prepared, as well as a control phantom gel using an equivalent volume of 1X PBS.
Gel aliquots were kept inside the coil of the MRI (9.4 T MRI 94/20, Bruker BioSpin Corporation, Billerica, MA) scanner for simultaneous imaging. Paravision 3.0 software (Bruker BioSpin Corporation) was used for data acquisition, reconstruction and visualization/analysis of images. Images were acquired at various echo time (TE) from 10 ms to 340 ms with repetition time (TR) of 10,000 ms. Axial slice images of thickness 2.0 mm were used to measure the signal intensity. Intensities were measured within the manually drawn regions of interest (ROI). Transverse relaxation rate R2 (R2=1/T2) was calculated by curve fitting the plot of signal intensity vs. TE. The R2 was calculated for gels with different iron concentration and slope (T2 relaxivity) was calculated to compare different formulations for sensitivity of the contrast enhancing properties. Similarly, for calculating R1 (the longitudinal relaxation rate 1/T1) for the corresponding samples, TE was kept fixed at 10 ms and TR was varied between 15.4 ms to 10,000 ms and T1 relaxivity was calculated.
Clearance of Magnetic Nanoparticles In Vivo
All experimental procedures involving live animals were approved by the Cleveland Clinic Foundation Institutional Animal Care and Use Committee, Cleveland, Ohio. Athymic nude mice (male, nu/nu, 30–40 g; Charles River Laboratories, Wilmington, MA) were used for in vivo experiments. The mice were maintained on isoflurane anesthesia throughout the experiments. Feridex IV and OE-80 MNP suspensions in mannitol citrate buffer were injected via tail vein over 40 s using a 30 gauge needle connected to PE-20 tubing at a dose of 7 mg Fe/kg mouse. Dynamic scanning of mice pre- and post-injection of the MNPs was carried out using the 9.4 T MRI to detect the changes in the signal intensities in the carotid arteries. A FLASH sequence (TR/TE = 12.4/3.5 ms, FOV = 2.0 × 2.0 cm, matrix = 128 × 128, α = 30°, slice thickness = 1 mm) was employed to acquire axial images of the carotid arteries for each mouse. Signal intensities within ROIs drawn inside the carotid arteries were measured at different time intervals. Relative concentrations of OE-80 MNPs and Feridex IV particles were calculated from the signal intensity changes by the following equation, a simplified version of the Bloch equations for the FLASH acquisitions with short TR and T2* changes caused by the particle suspensions (9):
| (2) |
where So is the signal intensity before MNP injection, S(t) is the signal intensity at time “t” post injection. Echo time (TE) was kept constant for each acquisition.
Drug Loading of Magnetic Nanoparticles
Doxorubicin (DOX) was used as a model anticancer drug. Doxorubicin hydrochloride was first converted to a water-insoluble base (DOX) (10), so that it could partition into the OA layer. An ethanolic solution of DOX (200 μL, 5 mg/mL) was added drop-wise while stirring to an aqueous dispersion of PEG-OA-MNPs (10 mg of particles in 3 mL water) and stirred overnight (~16 h). The DOX loaded PEG-OA-MNPs (DOX-MNPs) were separated from the free drug by washing three times using magnetic separation, and dispersed in 2 mL of sterile distilled water. Doxorubicin loaded MNPs were characterized using DLS for size and zeta potential, and FTIR for chemical composition.
To determine DOX loading, a 200 μL aliquot of the suspension of DOX-MNPs was lyophilized for 2 days, and the weight of each lyophilized sample was determined. To each lyophilized sample, 2 mL of 12.5% v/v methanol in chloroform was added to extract the drug. The samples were kept on a shaker rotating at 100 rpm at room temperature for 24 h, then centrifuged for 10 min at 14,000 g. A 100 μL aliquot of the supernatant from each sample was diluted to 2 mL with 12.5% v/v methanol in chloroform, and the DOX concentration determined using a fluorescence spectrophotometer (LS55 Fluorescence Spectrophotometer, PerkinElmer LLC, Shelton, CT) at λex = 485 nm and λem = 591 nm. A standard plot was prepared under identical conditions in the concentration range of 0.5–10 μg/mL. The supernatant from the control, i.e., PEG-OA-MNPs without drug, was used as a blank.
Drug Release from OE-80 DOX-MNPs
Drug release studies were carried out in PBS containing 0.1% w/v Tween-80 at 37 °C in a shaker at 100 rpm. Dialysis tubing (MWCO 12-kD, Spectrum®, Laguna Hills, CA) containing 1 mL of 155 μg drug/mL of OE-80 DOX-MNPs was placed in a 50 mL falcon tube containing 3 mL PBS. To monitor the drug release from the MNPs, the outside buffer was completely removed and replaced with fresh PBS solution at varying time intervals. Throughout the experiment, the concentration gradient was maintained and the drug concentration at any time point in the outer buffer did not exceed 20 % of the drug solubility in the buffer. The DOX present in the collected samples was determined using a fluorescence spectrophotometer as described above with standards of DOX prepared in Tween-PBS (0.38–1.55 μg/mL).
Transferrin Antibody Conjugation
Magnetic nanoparticles used for antibody conjugation were synthesized through the addition of OA with continued cooling of the solution to 4 °C. A cooled solution of OA-PEG-80 with NHS functional group (200 mg in 5 mL of water) was added over 5 min to the OA-MNPs while stirring at 400 rpm at 4 °C. The stirring was continued for an additional 24 h at 4 °C. Conjugation of transferrin antibody FITC to the OE-80 MNPs was carried out at 4 °C in PBS. A suspension of OE-80 MNPs (10 mg in PBS) and 500 μg antibody were mixed together to a final volume of 3 mL in PBS, and shaken overnight at 4 °C. FITC-labeled antibody was used to quantify the amount bound to MNPs. The antibody conjugated particles (OE-80-Ab MNPs) were washed with 2 mL of PBS buffer 3 times by magnetic separation, reconstituted in 3 mL of mannitol buffer after the final wash, and stored at 4 °C. Washings containing unconjugated antibody were analyzed in a 96-well plate using a spectrophotometer at λex = 495 nm and λem = 528 nm. Antibody standards (1–8 μg/mL) were prepared in PBS at 4 °C. The antibody conjugated to the particles was calculated from the difference between the antibody used for conjugation and that present in the washings. The average number of antibody molecules conjugated onto OE-80 MNPs, was calculated by dividing the number of antibody molecules bound to MNPs by the calculated average number of MNPs (n) using equation (3) (11):
| (3) |
where m is the weight of OE-80 particles, D is mean diameter of the OE-80 MNPs determined by dynamic light scattering (DLS) and ρ is density of the OE-80 MNPs (weight/volume), estimated as ~ 4.2 g/cm3 based on OE-80 (MNP:74.88%, OA:12.78% and OA-PEG 5.83%) composition.
Antibody Orientation on OE-80-Ab MNPs
Antibody orientation following conjugation to OE-80 MNPs was determined by adding 100 μL of OE-80-Ab MNPs (105 nM of antibody; of 157.61 μg antibody/mL OE-80-Ab MNPs) to varying concentrations of transferrin antigen or control BSA solutions (129.87 nM to 25.97 μM). The solutions were combined in 1.5 mL Eppendorf tubes, incubated at 37 °C for 2 h, cooled to room temperature and the size determined by DLS as described above.
Antiproliferative Activity of Drug-Loaded MNPs
Breast cancer cells (MCF-7) from American Type Culture Collection (ATCC, Manassas, VA) were maintained in DMEM medium supplemented with 10% fetal bovine serum, 100 μg/mL penicillin G, and 100 μg/mL streptomycin at 37 °C in a humidified and 5% CO2 atmosphere. Cells were seeded at 3,000 cells per well in 96 well plates and were allowed to attach for 24 h. Doxorubicin in solution and DOX-MNPs were prepared and diluted to different concentrations in culture medium to study the dose-response effect. Medium from wells was replaced with either DOX solution or the suspension of DOX-MNPs and the cells were returned to the incubator for 24 h. The medium in the wells was changed at 2 and 4 days after the treatment and no further dose of the drug was added. Medium and control MNPs (without drug) acted as the respective controls for DOX solution and DOX-MNPs. Cell viability was determined on day 5 using an MTS assay (CellTiter 96 AQueous, Promega, Madison, WI). Prior to the assay, the medium from each well was replaced with 100 μL fresh medium and 20 μL of MTS reagent was added. The plates were incubated at 37 °C in CO2 incubator for 90 min, and the absorbance was measured at 490 nm using a microplate reader (BioTek EL800, BioTek Instrument Inc., Winooski, VT). Percentage proliferation of cells in the presence of drug was calculated with respect to the respective controls; i.e., medium control for cells treated with drug in solution and cells treated with DOX-MNPs with control MNPs. The following equation was used to calculate the IC50 values:
| (4) |
where y is % growth, x is the added concentration of Doxorubicin content, A1 is the % growth on the top plateau of the curve, A2 is the % growth on the bottom plateau of the curve, x0 is the inflection point, and p is the slope factor. Experimental data points were fitted to this equation and the IC50 was determined by setting y equal to 50 in the above equation and solving for x.
Statistical Analysis
The least square method was used for linear curve fitting. Origin 7.5 (OriginLab Corporation, Northampton, MA) was used to curve fit the dose-response equation, and exponential equations to calculate the IC50 values and relaxation times (T1 and T2).
RESULTS
Particle Characterization
The mean hydrodynamic diameter of the poly(ethylene) glycol coated oleic acid magnetic nanoparticles (PEG-OA-MNPs) depends on the amount and molecular weight of the OA-PEG polymers used for coating, and the buffer used to disperse the MNPs. An optimal concentration of 200 mg of the polymer produced the smallest particle diameter with a low polydispersity index of 0.009–0.11 for all three of the OA-PEG polymers tested in water (Figure 2A), with an iron oxide core of ~8 nm (Figure 2B). When coated with 200 mg of OA-PEG polymer the particle size and aggregation increased when the particles were dispersed in saline or 1X PBS but did not change in water or mannitol citrate buffer (Figure S1).
Figure 2.

Magnetic nanoparticle size changes with OA-PEG polymer coating. (A) Mean particle size in water after modification with 25–300 mg of OE-20, OE-40, or OE-80 polymers. (B) Representative transmission electron micrograph of OE-40 polymer coated MNPs. Diameter of iron-oxide core ~8 nm.
Presence of the magnetic core, OA, and polymer coating of the MNPs was confirmed by Fourier transform infrared (FTIR) spectroscopy. Bare MNPs had a strong FTIR peak in the region of 550 cm−1 from the iron oxide skeleton (–O–Fe), and a broad peak between 3500–3000 cm−1 due to surface –OH and NH2 groups (Figure 3A). Intense bands appeared in the OA-MNPs at 2914/2848 and 1465 cm−1 due to -CH2 asymmetric/symmetric stretching and bending vibrations of the hydrophobic OA chain. The hydrophilic carboxyl group on OA caused asymmetric vibration of the C=O at 1708 cm−1. After the addition of OA-PEG polymers to the OA-MNP core, prominent peaks appeared in the OE-20, OE-40, and OE-80 MNPs at 1354 and 1102 cm−1 indicating asymmetric and symmetric stretching of C–O–C, and out-of-plane bending of the –CH of the PEG chains at 960 cm−1. Elemental composition of the MNPs was determined by thermogravimetric analysis (TGA), with weight loss of 5.94, 4.56 and 5.71 wt.% of polymers OE-20, OE-40 and OE-80 from the MNPs and 42.55% dextran polymer coating on Feridex IV (Figure 3B).
Figure 3.

Polymer modifications to OA-MNP core confirmed by (A) FTIR and (B) TGA.
Elemental surface composition of the MNPs was determined using X-ray photoelectron spectroscopy. Bare MNPs had peaks at 848 eV and 709 eV from Fe 2s and 2p3/2 electrons, 528 eV from O 1s electrons, 283 eV from C 1s electrons, and at 54 eV from Fe 3p3/2 electrons (Figure 4). The additional carbon atoms on the surface of the OA-MNPs and OE-80 MNPs increased the peak intensity at 283 eV. Atomic concentrations of iron, carbon, and oxygen were calculated from the integrated area of the respective photoemission peaks of the atoms and compared with Feridex IV (Table SI). Superparamagnetic behavior determined by the magnetization saturation (Ms) values of the PEG-OA-MNPs and Feridex IV range from 66.08–85.66 emu/g Fe (Table SI).
Figure 4.

X-ray photoelectron spectra with plain, OA, and OA and PEG-OE-80 coatings.
Protein binding and macrophage uptake
The OE-80 MNPs have a lower protein binding constant (Kb) and fewer binding sites than the OE-20 MNPs, OE-40 MNPs, and Feridex IV when measured in mannitol buffer. In PBS, OE-20 MNPs have the lowest Kb and fewer binding sites than all other formulations (Table I). Feridex IV had less macrophage iron uptake than the PEG-OA-MNPs per milligram of protein (Figure 5). The levels of macrophage iron uptake generally increased as the overall diameter of the particles (Figure 5, Inset).
Table I.
Protein binding constant (Kb) and number of binding sites (n) of MNPs in mannitol citrate buffer and PBS
| Formulation | Mannitol Citrate Buffer | PBS | ||
|---|---|---|---|---|
| Kb (μg/μg) | n | Kb (μg/μg) | n | |
| OE-20 MNPs | 12.2 × 10−2 | 1.3 | 5.6 × 10−2 | 1.2 |
| OE-40 MNPs | 10.9 × 10−2 | 1.4 | 6.6 × 10−2 | 1.6 |
| OE-80 MNPs | 7.2 × 10−2 | 1.3 | 5.8 × 10−2 | 1.4 |
| Feridex IV | 11.1 × 10−2 | 1.3 | 16.7 × 10−2 | 1.9 |
Figure 5.
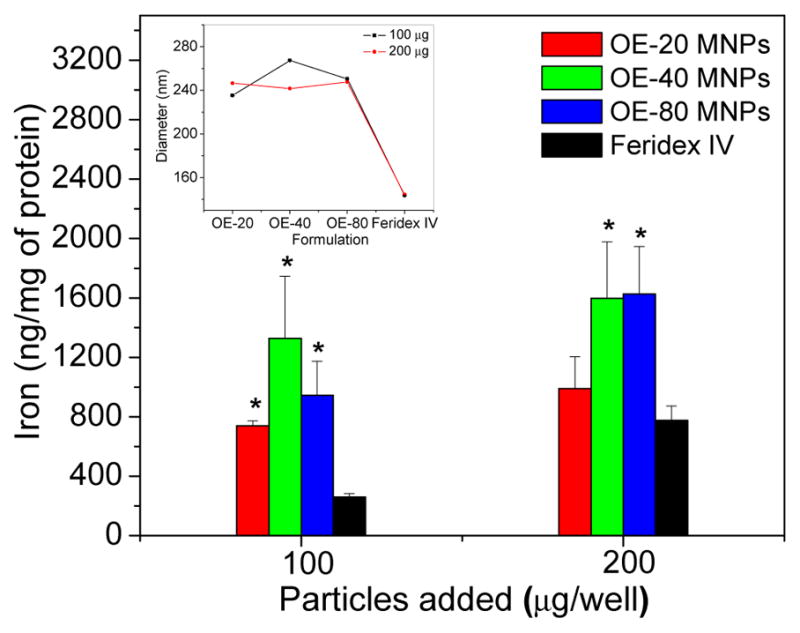
Macrophage uptake of particles with respect to iron level in protein. (Inset) Particle size in RPMI media (data is represented as mean ± SEM, n=3, *p<0.05 compared to Feridex IV).
Magnetic Resonance Imaging
An increase in the particle concentrations (μg Fe/mL) in phantom gels significantly decreased the MR signal intensities, particularly in OE-80 MNPs at 20 and 50 μg Fe/mL gel (Figure 6A). The relaxivity rates r1 (T1 relaxivity) and r2 (T2 relaxivity) (Figure 6B and C), varied among the different formulations calculated from 1/T1 and 1/T2 (Table II). Feridex IV had higher T1 relaxivity, and OE-80 MNPs had highest T2 relaxivity. The strong T2 contrast characteristics of our OE-80 MNPs in vitro led to our testing its’ application in vivo. The MR signal intensity in the mouse carotid artery following tail vein injection of Feridex IV or OE-80 MNPs is graphed as the relative concentration vs. time. (Figure 7). The relative concentration of Feridex IV decreased steadily for 26.5 min, at which point, most of the particles were cleared from circulation. The OE-80 MNPs had a sharp initial drop in the relative concentration, followed by a slow rise in the relative concentration for 45 min. However, approximately 30% of the OE-80 MNPs were in circulation after 52 min. Feridex IV had 30% of particles remaining in circulation at about 10 min, with almost all particle cleared by 50 min.
Figure 6.

Magnetic resonance imaging. (A) Contrast at varying iron concentrations for OA-PEG-MNPs and Feridex IV in agar gels. T2 relaxivity (B) and T1 relaxivity (C) for OE-80 MNPs. (Data as values obtained from curve fitting and standard errors are uncertainties in fitting.)
Table II.
T1 and T2 relaxivity of MNPs in phantom gels (0–20 μg Fe/ml)
| Formulation | T1 relaxivity (r1) (s−1μg−1mL) | T2 relaxivity (r2) (s−1μg−1mL) |
|---|---|---|
| OE-20 MNPs | 3.4 × 10−3 | 4.2 |
| OE-40 MNPs | 5.2 × 10−3 | 3.5 |
| OE-80 MNPs | 7.9 × 10−3 | 9.0 |
| Feridex IV | 13.9 × 10−3 | 4.8 |
Figure 7.
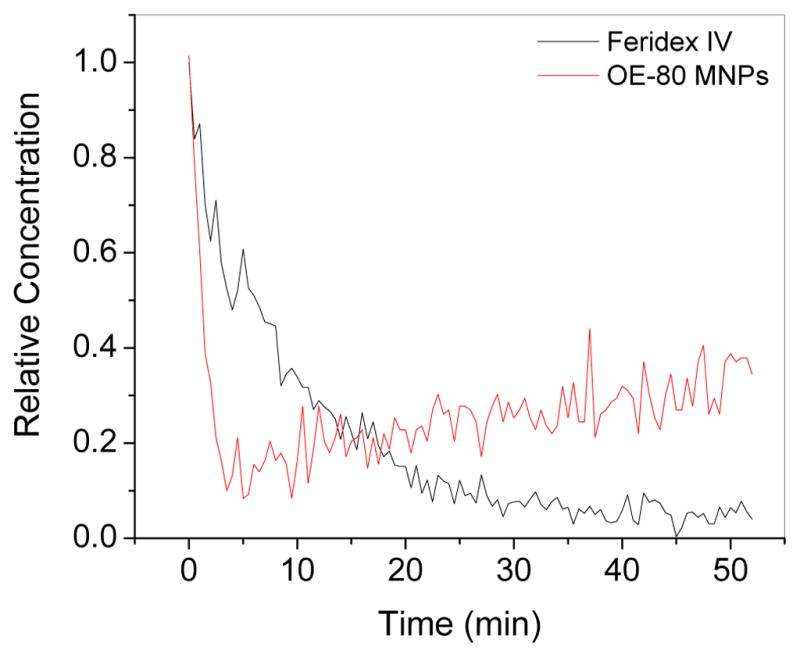
Calculated relative iron concentration vs. time profiles of magnetic nanoparticles in a mouse carotid artery. Shown is the change in one carotid artery but both carotid arteries, and MNP injections in additional mice, showed an almost identical pattern.
Doxorubicin Loading, Characterization, and Drug Release
Loading efficiencies of the anti-cancer drug doxorubicin (DOX) into the MNPs in water was similar in the three OA-PEG-MNPs. The particle size after DOX loading increased from about 184 nm to 250 nm, while cationic charge of DOX increased zeta potential of the OA-PEG-MNPs (Table III). The DOX and MNP interaction was confirmed by FTIR (Figure 8A). Peaks in the DOX-MNPs at 1706, 1614, and 1577/1550 cm−1 were also observed for DOX alone. Stretching of PEG C-O-C was maintained at 1352 and 1102 cm−1, with broadening of C-O-C indicating some hydrogen bonding between the drug molecules and the PEG chains. Peaks between 1300 and 1220 cm−1, 1018 and 986 cm−1, and broadening of the peaks at 1088 and 839 cm−1 further confirm the presence of DOX in the DOX-MNP formulations. Drug release from OE-80-DOX MNPs was sustained, with about 14.9% cumulative release occurring in the first 7 days and 26.8% in 32 days (Figure 8B).
Table III.
Characterization of DOX-loaded MNPs
| Formulation | Size (nm) and (Polydispersity Index) | Zeta Potential (mV) | Drug Loading (%) | Loading Efficiency (%) |
|---|---|---|---|---|
| OE-20-DOX MNPs | 260.0 (0.144) | − 0.4 | 5.4 | 53.9 |
| OE-40-DOX MNPs | 252.5 (0.076) | − 5.7 | 6.4 | 63.9 |
| OE-80-DOX MNPs | 250.7 (0.066) | − 16.7 | 5.2 | 52.2 |
Figure 8.

Characterization and drug release of DOX-MNPs. (A) Polymer formulations modified with DOX and (B) release profile of DOX from DOX-OE-80 MNPs (data is represented as mean ± SEM, n=3).
Antibody-Conjugated OE-80 MNPs and DOX Loading
Transferrin antibody conjugation to the OE-80 MNPs was confirmed with FTIR, and proper orientation of the antibody molecules was tested by adding transferrin to the MNPs and determining the particle size. Intensified peaks for OE-80-DOX MNPs and OE-80-Ab-DOX MNPs at 2919, 1723, 1615, 1578, and 1407 cm−1 compared to OE-80 MNPs and the appearance of additional peaks at 1285, 1068, and 974 cm−1 were attributed to DOX in the formulations (Figure 9A). Broadening of the peak between 3610-3010 cm−1 in OE-80-Ab MNPs is due to hydrogen bonding of the antibody molecules with the PEG chains, and a peak at 1732 cm−1 is from the amide bonds of the antibody.
Figure 9.

Transferrin antibody conjugation to OE-80 MNPs. (A) Antibody conjugation to plain and DOX loaded OE-80 MNPs. (B) Transferrin and BSA binding to OE-80-Ab MNPs indicates proper orientation of transferrin antibody antigen binding sites (data is represented as mean ± SEM, n=3).
Transferrin antigen significantly increased the size of the OE-80-Ab MNPs but not the OE-80 MNPs (Figure 9B). Bovine serum albumin exhibited a similar trend as transferrin, but at higher concentrations, likely due to non-specific interactions. The increase in size after the addition of transferrin antigen or BSA indicates that the antibody is binding to the OE-80 MNPs with the proper outward orientation of the antigen binding sites. Drug loading into the OE-80 MNPs at 4 °C was less than the drug loading at room temperature and it was reduced further after antibody conjugation (Tables III and IV). The drug loading did not change the amount of antibody present in the formulations.
Table IV.
Characterization of MNPs after DOX loading and Antibody Conjugation
| Formulation | Size (nm) and (Polydispersity Index) | Drug Loading (%) | IC50 (ng/mL) |
|---|---|---|---|
| Doxorubicin | - | - | 40.6 |
| OE-80 MNPs | 234.2 (0.17) | - | -b |
| OE-80-DOX MNPs | 288.9 (0.20) | 3.1 | 952.6 |
| OE-80-Ab MNPsa | 242.4 (0.17) | - | -b |
| OE-80-Ab-DOX MNPsa | 288.8 (0.21) | 2.3 | 530.6 |
An average of 3.4 antibody molecules bound to each MNP with 88.2% conjugation efficiency.
The OE-80 MNPs and OE-80-Ab MNPs were non-toxic at all concentrations tested.
Antiproliferative Activity
To further address the applicability of drug targeting and controlled drug release behavior of our formulation, we determined the IC50 for the DOX-OE-80 and DOX-OE-Ab MNP formulations in treating the MCF-7 breast cancer cell line in vitro. The OE-80 MNPs and OE-80-Ab MNPs did not have cytotoxic effects on the MCF-7 cells at the drug concentrations tested. Treatment of MCF-7 cells with DOX solution, DOX-OE-80 MNPs, and DOX-OE-80-Ab MNPs resulted in dose-dependent antiproliferation. Doxrubicin solution was most cytotoxic. Antibody conjugated MNPs loaded with DOX increased the amount of drug uptake within cells and decreased the IC50 by almost two-fold compared to DOX-OE-80 MNPs (Table IV).
DISCUSSION
There are several major obstacles when modifying MNPs for chemotherapeutic applications that affect the magnetic properties for MRI and the drug release from the particles. Conventional magnetic particles such as magnetic microspheres, micro- and nano-gels, dendrimers (12–16); and natural polymer coated MNPs (17–22) may have less morphological control and lower magnetization saturation (Ms), when used as a drug carrier compared to synthetic polymer coated MNPs. Additional challenges for drug delivery include low loading of therapeutic agents especially hydrophobic drugs, fast release of the loaded drug molecules within a few days from the formulations, aggregation of particles, and complexity of drug and biomacromolecular conjugation. To address these common problems, we designed a multifunctional MNP with PEG, a biocompatible synthetic polymer that can be used for drug delivery, targeting, and imaging applications.
Functionalized Poly(ethylene glycol) based polymers stabilize nanoparticles and offer advanced features to control the chemical structure and tailor the surface or functional groups on the nanoparticles (23–26). We tested three different molecular weight OA-PEG polymers at varying concentrations and found the stability and size of PEG-OA-MNPs depends on both the molecular weight and amount of polymer added to the MNP. Plain OA-MNPs aggregate and cluster due to the interaction of the hydrophobic OA layers. The high hydrophilic content of OA-PEG reduced aggregation and decreased particle size, while the increased molecular weight of the different OA-PEG polymers tested likely led to greater size with increasing molecular weight of OA-PEG polymers (26, 27). Interestingly, adding up to 100 mg of the OE-80 PEG polymer initially increased the particle size; the size declined when up to 200 mg of coating was added (Figure 2A). The initial increase in size of OE-80 polymer coated MNPs may be due to the longer hydrophilic PEG chains of the OE-80 polymer which led to insufficient coating and aggregation of particles. Further increasing the OE-80 concentration could have caused the OA group from the OA-PEG-80 to align along the OA coated iron-oxide core, thus stabilizing the particle dispersion. (Figure 1B). Since the hydrophilic PEG chains are smaller in OE-20 and OE-40 polymers than in the OE-80 polymer, the balance between the hydrophilic PEG and hydrophobic OA could have caused these to anchor onto the OA coated around the iron-oxide core.
The hydrodynamic diameter of the OA-PEG MNPs is larger than typical PEG, PVA, dextran, and starch coated MNPs due to the OA-OA-PEG layers formed around the magnetic core. However, most of the superparamagnetic formulations have a greater polymeric content (40–50 wt%) on their surface for stability (20, 24, 28), but our formulations were stable with a low polymer content of 4.56–5.94 wt% (Figure 3B). The low polymer content of our MNP formulation with –NHS functional groups facilitates further chemical modification, and the polymer acts as a shield over the magnetic core. Our OA-PEG polymer coatings lower the magnetization saturation of the MNPs compared to bare particles and OA-MNPs, but our formulation has a close Ms value to the commercial contrast agent Feridex IV.
A critical parameter in nanoparticle design is to prevent phagocytosis and avoid rapid clearance by the RES after IV administration. PEG helps reduce the protein adsorption in vitro (Table I) by increasing the hydrophilic content of the particles. In particular, the longer hydrophilic chain length in the OE-80 formulation lowered BSA binding compared to the remaining formulations with lower or no hydrophilic domains (Figure 1B). Increased protein binding to Feridex IV may be a result of electrostatic interactions between BSA and the iron-oxide core as dextran equilibrates with the surrounding medium (29). Such interactions could have been prevented in our MNP formulation because of PEG forming the corona. Despite higher protein binding, Feridex IV showed lower phagocytic uptake in vitro than our formulations, which could be the effect of smaller size of Feridex IV than our formulation. Particle uptake by macrophages is dependent on both particle size and surface coating of the particles (26). Thus, while protein binding is known to lead to rapid clearance in vivo, there appears to be no correlation in our in vitro experiments between protein binding and phagocytic uptake of MNPs because Feridex IV, despite higher protein binding, shows lower phagocytic cell uptake (Figure 5 and Table I).
In general, the process of T1 shortening (T1 relaxivity) requires the direct interaction between protons and the magnetic component of the contrast agents (30). The greater hydrophilic content of dextran coated Feridex IV and of OE-80 MNPs compared to OE-40 and OE-20 MNPs may increase the proximity of the contrast agent to water molecules (protons) and lower the spin-lattice relaxation time (T1 relaxivity). The hydrophobic domains from the OA layer in the OE-20, OE-40 and OE-80 MNPs may have decreased the T1 relaxivities (Table II and Figure 6). Our OE-80 MNPs show closer T1 relaxivity (lower T1 relaxivity value) to the Feridex IV formulation than the OE-20 and OE-40 MNPs, likely due to the higher PEG chain lengths and greater hydrophilicity of the formulation.
The large particle size, magnetization saturation (Ms), and long polymer chain length can each enhance the T2 MRI contrast of magnetic nanoparticles. Particle aggregation changes the magnetic relaxation properties of near by water protons, reducing the T2 relaxation times (31, 32), while increasing Ms increases in the T2 relaxivities and contrast. Each of these properties may support the relative increase in T2 relaxivity and better contrast properties of our OE-80 formulation compared with Feridex IV (Table II) and our similar formulations with Pluronic® coated OA-MNPs (3.8 × 10−3 s−1μg−1mL) (33). The shorter chain lengths of the OE-20 and OE-40 may increased the aggregation altering the local magnetic heterogeneity in the agarose gels and T2 contrast properties (34).
The significance of formulating MNPs with various polymer coatings is to minimize macrophage uptake and improve blood circulation time (34–38). There are several commercial dextran or polymer coated MNP formulations with available for diagnostics, but the low in vivo half life and rapid capture of the particles by cells and organs of the RES limits their application to spleen and liver diagnostics (2). We observed an initial steep drop in contrast with our formulation followed by a gradual increase, compared to Feridex IV, which gradually decreased over time following injection. The initial sharp decline with our OE-80 MNP formulation may be due to particles initially being removed from circulation into body compartments such as the liver, and then returning to circulation instead of being broken down and cleared by the liver. The long chain length of our PEG coated MNPs may not be recognized as foreign particles in vivo as easily as dextran coated Feridex, and our MNPs may slowly equilibrate with the vascular compartment leading to a gradual increase in the signal intensity detected. We observed a similar pattern of change in the dynamics of iron content in different tissue with time in our previous studies with similarly formulated pluronic stabilized MNPs (39). Conversely, Feridex IV may decline gradually as the dextran, which is in equilibrium with the surrounding environment and weakly associated with the iron-oxide core (29), dissociates and the particles aggregate. This observation is similar our in vitro observation of protein binding, where we saw greater protein binding on Feridex than our MNP formulation (Table I). The prolonged circulation time of our OE-80 formulation in the mouse carotid arteries compared to both Feridex IV (Figure 7) and Pluronic® based formulations may be due to the longer PEG chain length and high hydrophilic content of the OE-80 MNPs (40). We observed less macrophage uptake for the smaller Feridex IV formulation in vitro, thus reducing the size further and increasing the hydrophilic content of the current formulation may result in an even longer circulation time in vivo (36, 41).
A major advantage in using nanoparticles for chemotherapeutic applications is to prevent systemic drug toxicity by encapsulating the drug. In addition, the particles can allow for sustained drug release over time, potentially lowering the dosing frequency and improving patient compliance. The OA layer in the current formulation entraps the hydrophobic drug Doxorubicin through physical interaction. This physical interaction is a significant advantage over nanoparticle-drug conjugates, magnetic liposomes, and drug encapsulated formulations because easy drug loading, high loading efficiency, and sustained release as opposed to burst release. In particular, the release of hydrophilic drugs from hydrophilic compounds is rapidly reversible and the drug dissociates within hours (4). The sustained release of doxorubicin from the MNPs over several days likely occurs through two mechanisms, drug release from the surface of the MNPs and release through diffusion from OA layers due to concentration gradient with respect to the surrounding environment.
The versatility of the OE-80 MNP formulation allowed us to investigate antibody conjugation and thus targeted drug delivery in addition to the MRI. The OA-PEG-80 chain has an active – NHS end group which offers high and direct conjugation capacity to the amine group on antibodies without requiring a cross-linker like some polymer coated MNP formulations used for conjugation (42). For antibody conjugation, the reaction and drug loading was carried out at 4 °C in PBS. The hydrophilicity of the particle after antibody conjugation and a the low temperature reduced the drug loading capacity of the formulation compared to drug loading at room temperature because the parent OA freezing point is 16.4 °C. In addition, drug loading in PBS may have further reduced the loading capacity due to some aggregation of the particles which was not observed when drug was added to particles suspended in water (Table S1 and Table III). However, the drug loading combined with antibody conjugation was still sufficient to provide a two-fold decrease in drug required to inhibit 50% of cell growth in vitro (Table IV). In addition, drug is released over several weeks from the MNPs, whereas 100% of the free drug is available to inhibit cell growth. Based on the drug release profile (Figure 8B), about 13.68% of the drug is released over five days, the length of the in vitro study. Thus, the drug released may be around 72.6 ng/mL for OE-80-Ab-DOX MNPs, and 130.3 ng/mL OE-80-Ab MNPs, which is much closer to the 40.6 ng/mL DOX required to inhibit 50% of the cell growth in vitro. Confocal scanning laser microscopy of DOX MNPs confirms that drug is present within the cytoplasm and likely still associated with the MNPs (43). There is a slow increase in accumulation of drug detected in the nucleus, the site action, as the drug is released from the MNPs. Plain drug, alternatively, is visible almost exclusively in the nucleus; this likely accounts for the higher toxicity observed of free DOX compared to that of DOX MNPs. Our formulation can be further optimized for the drug and also for specific targeting ligand and its density around MNPs to further improve the efficacy.
Our MNPs have potential to be developed as an imaged-guided drug therapy because these can be imaged using MRI and simultaneously deliver drugs to the target tissue. Image-guided drug therapy is particularly important in cancer therapy because of significant variations in tumor vascularity, heterogeneous and diverse expression of cell surface receptors, and genetic make-up of cancer cells (44), as these factors could significantly influence the efficacy of nanocarrier-mediated drug delivery as well as drug response. Nanocarriers with imaging capacity can ensure drug delivery to the target tissue.
CONCLUSION
We show that our MNP formulation has dual functionality, i.e. drug delivery and MRI properties. In the future, our MNPs can potentially be used as a theranostic agent for image-guided cancer therapy to maximize therapeutic effect while minimizing drug toxicity.
Supplementary Material
Acknowledgments
The study reported here is funded by grant R01 EB005822 (to VL) from the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health. SPF is a predoctoral student in Cleveland Clinic’s Molecular Medicine Ph.D. Program, which is funded by the “Med into Grad” initiative of the Howard Hughes Medical Institute [http://www.lerner.ccf.org/molecmed/phd/].
References
- 1.Bulte JW, Kraitchman DL. Monitoring cell therapy using iron oxide MR contrast agents. Curr Pharm Biotechnol. 2004;5:567–584. doi: 10.2174/1389201043376526. [DOI] [PubMed] [Google Scholar]
- 2.Josephson L. Magnetic Nanoparticles for MR Imaging. Springer; US: 2007. [Google Scholar]
- 3.Bulte JW, Kraitchman DL. Iron oxide MR contrast agents for molecular and cellular imaging. NMR Biomed. 2004;17:484–499. doi: 10.1002/nbm.924. [DOI] [PubMed] [Google Scholar]
- 4.Alexiou C, Arnold W, Klein RJ, Parak FG, Hulin P, Bergemann C, Erhardt W, Wagenpfeil S, Lubbe AS. Locoregional cancer treatment with magnetic drug targeting. Cancer Res. 2000;60:6641–6648. [PubMed] [Google Scholar]
- 5.Beaven GH, Chen SH, d’Albis A, Gratzer WB. A spectroscopic study of the haemin--human-serum-albumin system. Eur J Biochem. 1974;41:539–546. doi: 10.1111/j.1432-1033.1974.tb03295.x. [DOI] [PubMed] [Google Scholar]
- 6.Lehrer SS, Fasman GD. The fluorescence of lysozyme and lysozyme substrate complexes. Biochem Biophys Res Commun. 1966;23:133–138. doi: 10.1016/0006-291x(66)90517-1. [DOI] [PubMed] [Google Scholar]
- 7.Chipman DM, Grisaro V, Sharon N. The binding of oligosaccharides containing N-acetylglucosamine and N-acetylmuramic acid to lysozyme. The specificity of binding subsites. J Biol Chem. 1967;242:4388–4394. [PubMed] [Google Scholar]
- 8.Jeffery GH, Bassett J, Mendham J, Denny RC. Vogel’s text book of quantitative chemical analysis. John Wiley & Sons Inc; New York: 1989. [Google Scholar]
- 9.Moffat BA, Reddy GR, McConville P, Hall DE, Chenevert TL, Kopelman RR, Philbert M, Weissleder R, Rehemtulla A, Ross BD. A novel polyacrylamide magnetic nanoparticle contrast agent for molecular imaging using MRI. Mol Imaging. 2003;2:324–332. doi: 10.1162/15353500200303163. [DOI] [PubMed] [Google Scholar]
- 10.Yolles S, Aslund B, Morton JF, Olson OT, Rosenberg B. Timed-released depot for anticancer agents. II. Acta Pharm Suec. 1978;15:382–388. [PubMed] [Google Scholar]
- 11.Olivier JC, Huertas R, Lee HJ, Calon F, Pardridge WM. Synthesis of pegylated immunonanoparticles. Pharm Res. 2002;19:1137–1143. doi: 10.1023/a:1019842024814. [DOI] [PubMed] [Google Scholar]
- 12.Gou ML, Qian ZY, Wang H, Tang YB, Huang MJ, Kan B, Wen YJ, Dai M, Li XY, Gong CY, Tu MJ. Preparation and characterization of magnetic poly(epsilon-caprolactone)-poly(ethylene glycol)-poly(epsilon-caprolactone) microspheres. J Mater Sci Mater Med. 2007;19:1033–1041. doi: 10.1007/s10856-007-3230-3. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Kaminski MD, Chen H, Torno M, Taylor L, Rosengart AJ. Synthesis and characterization of highly-magnetic biodegradable poly(d,l-lactide-co-glycolide) nanospheres. J Control Release. 2007;119:52–58. doi: 10.1016/j.jconrel.2006.11.031. [DOI] [PubMed] [Google Scholar]
- 14.Okassa LN, Marchais H, Douziech-Eyrolles L, Herve K, Cohen-Jonathan S, Munnier E, Souce M, Linassier C, Dubois P, Chourpa I. Optimization of iron oxide nanoparticles encapsulation within poly(d,l-lactide-co-glycolide) sub-micron particles. Eur J Pharm Biopharm. 2007;67:31–38. doi: 10.1016/j.ejpb.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 15.Hamoudeh M, Al Faraj A, Canet-Soulas E, Bessueille F, Leonard D, Fessi H. Elaboration of PLLA-based superparamagnetic nanoparticles: characterization, magnetic behaviour study and in vitro relaxivity evaluation. Int J Pharm. 2007;338:248–257. doi: 10.1016/j.ijpharm.2007.01.023. [DOI] [PubMed] [Google Scholar]
- 16.Bhattacharya S, Eckert F, Boyko V, Pich A. Temperature-, pH-, and magnetic-field-sensitive hybrid microgels. Small. 2007;3:650–657. doi: 10.1002/smll.200600590. [DOI] [PubMed] [Google Scholar]
- 17.Shen F, Poncet-Legrand C, Somers S, Slade A, Yip C, Duft AM, Winnik FM, Chang PL. Properties of a novel magnetized alginate for magnetic resonance imaging. Biotechnol Bioeng. 2003;83:282–292. doi: 10.1002/bit.10674. [DOI] [PubMed] [Google Scholar]
- 18.Bonacchi D, Caneschi A, Dorignac D, Falqui A, Gatteschi D, Rovai D, Sangregorio C, Sessoli R. Nanosized iron oxide particles entrapped in pseudo-single crystals gamma-cyclodextrin. Chemistry of Materials. 2004;16:2016–2020. [Google Scholar]
- 19.Bonacchi D, Caneschi A, Gatteschi D, Sangregorio C, Sessoli R, Falqui A. Synthesis and characterisation of metal oxides nanoparticles entrapped in cyclodextrin. Journal of Physics and Chemistry of Solids. 2004;65:719–722. [Google Scholar]
- 20.Mikhaylova M, Kim DK, Bobrysheva N, Osmolowsky M, Semenov V, Tsakalakos T, Muhammed M. Superparamagnetism of magnetite nanoparticles: dependence on surface modification. Langmuir. 2004;20:2472–2477. doi: 10.1021/la035648e. [DOI] [PubMed] [Google Scholar]
- 21.Kim DK, Mikhaylova M, Wang FH, Kehr J, Bjelke B, Zhang Y, Tsakalakos T, Muhammed M. Starch-coated superparamagnetic nanoparticles as MR contrast agents. Chemistry of Materials. 2003;15:4343–4351. [Google Scholar]
- 22.Pardoe H, Chua-anusorn W, St Pierre TG, Dobson J. Structural and magnetic properties of nanoscale iron oxide particles synthesized in the presence of dextran or polyvinyl alcohol. J Magn Magn Mater. 2001;225:41–46. [Google Scholar]
- 23.Lee H, Yu MK, Park S, Moon S, Min JJ, Jeong YY, Kang HW, Jon S. Thermally cross-linked superparamagnetic iron oxide nanoparticles: synthesis and application as a dual imaging probe for cancer in vivo. J Am Chem Soc. 2007;129:12739–12745. doi: 10.1021/ja072210i. [DOI] [PubMed] [Google Scholar]
- 24.Wan S, Huang J, Guo M, Zhang H, Cao Y, Yan H, Liu K. Biocompatible superparamagnetic iron oxide nanoparticle dispersions stabilized with poly(ethylene glycol)-oligo(aspartic acid) hybrids. J Biomed Mater Res A. 2007;80:946–954. doi: 10.1002/jbm.a.31022. [DOI] [PubMed] [Google Scholar]
- 25.Lutz JF, Stiller S, Hoth A, Kaufner L, Pison U, Cartier R. One-pot synthesis of pegylated ultrasmall iron-oxide nanoparticles and their in vivo evaluation as magnetic resonance imaging contrast agents. Biomacromolecules. 2006;7:3132–3138. doi: 10.1021/bm0607527. [DOI] [PubMed] [Google Scholar]
- 26.Xie J, Xu C, Kohler N, Hou Y, Sun S. Controlled PEGylation of monodisperse Fe3O4 nanoparticles for reduced non-specific uptake by macrophage cells. Adv Mater. 2007;19:3163–3166. [Google Scholar]
- 27.Ditsch A, Laibinis PE, Wang DI, Hatton TA. Controlled clustering and enhanced stability of polymer-coated magnetic nanoparticles. Langmuir. 2005;21:6006–6018. doi: 10.1021/la047057+. [DOI] [PubMed] [Google Scholar]
- 28.Weissleder R, Elizondo G, Wittenberg J, Lee AS, Josephson L, Brady TJ. Ultrasmall superparamagnetic iron oxide: an intravenous contrast agent for assessing lymph nodes with MR imaging. Radiology. 1990;175:494–498. doi: 10.1148/radiology.175.2.2326475. [DOI] [PubMed] [Google Scholar]
- 29.McCarthy JR, Weissleder R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv Drug Deliv Rev. 2008;60:1241–1251. doi: 10.1016/j.addr.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okuhata Y. Delivery of diagnostic agents for magnetic resonance imaging. Adv Drug Deliv Rev. 1999;37:121–137. doi: 10.1016/s0169-409x(98)00103-3. [DOI] [PubMed] [Google Scholar]
- 31.Yigit MV, Mazumdar D, Lu Y. MRI detection of thrombin with aptamer functionalized superparamagnetic iron oxide nanoparticles. Bioconjugate Chem. 2008;19:412–417. doi: 10.1021/bc7003928. [DOI] [PubMed] [Google Scholar]
- 32.Jun YW, Huh YM, Choi JS, Lee JH, Song HT, Kim S, Yoon S, Kim KS, Shin JS, Suh JS, Cheon J. Nanoscale size effect of magnetic nanocrystals and their utilization for cancer diagnosis via magnetic resonance imaging. J Am Chem Soc. 2005;127:5732–5733. doi: 10.1021/ja0422155. [DOI] [PubMed] [Google Scholar]
- 33.Jain TK, Richey J, Strand M, Leslie-Pelecky DL, Flask CA, Labhasetwar V. Magnetic nanoparticles with dual functional properties: Drug delivery and magnetic resonance imaging. Biomaterials. 2008;29:4012–4021. doi: 10.1016/j.biomaterials.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bulte JWM, Cuyper MD, Despres D, Frank JA. Preparation, relaxometry, and biokinetics of PEGylated magnetoliposomes as MR contrast agent. J Magn Magn Mater. 1999;194:204–209. [Google Scholar]
- 35.Zhang Y, Kohler N, Zhang MQ. Surface modification of superparamagnetic magnetite nanoparticles and their intracellular uptake. Biomaterials. 2002;23:1553–1561. doi: 10.1016/s0142-9612(01)00267-8. [DOI] [PubMed] [Google Scholar]
- 36.Arruebo M, Fernandez-Pacheco R, Ibarra MR, Santamaria J. Magnetic nanoparticles for drug delivery. Nano Today. 2007;2:22–32. [Google Scholar]
- 37.Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat Biotechnol. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 38.Seo SB, Yang J, Hyung W, Cho EJ, Lee TI, Song YJ, Yoon HG, Suh JS, Huh YM, Haam S. Novel multifunctional PHDCA/PEI nano-drug carriers for simultaneous magnetically targeted cancer therapy and diagnosis via magnetic resonance imaging. Nanotechnology. 2007;18:1–8. [Google Scholar]
- 39.Jain TK, Reddy MK, Morales MA, Leslie-Pelecky DL, Labhasetwar V. Biodistribution, clearance, and biocompatibility of iron oxide magnetic nanoparticles in rats. Mol Pharmaceutics. 2008;5:316–327. doi: 10.1021/mp7001285. [DOI] [PubMed] [Google Scholar]
- 40.Jain TK, Foy SP, Erokwu B, Dimitrijevic S, Flask CA, Labhasetwar V. Magnetic resonance imaging of multifunctional pluronic stabilized iron-oxide nanoparticles in tumor-bearing mice. Biomaterials. 2009;30:6748–6756. doi: 10.1016/j.biomaterials.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neuberger T, Schopf B, Hofmann H, Hofmann M, von Rechenberg B. Superparamagnetic nanoparticles for biomedical applications: Possibilities and limitations of a new drug delivery system. Journal of Magnetism and Magnetic Materials. 2005;293:483–496. [Google Scholar]
- 42.Jun YW, Lee JH, Cheon J. Chemical design of nanoparticle probes for high-performance magnetic resonance imaging. Angew Chem Int Ed Engl. 2008;47:5122–5135. doi: 10.1002/anie.200701674. [DOI] [PubMed] [Google Scholar]
- 43.Jain TK, Morales MA, Sahoo SK, Leslie-Pelecky DL, Labhasetwar V. Iron oxide nanoparticles for sustained delivery of anticancer agents. Mol Pharmaceutics. 2005;2:194–205. doi: 10.1021/mp0500014. [DOI] [PubMed] [Google Scholar]
- 44.Kosaka N, Ogawa M, Longmire MR, Choyke PL, Kobayashi H. Multi-targeted multi-color in vivo optical imaging in a model of disseminated peritoneal ovarian cancer. J Biomed Opt. 2009;14:014023. doi: 10.1117/1.3083449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.