

Abstract
Most physiological processes are subjected to molecular regulation by growth factors, which are secreted proteins that activate chemical signal transduction pathways through binding of specific cell-surface receptors. One particular growth factor system involved in the in vivo regulation of blood vessel growth is called the vascular endothelial growth factor (VEGF) system. Computational and numerical techniques are well-suited to handle the molecular complexity (the number of binding partners involved, including ligands, receptors, and inert binding sites) and multi-scale nature (intra-tissue vs. inter-tissue transport and local vs. systemic effects within an organism) involved in modeling growth factor system interactions and effects. This paper introduces a variety of in silico models that seek to recapitulate different aspects of VEGF system biology at various spatial and temporal scales: molecular-level kinetic models focus on VEGF ligand-receptor interactions at and near the endothelial cell surface; meso-scale single-tissue 3D models can simulate the effects of multi-cellular tissue architecture on the spatial variation in VEGF ligand production and receptor activation; compartmental modeling allows efficient prediction of average interstitial VEGF concentrations and cell-surface VEGF signaling intensities across multiple large tissue volumes, permitting the investigation of whole-body inter-tissue transport (e.g., vascular permeability and lymphatic drainage). The given examples will demonstrate the utility of computational models in aiding both basic science and clinical research on VEGF systems biology.
1. Background
1.1 Biology of growth factor systems
Growth Factor Systems in Angiogenesis
At the molecular and cellular levels, growth factors are extracellularly secreted polypeptides which, upon binding to specific cell-surface target receptors, trigger intracellular signal transduction pathways that regulate cell proliferation, differentiation and survival (Lodish et al., 2004). At the tissue and organ levels, growth factors are responsible for orchestrating many physiological processes in complex multicellular organisms.
Of utmost importance in human physiology and pathology is the process known as angiogenesis – the growth of new capillaries or microvessels from pre-existing blood vasculature – which critically supports organogenesis during embryonic development (Haigh, 2008); physiological growth and repair in adult tissues (such as in wound healing (Bao et al., 2009), muscular adaptation to exercise (Brown and Hudlicka, 2003), or endometrial regeneration (Girling and Rogers, 2005)); as well as the malignant growth of tumor tissues (Kerbel, 2008).
Sprouting angiogenesis is a well-coordinated and complex cascade of molecular, cellular, and tissue-level events (Qutub et al., 2009): First, tissue ischemia is converted into a chemical cue for angiogenesis, as hypoxic cells (e.g., cancer cells in tumor tissue or myocytes in ischemic muscle) transcribe and secrete growth factors in response to hypoxia-inducible factor 1 (HIF1) activation. The growth factor ligands then diffuse throughout the extracellular fluid, where some become sequestered to matrix proteoglycans and others bind cell-surface receptors on the capillary endothelium. Cell surface receptor-bound ligands initiate vessel sprouting by turning quiescent endothelial cells into migratory tip cells. Extracellular matrix-bound and freely diffusing ligands form chemotatic gradients that guide the migration of tip cell filopodia in the capillary sprout. In the next two sections, we further explore the multi-scale nature and molecular complexity of angiogenic growth factor interactions.
Systems Biology of VEGF: Interaction Networks and Molecular Cross-Talk
Many growth factor systems are involved in angiogenic regulation, including the vascular endothelial growth factor (VEGF) system of at least 5 ligands (VEGF-A, PlGF, VEGF-B, VEGF-C, VEGF-D) and 3 receptors (VEGFR1, VEGFR2, VEGFR3) (Roy et al., 2006; Mac Gabhann and Popel, 2008); the fibroblast growth factor (FGF) system of at least 18 ligands (FGF1 to FGF10 and FGF16 to FGF23) and 4 receptors (FGFR1 to FGFR4) (Beenken and Mohammadi, 2009); the angiopoietin (Ang) system of at least 4 ligands (ANG1 to ANG4) and 2 receptors (TIE1 and TIE2) (Augustin et al., 2009); the platelet-derived growth factor (PDGF) system of at least 4 ligands (PDGF-A to PDGF-D) and 2 receptors (PDGFR-α and PDGFR-β) (Andrae et al., 2008); and the insulin-like growth factor (IGF) system of at least 2 ligands (IGF1 and IGF2) and 2 receptors (IGF1R and IGF2R) (Mazitschek and Giannis, 2004; Pollak, 2008).
There are organizational similarities between these growth factor systems – all of the above receptors except for the IGF2R are transmembrane receptor tyrosine kinases (RTKs) that are activated by ligand-induced dimerization and transphosphorylation of tyrosine residues (Gschwind et al., 2004); co-receptors (e.g., neuropilin-1 (NRP1) for VEGFRs and syndecan-4 for FGFRs) and endothelial integrins (e.g., αvβ5 for VEGFRs and αvβ3 for FGFRs) often modulate receptor signaling (Simons, 2004); heparan sulfate proteoglycans (HSPGs) are involved in the extracellular matrix sequestration of ligands in the VEGF, PDGF, FGF systems (Roy et al., 2006; Andrae et al., 2008; Beenken and Mohammadi, 2009).
Intracellularly, details are also emerging on the convergent and integrative crosstalk between and within growth factor systems in angiogenic signaling. Among the overlap in their transcriptional profiles downstream of RTK activation, all aforementioned growth factor systems can activate the canonical Ras-MAPK signaling pathway (Simons, 2004). VEGF and FGF2 were observed to induce the expression of each other (Simons, 2004). Within the VEGF system, the existence of heterodimeric ligands (e.g., VEGF-A/PlGF and VEGF-A/VEGF-B) and heterodimeric receptors (e.g., VEGFR1/VEGFR2) is expected to introduce new signal transduction pathways in addition to those downstream of classic homodimeric ligand-receptor activation (Mac Gabhann and Popel, 2008; Cao, 2009). While VEGFR1 is mainly a negative regulator of angiogenesis, its possible pro-angiogenic roles are suspected to involve the intermolecular transphosphorylation of VEGFR2 by PlGF-activated VEGFR1 (Autiero et al., 2003).
In this paper, we will introduce computational frameworks that are well suited for the quantitative modeling of highly complex molecular interaction networks such as that of the VEGF system in angiogenesis. While our examples focus on the VEGF system, the mathematical frameworks are generally adaptable for any of the organizationally similar growth factor systems introduced above, with the potential of further integration between the VEGF, FGF, PDGF, IGF, Ang-Tie system modules themselves.
Multi-scale Biology of VEGF: Transport and Signaling Range
In understanding the biology of angiogenic growth factors, it is of equal importance to identify the key molecular players and to distinguish where in the body the molecular interactions take place. The spatial range of activity can vary between growth factors (or between isoforms of the same growth factor) depending on their propensity for intra-tissue and inter-tissue transport.
The intra-tissue transport of a secreted growth factor – i.e., its diffusive and convective transport within the extracellular matrix (ECM) – is dependent on the growth factor’s molecular size, the pore sizes of the ECM, and its chemical affinity for ECM proteoglycans. For instance, heparin-binding affinity of VEGF, which determines the extent of its sequestration by ECM heparan sulfate proteoglycans, is traditionally thought to be encoded in the VEGF-A gene on exons 6 and 7 (Harper and Bates, 2008). Hence, the pro-angiogenic VEGF121 and anti-angiogenic VEGF121b splice isoforms which skip exons 6 and 7 are mostly freely diffusible once secreted into the extracellular fluid; while the higher molecular-weight isoforms VEGF145(b), VEGF148, VEGF165(b), VEGF183(b), VEGF189(b), and VEGF206 have progressively higher heparin-binding affinity due to their inclusion of increasingly greater portions of exons 6 and 7 (Harper and Bates, 2008). Yet these higher molecular-weight isoforms in their matrix-bound state can be subjected to proteolytic cleavage by plasmin or matrix metalloproteinases (MMPs), which releases active fragments of 110 to 113 amino acids in length and with similar angiogenic properties as VEGF121 (Ferrara and Davis-Smyth, 1997; Lee et al., 2005; Qutub et al., 2009).
On a larger scale, the inter-tissue transport of a growth factor is first affected by its rate of entry into or exit from the blood or lymphatic vasculature, i.e., its permeability through the blood capillary endothelium or its lymphatic drainage rate from interstitial spaces. Once in the bloodstream or lymphatic fluid, the inter-tissue transport of a growth factor may be further facilitated by specific carrier proteins or circulating cells. For VEGF, its potential carriers in blood include soluble forms of its normal receptors (sVEGFR1, sVEGFR2, sNRP1) (Gagnon et al., 2000; Ebos et al., 2004; Sela et al., 2008), plasma fibronectin (Wijelath et al., 2006), as well as platelets (Verheul et al., 2007).
All together, these transport properties influence the distance over which a growth factor can signal. Autocrine signaling occurs when growth factors act upon the same cells that produced them; juxtacrine growth factors act upon adjacent cells after secretion; paracrine growth factors diffuse through the extracellular fluid and target cells within the same tissue but of a different cell type than that which secreted them; whereas growth factors that are transported through the bloodstream to distant target tissues act in an endocrine manner (Lauffenburger and Linderman, 1993; Lodish et al., 2004).
Angiogenic VEGF signaling occurs predominantly in a paracrine fashion: epithelial cells in fenestrated organs (e.g. glomerular podocytes), mesenchymal cells (e.g., skeletal myocytes in ischemic muscles), vascular stromal cells (e.g., pericytes and smooth muscle cells) and hypoxic tumor cells are all known to secrete VEGF, which then diffuses to and activates the VEGF receptors on nearby endothelial cell surfaces (Maharaj and D'Amore, 2007; Kerbel, 2008). However, there is also evidence of autocrine VEGF signaling loops in VEGF-producing endothelial cells (Martin et al., 2009). Furthermore, autocrine VEGF signaling involving intracellular VEGF receptors (“intracrine signaling”) has been documented in breast carcinoma cells (Lee et al., 2007) and haematopoietic stem cells (Gerber et al., 2002); although in these contexts, VEGF functions as a cell survival signal rather than a pro-angiogenic factor.
While there have not been formally established specific endocrine functions of VEGF in normal physiology, aberrantly high circulating levels of VEGF may have deleterious systemic effects. In clinical trials administering VEGF via intravascular infusion to stimulate therapeutic angiogenesis for ischemic muscle diseases, unintended side effects of the high systemic VEGF concentrations such as hypotension and macular edema have been transiently and sporadically observed (Collinson and Donnelly, 2004). The VEGF concentrations in the plasma of cancer patients are also known to be several-fold higher than healthy baseline levels, although it is uncertain whether the tumors are themselves the source of the elevated circulating VEGF, or whether conversely the elevated circulating VEGF triggered the malignant growth of tumors (Kut et al., 2007; Stefanini et al., 2008). Therefore, a complete understanding of VEGF biology – including its pathogenic role in cancer and its therapeutic potential in ischemic diseases – necessitates an appreciation of the dynamic distribution of VEGF in the human body (Kut et al., 2007).
The computational models presented in this paper are complementary, capturing the biology of VEGF interactions at different length scales: the molecular-level kinetic models can predict the local intensity of VEGF-VEGFR complex formation on endothelial cell surfaces as a marker of angiogenic activation or vessel sprouting initiation; the meso-scale single-tissue 3D models involving multiple cell types can predict the spatial gradients of VEGF in the extracellular space and simulate paracrine signaling distributions that guide capillary sprout migration; and the multi-tissue compartmental models can be used to simulate whole-body VEGF distributions and to investigate the possibility of endocrine VEGF effects.
1.2 Computational models of the VEGF system
Figure 1 summarizes the multi-scale nature and complexity of the molecular interactions involved in the systems biology of the VEGF ligand-receptor system. In the following sections, we introduce models for investigating emergent behavior at progressively higher spatial scales: from molecular (subcellular) level models, to meso-scale (intra-tissue) models, to whole-body (inter-tissue) models. The chosen examples also illustrate the versatility of computational modeling for investigating basic science questions (e.g., simulating the molecular mechanisms underlying the PlGF-VEGF synergy and NRP1-VEGFR2 synergy) and assisting in the design of translational medicine (e.g., comparing the therapeutic efficacy of cell-based versus protein delivery of VEGF; optimizing the dose of anti-VEGF therapy).
Figure 1. Multi-scale Systems Biology of the VEGF ligand-receptor system.
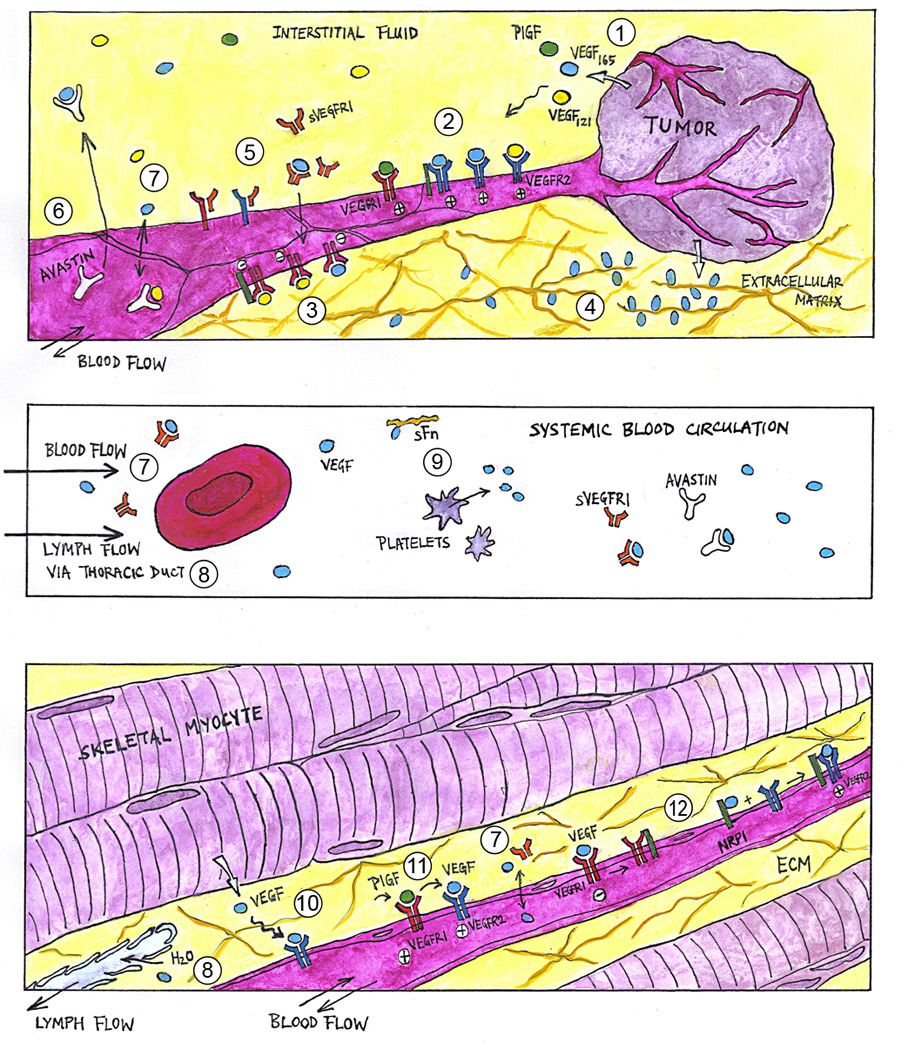
(1) Hypoxia, such as that in growing tumor tissues (top panel), trigger the expression and extracellular secretion of VEGF ligand proteins, e.g., VEGF-A (isoforms VEGF121 and VEGF165) and PlGF. At the cellular level, VEGF ligands diffuse towards nearby capillary surfaces, binding endothelial cell surface receptors (VEGFR1, VEGFR2) and co-receptors (NRP1) in various configurations to activate (2) pro-angiogenic and (3) anti-angiogenic downstream signaling. (4) At the tissue level, VEGF ligands with heparin-binding domains can be sequestered at heparan sulfate proteoglycan sites in the extracellular matrix (ECM), forming chemotactic gradients that guide capillary sprout migration. (5) Soluble VEGFR1 (sVEGFR1) potentially modulates angiogenic signaling via ligand trapping or dominant-negative heterodimerization with transmembrane VEGFR monomers. (6) Humanized anti-VEGF antibodies (e.g., Avastin®), through their capacity to sequester specific VEGF ligands, are being investigated as anti-angiogenic agents in cancer treatment. At the whole-organism level, macromolecules such as VEGF ligands and their soluble receptors may have systemic effects, as they enter the blood circulatory system (middle panel) through inter-tissue transport processes including (7) transcapillary vascular permeability and (8) lymphatic drainage of the interstitial fluid. (9) Other VEGF carriers in the blood include soluble fibronectin and platelets. (10) Similarly in skeletal muscle (bottom panel), VEGF ligand expression is upregulated in hypoxic myocytes. However in peripheral arterial disease, the angiogenic response is insufficient to alleviate muscle ischemia. Pro-angiogenic therapies under investigation include VEGF-A delivery through cell, gene, and protein therapy. Adjuvant therapeutic targets include (11) PlGF (thought to work synergistically through VEGFR1 signaling or ligand shifting) and (12) NRP1 (via presentation of VEGF to VEGFR2 or reducing anti-angiogenic VEGF·VEGFR1 complexes).
2. Molecular-level kinetics models: simulation of in vitro experiments
In our first two examples, models were developed to investigate specific molecular functions and interactions of key players in the VEGF ligand-receptor system – PlGF and NRP1 – by recapitulating in vitro experiments. The spatial scope of these models focused on molecular behaviors near the endothelial cell surface, including extracellular ligand diffusion and cell-surface ligand-receptor binding.
2.1 Mathematical Framework for Biomolecular Interaction Networks
Mathematical theory and formulations for kinetic modeling of cell surface ligand-receptor binding and cell-surface receptor/ligand trafficking have been presented in classical texts (Lauffenburger and Linderman, 1993). The standard description for the binding kinetics of ligand L to receptor R to form complex C involves characterization of the complex association and dissociation rate constants kon and koff:
| (1) |
Endocytotic internalization of free receptors and complexes are generally characterized by first-order rate constants:
| (2) |
Free receptor insertion rates are typically introduced through zero-order source terms; in the following models, they are chosen to maintain a steady total population (free and bound) of receptors in the absence of added ligand.
2.2 Case Study: Mechanism of PlGF synergy: shifting VEGF to VEGFR2 vs. PlGF-VEGFR1 signaling
Our first case study sought to decipher the molecular mechanisms behind PlGF’s observed ability to augment the angiogenic response to VEGF-A in in vitro assays for endothelial cell survival, proliferation and migration. Details and full references can be found in (Mac Gabhann and Popel, 2004). These two members of the VEGF family have different receptor-binding properties: VEGF-A (hereinafter referred to as simply “VEGF”) binds with both VEGFR1 and VEGFR2; while PlGF only binds VEGFR1. Two proposed mechanisms for the PlGF-VEGF synergy were: (a) “ligand shifting”, where PlGF displaces VEGF from VEGFR1, effectively freeing more VEGF to bind the more pro-angiogenic VEGFR2; and (b) PlGF-VEGFR1 signaling, where PlGF activation of VEGFR1 may transduce qualitatively different (pro-angiogenic) signals than that from VEGF activation (generally inhibitory of angiogenic signaling). An in silico model was thus constructed to quantify these mechanistic contributions to the VEGF-PlGF synergy.
The in silico model formulation mimicked in vitro assay geometry and conditions as illustrated in Fig. 2A. At the bottom of a cell culture well, a confluent layer of endothelial cells expressing receptors VEGFR1 and VEGFR2 on the surface (z=0) was exposed to the fluid media. Into the cell culture media (from z=0 to z=h), ligands were administered at time zero – either VEGF alone (“PlGF-” case) or VEGF and PlGF (“PlGF+” case) – to assess the synergistic effects of PlGF. Mathematically, each molecular species was represented by either a volumetric concentration (V for VEGF, P for PlGF) or cell-surface concentration (R1 for VEGFR1, R2 for VEGFR2, VR1 for VEGF·VEGFR1 complex, PR1 for PlGF·VEGFR1 complex, VR2 for VEGF·VEGFR2 complex), using the continuum approach. The initial value problem was essentially a single spatial dimension problem (z-direction) as molecular concentrations are assumed uniform in the plane parallel to the endothelial cell surface.
Figure 2. Molecular-level kinetics models.

Modeling of molecular interactions underlying PlGF-VEGF synergy: experimental setup (A) and sample results (B) based on data previously published in (Mac Gabhann and Popel, 2004). Modeling of NRP1’s role in the differential binding of VEGF isoforms to VEGFR2: experimental setup (C) and sample results (D) based on data previously published in (Mac Gabhann and Popel, 2005).
Coupled diffusion and reaction equations (Eqns. 3 and 4 respectively) were used to describe the time evolution of extracellular ligand transport and cell-surface molecular binding interactions. D represented ligand diffusivity (cm2/s); sR is the insertion rate or receptor R (mol/cm2/s); kint is the receptor or complex internalization rate (s−1); kon and koff are the rate constants of complex association (M−1s−1) and dissociation (s−1).
| (3) |
| (4) |
Boundary conditions were given by Eqn. 5. qV is the endothelial secretion rate of VEGF (mol/cm2/s).
| (5) |
The initial conditions describing exogenous ligand administration and total receptor densities were specified by Eqn. 6.
| (6) |
The predicted final concentration of cell-surface ligated VEGF·VEGFR2 complexes served as a surrogate marker for achieved angiogenic response.
Representative values used for the model parameters were based on experimental literature as detailed in (Mac Gabhann and Popel, 2004). Numerical solution of the coupled nonlinear differential equations (Eqns. 3–6) was achieved using an iterative implicit finite-difference scheme. Sample results are shown in Fig. 2B. Crucially, the simulations predicted that PlGF addition only increased VEGF·VEGFR2 complex formation up to 5% at the peak percentile change between PlGF+ and PlGF− cases; i.e., the “ligand shifting effect” is expected to be minimal. On the other hand, the transient increase in total ligated VEGFR1 complexes was as high as 43%, during which, the magnitude increase in PlGF·VEGFR1 complexes exceeded that of the decrease in VEGF·VEGFR1 complexes. This would suggest that “VEGFR1 signaling” plays a more prominent role in the observed PlGF-VEGF synergy: that PlGF critically alters the VEGFR1 signaling profile in both the absolute quantity of signaling VEGFR1 complexes and the signaling quality of VEGFR1 complexes (elevated pro-angiogenic PlGF·VEGFR1 signaling and reduced modulatory VEGF·VEGFR1 signaling). Experimental support for these computational predictions have been found (Autiero et al., 2003), as intermolecular crosstalk was reported to occur downstream of PlGF-VEGFR1 binding, leading to the transphosphorylation of VEGFR2 and amplification of pro-angiogenic VEGF·VEGFR2 signaling.
2.3 Case Study: Mechanism of NRP1-VEGFR2 coupling via VEGF165: effect on VEGF isoform-specific receptor binding
Our second case study investigated another molecular player in the systems biology of VEGF, neuropilin-1 (NRP1). As illustrated in Fig. 2C, NRP1-binding affinity is conferred to the higher molecular-weight isoforms of VEGF such as VEGF165 predominantly through transcription of exon 7; the VEGF121 isoform, which lacks exon 7, is generally considered to have negligible affinity for NRP1. Because the NRP1-binding domains of VEGF165 do not overlap with its VEGFR-binding domains, VEGF165 can act as a bridge in the formation of a ternary complex: VEGF165·VEGFR2·NRP1. A reduced interaction network model involving VEGF121, VEGF165, VEGFR2 and NRP1 (Fig. 2C) was thus constructed to quantify the role of VEGF165-bridged VEGFR2-NRP1 coupling in generating VEGF isoform-specific angiogenic responses. Details and full references can be found in (Mac Gabhann and Popel, 2005).
Geometrically, the experimental setup again involved a confluent layer of endothelial cells on the bottom of a cell culture well, as in Fig. 2A. As before, the initial value problem in one spatial dimension was formulated as a system of coupled diffusion and reaction equations (Eqns. 7 and 8–13 respectively). A new parameter, kC, represents the coupling rate between VEGFR2 and NRP1 via VEGF165.
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
Boundary conditions were given by Eqn. system 14.
| (14) |
The predicted final VEGF·VEGFR2 concentration again served as a marker for the strength of pro-angiogenic signal transduction.
The experimental and theoretical derivations of model parameter values are detailed in (Mac Gabhann and Popel, 2005). Numerical solution of the coupled nonlinear differential equations (Eqns. 7–14) was achieved using an iterative implicit finite-difference scheme. Sample results are shown in Fig. 2D. The in silico modeling of stepwise reaction kinetics (Fig. 2C) allowed the prediction of differential anti-angiogenic efficacies from therapeutic interference of two distinct aspects of NRP1 function: VEGF binding and VEGFR2 coupling. Simulated blockade of VEGF165 binding to NRP1 (blocking reactions ‘Rx1’ and ‘Rx2’ in Fig. 2C) resulted in the convergence of VEGF165 response to that of VEGF121 in terms of VEGFR2 activation; however, simulated blockade of NRP1-VEGFR2 coupling (blocking reactions ‘Rx1’ and ‘Rx3’ in Fig. 2C) converted NRP1 into a VEGF165 sink (through intact reaction ‘R2’ in Fig. 2C), further reducing VEGF165 response to below that of VEGF121 (Fig. 2D).
3. Meso-scale single-tissue 3D models: simulation of in vivo tissue regions
The study of VEGF binding to receptors on cells in vitro, and the validation of the VEGF kinetic interaction network between multiple ligands and multiple receptors, leads us to ask the question: how does this network behave in vivo? In sections 4 and 5 we will discuss the transport of VEGF between tissues and around the body, but here we will focus first on the behavior of VEGF in a local volume of tissue. This multicellular milieu requires significant additions to our model in order to accurately simulate the local transport of VEGF, including diffusion of VEGF ligands over significant distances, extracellular matrix sequestration and variable production rates of VEGF throughout the tissue. We place these all in an anatomically-based 2D or 3D multicellular tissue geometry.
The models can predict the creation of interstitial VEGF gradients due to the non-uniform nature of the tissue anatomy. This is of particular interest because VEGF is believed to be a chemotactic guiding agent for blood vessels, but also because local variability in VEGF concentration can lead to local variation in VEGF receptor ligation and signaling, allowing for focal activation of endothelial cells. The model framework can be adapted to most tissues; here we present a case with parameters specifically selected to represent a skeletal muscle experiencing ischemia (specifically, rat extensor digitorum longus, or EDL, for a rodent model of hindlimb artery ligation), and we describe how to computationally test several therapeutic interventions including gene therapy and exercise.
3.1 Mathematical Framework for Tissue Architecture, Blood Flow & Tissue Oxygenation
2D and 3D tissue geometry based on microanatomy
A cross-section (for 2D) or a volume of tissue (for 3D) is reconstructed from histological and other microanatomical information (Figure 3A–C). The major relevant features of the tissue are the blood vessels, the parenchymal cells (from here on, we will assume these are skeletal myocytes, i.e. long multinucleated cells) and the interstitial space between them. From a computational modeling point of view, the tissue comprised of volumes and surfaces, defined as those portions of the tissue where molecules can move in all directions (volumes) and those portions where the movement of molecules is restricted to a plane (e.g. receptors inserted in cell membrane can move only laterally). There are three major volumes of the tissue for our purposes: the vascular space (i.e. inside the blood vessels, determined by the density of blood vessels and their diameters); the intracellular space (whether inside of parenchymal cells or endothelial cells); and the interstitial space between cells, which is itself divided into three volumes (each of which is not contiguous), based on the density of the fibrous matrix present: the extracellular matrix, and basement membrane regions surrounding the endothelial cells and the myocytes.
Figure 3. Mesoscale modeling.
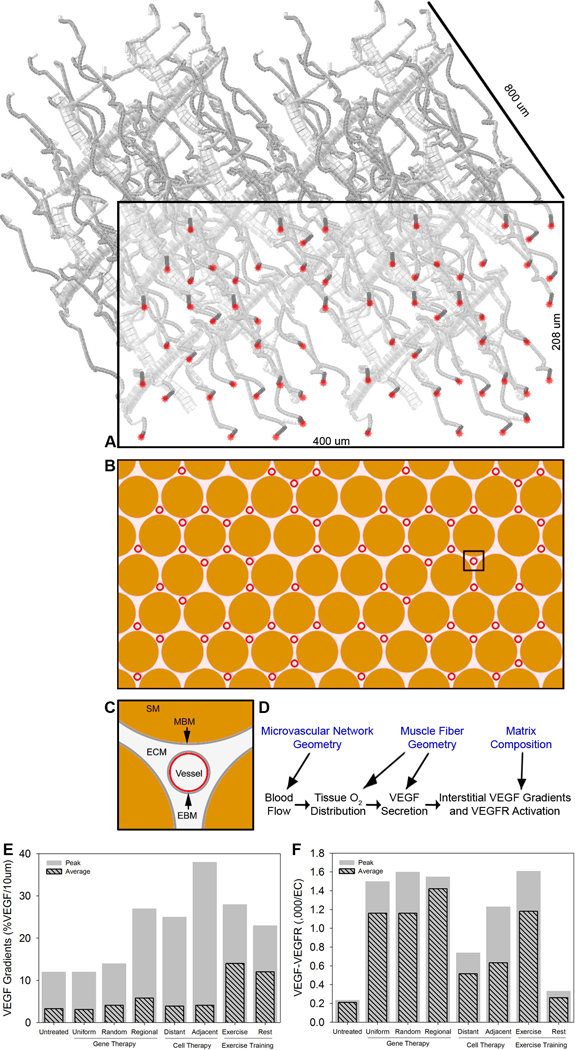
A, Schematic of generated microvascular network of capillaries, arterioles and venules, consistent with histological and other measurements of rat skeletal muscle. B and C, cross-section of muscle (indicated by box in A), showing the capillaries (red) and the muscle fibers or myocytes (brown; “SM”). Detail in C of myocytes, endothelial cells, and the extracellular matrix (ECM) and basement membranes (gray; “MBM” and “EBM”) between. For three-dimensional simulations, the full volume of tissue is used; for two-dimensional simulations the indicated cross-section is used. D, Schematic illustrating how the tissue microanatomy (top row) impacts on the calculation of blood flow, oxygen distribution, VEGF distribution and VEGFR binding. E, Local VEGF gradients within ligated EDL following treatment. The maximum within the tissue and the average across the tissue are reported. (F), VEGFR2 activation on vessels of ligated EDL following treatment. A–D adapted from (Mac Gabhann et al., 2007a). E–F based on results previously published in (Mac Gabhann et al., 2007a).
There are two major surfaces; again, these are not contiguous. First, the combined cell surfaces of the skeletal myocytes, which are assumed to be cylindrical (diameter 37.5 µm, consistent with rat histology), and arranged in a regular hexagonal grid formation, accounting for almost 80% of total tissue cross-sectional area. VEGF is secreted from the myocytes’ surfaces. Second, the surface of the endothelial cells that make up the blood vessels, specifically, the abluminal surface (the luminal surface faces the blood stream and we neglect it for now). Again, the blood vessels are assumed to be cylindrical, and although most (but not all) are parallel to the muscle fibers, they do not occupy every possible position between fibers, but instead have a stochastic, nonuniform arrangement (based on experimentally measured capillary-to-fiber ratios, capillary-to-fiber distances and histology) occupying 2.5% of total tissue volume (leaving ~18% as interstitial space). On this endothelial surface, VEGF receptors are expressed. Thus, VEGF must diffuse from the myocyte surface where it is secreted, through basement membranes and extracellular matrix, to the endothelial surface where it ligates its cognate receptors.
To model tissues at the meso-scale, we use the above microanatomical information as an input to a set of integrated models of blood flow, oxygen transport, and VEGF transport (Fig. 3D).
Volumes: Blood Flow
Blood flow and hematocrit value calculations are based on Pries et al.’s two-phase continuum model (Pries and Secomb, 2005), and reduces to a system of nonlinear algebraic equations (two per vessel) that are solved iteratively (Ji et al., 2006). The Fahraeus-Lindqvist effect and non-uniform hematocrit distribution at vascular bifurcations are included in the blood flow model. Higher blood flow rates are used for exercising conditions, to represent the increased perfusion (and enhanced oxygen delivery) to exercising muscles. In addition, exercise-trained rats have higher average capillary blood velocity.
Volumes: Diffusion and consumption of Oxygen
Oxygen transport in the tissue in detailed in (Ji et al., 2006) and (Goldman and Popel, 2000). Oxygen arrives in the tissue via the blood vessel network, and the partial pressure of oxygen in the vessels, Pb, is described by
| (15) |
where vb is mean blood velocity; αb is oxygen solubility in blood; HD is the discharge hematocrit; are the oxygen binding capacity and oxygen saturation of the red blood cell; ξ is the longitudinal position in the vessel; R is vessel radius; Jwall is the oxygen flux across the vessel walls (i.e. into the tissue). Oxygen diffuses across the endothelial cells, and freely throughout the tissue (both interstitial and intracellular). Within the cells, it may also be consumed by binding to Myoglobin (Mb). The local partial pressure of oxygen in the tissue, P, is described by
| (16) |
where DO2 and DMb are the diffusivities of oxygen and myoglobin; α is the oxygen solubility in tissue; CMbbind and SMb are the oxygen binding capacity and oxygen saturation of myoglobin; and M(P) represents Michaelis-Menten kinetic consumption of oxygen.
Volumes: Diffusion of VEGF and sequestration by ECM
The VEGF ligands, VEGF120 (rodent form of VEGF121) and VEGF164 (rodent form of VEGF165), can both diffuse through the interstitium following secretion, however the longer isoform also binds to glycoproteins in the extracellular matrix, becoming reversibly sequestered. The equations are thus identical to those of section 2, with the addition of binding and unbinding terms:
| (17) |
where Ci and Cj are concentrations of two interstitial molecules, i and j. In the rat EDL model, Ci=[V120] or [V164]; Cj=[GAG]. Concentration of proteins in the thin endothelial or myocyte basement membranes is given by an equation of the form:
| (18) |
where si is the secretion rate from the cell (typically from myocytes); Rm and Rim are the concentrations of receptor m and of the i-m complex on the cell surface (typically on endothelial cells); Jout is the Fickian diffusive flux from BM to ECM of VEGF; and dBM is the basement membrane thickness.
Surfaces: Receptor-ligand interactions
The ligand-receptor interactions that take place are precisely those that were outlined in section 2, and that will be used in Sections 4 and 5: VEGF120 and VEGF164 bind to VEGFR1 and VEGFR2, while only the longer isoform binds Neuropilin-1 and extracellular matrix. The general form of the receptor and receptor complex equations is therefore:
| (19) |
where sm and kint,m are the membrane insertion rate and internalization rate of receptor m; kcouple,m,n and kdissoc,mn are the kinetic rate of binding and unbinding of two surface receptors m and n to each other. Note in particular that the concentration of the ligand (Ci) in each case is the concentration in the basement membrane region closest to the receptor. Thus, the receptor occupancy varies from cell to cell across the capillary network. Examples of specific individual equations can be found in section 2.
Surfaces: VEGF production/secretion rates
The production and secretion of VEGF has been observed to be inducible by hypoxia (Forsythe et al., 1996; Qutub and Popel, 2006). Here, we use an empirical relationship (Mac Gabhann et al., 2007b) for the increase in the baseline secretion rate of VEGF (S0) based on the observed upregulation of VEGF mRNA and protein during hypoxia in cells and tissues (Jiang et al., 1996; Tang et al., 2004):
| (20) |
What is not included in these models?
Intracellular VEGF is not included in these simulations; that includes both post-internalization VEGF and pre-secretion VEGF. In addition, we neglect the intravasation of VEGF into the bloodstream, either by endothelial cell secretion or through paracellular routes, e.g. permeability. Lymphatic transport of VEGF is also neglected. These additional transport routes could be accommodated in the above model structure with the addition of new surfaces or terms. Although endothelial VEGF production and parenchymal VEGFR expression have been observed in recent years (Lee et al., 2007; Bogaert et al., 2009), these are not included as part of these simulations; there is no technical obstacle to doing so.
Relationship to single-compartment models
It is important to note that the spatial averages of VEGF concentrations at the endothelial cell surface and of VEGFR activation in the meso-scale models match well with the values in single-compartment models (section 4) that do not include diffusion or VEGF gradients. Thus, it may be possible to calculate the average receptor activation using less computationally intensive compartment models, and use the meso-scale models to estimate the spatial gradients.
3.2 Case Study: Pro-angiogenic VEGF gene therapy for muscle ischemia
In order to improve the perfusion and healing of ischemic muscle tissue with impaired angiogenic response, several therapies have been suggested, typically involving the delivery of VEGF (one or more isoforms) to the muscle.
The first of these, gene therapy, increases the VEGF secretion by adding additional VEGF-encoding genes to the cells that are transfected. By transfecting multiple copies, or by judicious choice of VEGF promoters and enhancers in the new construct, significant increases in VEGF secretion can be obtained. We have modeled both uniform upregulation of VEGF (increasing VEGF secretion at every myocyte surface point in the model) and stochastic upregulation, in which each cell has a randomly increased VEGF production within a certain range (using the myonuclear density, we know the size of the myocyte surface that is under the control of each nucleus; thus, we can assign a random number to each region, that stays constant through the simulation) (Mac Gabhann et al., 2007a). These increases in VEGF production result in increased VEGFR2 activation, however the VEGF gradients are not significantly increased (Fig. 3E,F); in this case blood vessels might be induced to sprout, but have no directional cues. Further simulations restricting the VEGF transfection to a specific region of the muscle demonstrates increased VEGFR2 activation coupled with very high VEGF gradients towards the transfected tissue, but only in a narrow region between transfected and nontransfected tissue (Mac Gabhann et al., 2007a). This suggests that VEGF gene delivery needs to be effectively localized with a high degree of spatial accuracy to allow the gradients of VEGF to bring the new vessels to the affected volume.
3.3 Case Study: Pro-angiogenic VEGF cell-based therapy for muscle ischemia
Another route to bringing more VEGF to the tissue, and one which may allow for more spatial specificity, is the delivery of VEGF-overexpressing cells, e.g. myoblasts that will effectively integrated into the existing muscle and produce excess VEGF locally. To simulate this, we select specific myocytes in the model to overexpress VEGF, and distribute these distantly or close together (Mac Gabhann et al., 2006; Mac Gabhann et al., 2007a). That is, since the secretion rate of VEGF can have a different value for every spatial location on the myocytes surface in our model, we can upregulate VEGF in a specific subset of these cells.
For this therapy, we observe in the simulations both increased VEGFR2 binding and increased VEGF gradients (Fig. 3E,F), but only within approximately one to two myocyte diameters from the new VEGF overexpressing cells (Mac Gabhann et al., 2006; Mac Gabhann et al., 2007a). In addition, cells close together synergize while distant ones do not. In this way, we can see that a small number of cells, or cells distributed too broadly, would have a low probability of attracting perfusion from a neighboring region; however, a large mass of cells, at the right location, could serve as a local chemoattractant.
The results described in sections 3.2 and 3.3, for therapies reliant on VEGF upregulation alone, mirror the outcome of the several clinical trials of VEGF isoforms in humans for coronary artery disease (CAD) or peripheral artery disease (PAD); these trials have not had the success that was expected of them. Instead, the standard of care for PAD continues to be exercise, and it is this therapy that we consider next.
3.4 Case Study: Pro-angiogenic exercise therapy for muscle ischemia
Exercise training in rats has been shown to not only restore the ability of hypoxic, ischemic tissue to upregulate VEGF following injury, but also increases the expression levels of the VEGF receptors (Lloyd et al., 2003). Thus, we used our model to simulate the exercise-dependent upregulation of both the ligands and the receptors, using experimentally measured increases (Ji et al., 2007; Mac Gabhann et al., 2007a; Mac Gabhann et al., 2007b).
In this case, we increase the secretion rate of VEGF isoforms from each point on the myocyte surface, during exercise; in addition, we increase the insertion rate of the VEGF receptors at every point on the endothelial cell surface at all times (as a result of exercise training). The results of these simulations are quite different from those before: first, during exercise, both the VEGFR2 activation and the VEGF gradients are increased, not just locally but across the upregulated tissue (Ji et al., 2007; Mac Gabhann et al., 2007a); second, during rest periods, while VEGF upregulation ceases and the occupancy of VEGFR2 returns to lower levels, the high VEGF gradients are maintained (Fig. 3E,F). This suggests that the activation step for attracting new blood vessels may be during a smaller window of time, while the guidance of the new vessel to its destination can take place continuously.
This observation that our current best strategy for PAD, exercise, increases both ligand expression and receptor activation, leaves us with the possibility of developing combined ligand-receptor therapy (especially for those who cannot exercise).
4. Single-tissue compartmental models: simulation of in vivo tissue
From section 3, we saw that the investigative use of VEGF models for therapeutic applications require larger spatial scale modeling of in vivo growth factor transport and effects at the tissue level. However, the full spatial resolution afforded by 3D modeling becomes computationally intensive when the volume of interest grows from tissue regions to whole tissues and organs. Here we demonstrate the strategy of compartmental modeling – where tissue fluid volumes (e.g., interstitial fluid volume) are approximated as well-mixed compartments of uniform protein concentrations, based on the assumption that diffusion occurs on faster timescales than that of molecular binding kinetics (Damkohler number <1 (Mac Gabhann and Popel, 2006)). While compartmental models cannot predict diffusion-limited VEGF gradients, they allow the prediction of average interstitial VEGF levels and average cell-surface VEGF-VEGFR binding within single tissues (section 4) or in multiple interconnected tissues (section 5).
4.1 Mathematical Framework for Tissue Porosity and Available Volume Fractions
In converting interstitial volumetric protein concentrations to their appropriate per-tissue-volume basis, it is useful to consider extravascular regions as porous media (Truskey et al., 2004) and to use the following standard definitions. ε is the porosity, defined as the fractional void space within the total tissue volume. Φ is the partition coefficient, representing the fractional interstitial fluid volume that is available or accessible to macromolecules, i.e., excluding all isolated or impenetrable pores. Together, they define the available volume fraction, KAV, which is used to convert interstitial concentrations from per-interstitial-fluid-volume (M) to per-tissue-volume (mol/(cm3 tissue)) basis:
| (21) |
Endothelial cell-surface receptor and complex densities also have to be converted from their per-surface-area units (moles per cm2 cell surface area) to per-tissue-volume (moles per cm3 tissue) basis for consistency within the mathematical equations given below. For this purpose, other geometrical attributes (e.g., microvessel density and diameters, surface area per endothelial cell, and endothelial surface area per tissue volume) have to be quantified to reflect the particular tissue architecture modeled, as documented in (Mac Gabhann and Popel, 2006) for breast tumor tissue geometry.
4.2 Case Study: Pharmacodynamic mechanism and tumor microenvironment affect efficacy of anti-NRP1 therapy in cancer
In this example of a single-tissue compartment model, three strategies of inhibiting different aspects of NRP1 functionality were simulated to predict their relative in vivo anti-angiogenic efficacy as cancer treatments in breast tumor tissues. The first strategy of inhibiting NRP1 expression (Fig. 4A), which could be achieved clinically through siRNA methods, was simulated by reducing the NRP1 insertion rate, sN. The second strategy of impeding VEGF binding to NRP1 (Fig. 4B), which could be implemented clinically through a PlGF fragment (P2Δ) that binds NRP1 exclusively, was modeled with additional equations representing the competition between VEGF and P2Δ for NRP1 (Eqn. 22) and corresponding modifications to the original NRP1 equation. The third strategy of blocking NRP1-VEGFR coupling while allowing NRP1-VEGF binding (Fig. 4C), which had been done experimentally using a NRP1 antibody, was modeled with additional equations for the antibody interactions (Eqn. 23) and corresponding modifications to the original VEGF165, NRP1, and VEGF165·NRP1 equations. Detailed references and full equations can be found in (Mac Gabhann and Popel, 2006).
| (22) |
| (23) |
Figure 4. Single-tissue compartmental model.

Simulation of three targeted strategies of blocking NRP1 functionality: (A) NRP1 expression; (B) VEGF-NRP1 binding; (C) VEGFR-NRP1 coupling. D, Inhibition of VEGF-VEGFR2 signaling by increasing doses of each of the three strategies outlined in A–C. Peak and average over the first 48 hours following therapeutic administration. Sample results (D) based on data previously published in (Mac Gabhann and Popel, 2006).
Numerical solution of the full system of coupled nonlinear ordinary differential equations was achieved using a Runge-Kutta integration scheme (Mac Gabhann and Popel, 2006). Sample results are shown in Fig. 4D. The results predict that the third strategy of blocking NRP1-VEGFR coupling will induce the most effective and sustained inhibition of VEGFR2 ligation and signaling, with the differential advantage being most discernible in tumor types where endothelial expression of VEGFR1 is low. This case study demonstrates that model incorporation of molecularly detailed interaction networks allow the sophisticated optimization of therapeutic strategies, from the fine-tuning of therapeutic agent dosing to the customization of targeted molecular mechanisms according to the diseased tissue type with its characteristic receptor expression levels.
5. Multi-tissue compartmental models: simulation of whole body
In this section we introduce models that compartmentalize the whole body into multiple tissue compartments. The two examples given below each have three compartments (Fig. 5): the blood; the tissue of interest (breast tumor tissue in section 5.2 and calf muscle in section 5.3); and the rest of the body, which we call the “normal compartment”. Future model extensions can further subdivide the normal compartment into individual organ compartments. In this paper we demonstrate that the modeling of two major inter-compartment transport processes – vascular permeability and lymphatic drainage – is essential in predicting whole-body pharmacokinetics of macromolecules.
Figure 5. Whole-body multi-tissue compartmental model.

Schematic of experimental setup for investigation of whole-body pharmacokinetics of sVEGFR1 and anti-VEGF, as well as their effects on VEGF signaling in the calf muscle and breast cancer (“tissues of interest”) respectively. Endogenous sources of sVEGFR1 include endothelial production. Intravascular infusion of exogenous sVEGFR1 and anti-VEGF were also simulated. Figure adapted from (Wu et al., 2009b).
5.1 Mathematical Framework of Inter-tissue Transport
Macromolecular Vascular Permeability
Unlike small molecules that can easily traverse microvessel endothelia through interendothelial cleft junctions (Fu and Shen, 2003), the transendothelial permeation of macromolecules such as peptide growth factors occurs through caveolar structures and vesiculo-vacuolar organelles at much slower rates that are dependent on protein size and VEGF-induction (VEGF is also known as VPF, vascular permeability factor) (Garlick and Renkin, 1970; Feng et al., 2002; Fu and Shen, 2003). The equations relevant to protein transport between the available fluid volumes of the tissue compartment and the blood compartment via vascular permeability are:
| (24) |
where represents the microvascular permeability rate for the protein of interest going from the tissue (T) to the blood (B) compartment (cm/s) across STB (cm2), the total endothelial surface area (i.e. the tissue-blood interface).
Microvascular permeability is modeled as a passive bidirectional transport process, represented on Figure 5 by a double-arrow, with identical intravasation and extravasation rates, . The in vivo value of kp for a given protein is rarely found in the literature. Instead, we extrapolate from calibration curves (Garlick and Renkin, 1970) correlating permeability rates with protein size (Stokes-Einstein radius of the molecule). For instance, the calculated Stokes-Einstein radius (ae) of the 45kDa globular VEGF protein is 30.2 Å according to 0.483 × (Molecular weight in Da)0.386 as given in (Venturoli and Rippe, 2005). Our extrapolation methods yield a baseline permeability rate of 4.3 × 10−8 cm/s for VEGF (Stefanini et al., 2008). The microvascular permeability for VEGF in tumor tissue (for use in modeling breast tumor as the tissue of interest in section 5.2) can be estimated from corresponding values for similar-sized molecules, e.g., ovalbumin (45 kDa; ae = 30.8 Å) was measured to permeate at about 5.77 × 10−7 cm/s (Yuan et al., 1995). Details can be found in (Stefanini et al., 2008).
Lymphatic Drainage
Lymphatic drainage is a major route by which interstitial proteins are transported to the blood because size-dependent transendothelial permeability restricts their intravasation into blood capillaries. In contrast, there is no macromolecular impedance in the filling of the initial lymphatics, hence the protein concentrations drained through the lymphatics are assumed to be continuous with interstitial concentrations at the lymphatic entrance. Mathematically, we describe unidirectional lymphatic drainage as:
| (25) |
where kL is the lymphatic drainage rate in cm3/s. Detailed derivation and parameterization can be found in (Wu et al., 2009b).
5.2 Case Study: Pharmacokinetics of anti-VEGF therapy in cancer
The pharmacokinetics of anti-VEGF therapy can be studied via whole-body compartmental modeling based on the detailed biochemical reactions between VEGF ligands and their receptors described above. Specifically, we can simulate and study the VEGF isoform specificity of anti-VEGF agents, ligand-agent binding configurations (e.g., whether such binding is monomeric or multimeric), agent biodistribution (if the anti-VEGF agent is confined to one or several compartments), as well as various therapeutic regimen designs (varying dosage, frequency of administration, the site of injection).
This example models bevacizumab, a humanized monoclonal antibody to VEGF, the characteristics and properties of which have been reported (150 kDa; Kd of 1.8 nM in (Presta et al., 1997); half-life = 21 days in (Gordon et al., 2001)). The diseased tissue of interest is a tumor. In the absence of the anti-VEGF agent, a total of 40 ordinary differential equations describes the compartmental system (19 ordinary differential equations (ODEs) for each tissue; 2 ODEs for the blood compartment). When the anti-VEGF agent is added and confined to the blood compartment (non-extravasating agent), 3 more equations are added to the model (blood compartment), representing the chemical interactions of the anti-VEGF with VEGF121 and VEGF165, as well as free anti-VEGF.
| (26) |
where KAV,blood,i is the available volume fraction of the molecule i (A=anti-VEGF, V=VEGF, VA=complex VEGF/anti-VEGF). Note that UAV = KAV U. The first term represents the concentration of anti-VEGF drug injected into the patient bloodstream. The second term represents the clearance of the anti-VEGF agent by the organs (for example, kidneys or liver). The equations related to the complex form of the anti-VEGF drug are of the form:
| (27) |
Figure 6 illustrates the transient dynamics resulting from the intravenous injection of the VEGF-antibody; in these simulations a tumor of 2 cm diameter is considered. In the single-dose treatment (10 mg/kg), intravenous injection leads to a rapid decrease in the free VEGF concentration (Fig. 6A). That level returns to baseline after about 3 to 4 weeks. For daily smaller doses of treatment or “metronomic” treatment (1 mg/kg for 10 days), a new lower pseudo-steady state for the plasma VEGF level emerges during the duration of treatment (Fig. 6B). Following treatment cessation (10 days), the plasma VEGF level returns to that before treatment after about 3 weeks. The metronomic injection also delays the peak of maximum formation of VEGF-antiVEGF complex as compared to a single-dose treatment (Figure 6C–D).
Figure 6. Compartmental model of whole-body anti-VEGF pharmacokinetics.

Comparison between single-dose (A) and metronomic (B) intravenously delivered anti-VEGF treatment (without extravasation of the anti-VEGF molecule). Each dose (intravenous infusion) takes place over 90 minutes. A,C,E. Single dose of 10 mg/kg, B,D,F. 1 mg/kg daily for 10 days.
Equations (26–27) describe an intravenous injection of anti-VEGF antibodies that would be confined to the plasma. If the anti-VEGF agent is injected intravenously and can extravasate, terms of the form of Eqn. (24) are added to equations (26–27) and additional equations corresponding to the free and bound antibody concentrations are needed for each tissue compartment in the systems of ODEs. Interestingly, the addition of extravasation to the model drastically changes the form of the response. Transiently after injection, the free VEGF level in plasma drops drastically (data not shown). This is due to the binding of the antibody to the free VEGF present in the plasma. Following this drop there is a several-fold increase of free VEGF concentration in plasma. The apparent “rebound” effect is due to the amount of drug delivered and its extravasation. Briefly, while some antibodies bind to the free VEGF in plasma, another portion extravasates and binds to the free VEGF present in the available interstitial fluid volume in the tissues (healthy tissue and breast tumor). Although some of the formed complexes subsequently dissociate within the same tissue, significant quantities are brought into the bloodstream (via microvascular permeability and lymphatic drainage) where the complex dissociates, leading to more free VEGF in the blood compartment. Such a counter-intuitive increase in serum VEGF following intravenous administration of anti-VEGF agents has been observed in experiments (Gordon et al., 2001; Willett et al., 2005; Segerstrom et al., 2006) and our model is, to our knowledge, the first to explain this phenomenon by an intrinsic mechanism of inter-tissue transport.
This example illustrates how computational models can provide useful insights that are not easily accessible by in vitro or in vivo experiments. This model can also be extended to examine the effects of drug treatment via alternate routes of anti-VEGF administration (e.g. intramuscular injection).
5.3 Case Study: Mechanism of sVEGFR1 as a ligand trap
In this last example of a multi-tissue compartmental model, we investigated the molecular mechanisms by which sVEGFR1, a truncated soluble variant of the endothelial cell-surface VEGFR1, inhibits VEGF signaling. The two prevailing postulated mechanisms are: direct VEGF ligand sequestration, reducing the VEGF available for VEGFR activation (Fig. 7A middle); and heterodimerization with cell-surface VEGFR monomers, rendering the receptor dimer non-functional as trans-phosphorylation of paired intracellular domains of full-length VEGFRs is necessary for activating signal transduction (Fig. 7A bottom). The model as described in detail in (Wu et al., 2009a) simulated the first mechanism, to assess the anti-angiogenic potential of sVEGFR1’s ligand trapping capacity alone.
Figure 7. Compartmental model of whole-body sVEGFR1 transport.

(A) sVEGFR1 has been postulated to have antagonistic effects on VEGF signaling complex formation (top row) through competitive binding of VEGF ligands (middle row) and dominant-negative heterodimerization with endothelial cell-surface VEGF receptors (bottom row). (B) Sample simulations of the ligand-trapping effects of intravascularly administered exogenous sVEGFR1 based on results previously published in (Wu et al., 2009a).
sVEGFR1 in its monomeric (110 kDa) and dimeric (220 kDa) forms are about 2–5 times larger than VEGF; thus free sVEGFR1 and the sVEGFR1·VEGF complex have lower vascular permeability rates than VEGF, while sharing the same lymphatic drainage rates as VEGF. Full mathematical equations describing these transport properties, along with the sVEGFR1-VEGF binding and sVEGFR1-NRP1 coupling interactions, can be found in (Wu et al., 2009a). Sample equations for the concentrations of free sVEGFR1 and sVEGFR1·VEGF165 in tissue j are given here:
| (28) |
| (29) |
In this model, calf muscle tissue was chosen as the “tissue of interest” compartment (Fig. 5), in order to investigate the effects of endogenous sVEGFR1 produced from the calf muscle on local (calf compartment) and global (normal compartment) VEGF signaling complex formation. Such predictions were expected to provide insight on whether pathological upregulation in sVEGFR1 expression in the calf may contribute to the dampened VEGF response observed in ischemic calf muscles in peripheral arterial disease. Therapeutic intravascular (IV) delivery of exogenous sVEGFR1 was also simulated to assess its efficacy in lowering systemic levels of VEGF (Wu et al., 2009a).
While, intuitively, intravascular sVEGFR1·VEGF complex formation following simulated IV infusion of sVEGFR1 would be expected to lead to a sustained reduction of plasma free VEGF, in fact sample results in Fig. 7B show that the permeability rates and lymphatic drainage rates of free sVEGFR1 and sVEGFR1·VEGF are predicted to critically determine whether this reduction will take place or not. In other words, these simulations suggest that prior to the clinical translation of administering exogenous sVEGFR1 to lower systemic VEGF levels in anti-angiogenic therapy, extensive experimental research is needed to exclude the computationally predicted possibilities where sVEGFR1 delivery counter-intuitively elevates plasma free VEGF. As in section 5.2, this computational study demonstrates that the inter-tissue transport properties of proteins significantly affect their whole-body pharmacokinetic effects.
Conclusions
In this paper, we summarized several computational models for investigating different spatial and temporal aspects of VEGF systems biology. In section 2, we described molecular network models that simulated in vitro endothelial cell-surface interaction experiments to investigate the roles of the PlGF ligand and NRP1 co-receptor within the VEGF system. In section 3, we presented meso-scale models for investigating the effects of in vivo tissue architecture on VEGF ligand and receptor interactions, and for predicting the intramuscular response and relative therapeutic efficacy of various modalities of pro-angiogenic therapies (gene vs. cell vs. exercise therapy) for ischemic muscle diseases. In sections 4 and 5, molecular-detailed compartmental modeling was introduced as a method for efficient prediction of average molecular concentrations within tissue subcompartments (e.g., interstitial or plasma VEGF concentrations; intramuscular cell-surface density of signaling complexes) and investigation of inter-compartment transport processes (e.g., microvascular permeability and lymphatic drainage). We described several compartmental model studies predicting in vivo effects of intra-tissue trafficking and whole-body pharmacokinetics on angiogenic response to treatments using NRP1, anti-VEGF and sVEGFR1 as therapeutic targets/agents. While the VEGF system models presented in this paper were limited to the representation of two ligand isoforms (VEGF121 and VEGF165) and three receptors (VEGFR1, VEGFR2, NRP1), these model frameworks can be readily extended to include other VEGF ligand isoforms and receptors (e.g.,VEGFR3, NRP2). Furthermore, similar computational modeling techniques are applicable and have contributed to the study of other growth factor systems, including that of FGF (Forsten et al., 2000; Filion and Popel, 2004; Filion and Popel, 2005) and EGF (Wiley et al., 2003).
Acknowledgements
Research described in this paper was supported by NIH grants R01 HL079653, R33 HL0877351, and R01 CA138264.
Contributor Information
Florence T.H. Wu, Email: florence.wu@jhu.edu.
Marianne O. Stefanini, Email: stefanini@jhmi.edu.
Feilim Mac Gabhann, Email: feilim@jhu.edu.
Aleksander S. Popel, Email: apopel@jhu.edu.
References
- Andrae J, Gallini R, Betsholtz C. Role of Platelet-Derived Growth Factors in Physiology and Medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin HG, Koh GY, Thurston G, Alitalo K. Control of Vascular Morphogenesis and Homeostasis through the Angiopoietin-Tie System. Nat Rev Mol Cell Biol. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, Kroll J, Plaisance S, De Mol M, Bono F, Kliche S, Fellbrich G, Ballmer-Hofer K, Maglione D, Mayr-Beyrle U, Dewerchin M, Dombrowski S, Stanimirovic D, Van Hummelen P, Dehio C, Hicklin DJ, Persico G, Herbert JM, Communi D, Shibuya M, Collen D, Conway EM, Carmeliet P. Role of PIGF in the Intra- and Intermolecular Cross Talk between the VEGF Receptors Flt1 and Flk1. Nat Med. 2003;9:936–943. doi: 10.1038/nm884. [DOI] [PubMed] [Google Scholar]
- Bao P, Kodra A, Tomic-Canic M, Golinko MS, Ehrlich HP, Brem H. The Role of Vascular Endothelial Growth Factor in Wound Healing. J Surg Res. 2009;153:347–358. doi: 10.1016/j.jss.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenken A, Mohammadi M. The FGF Family: Biology, Pathophysiology and Therapy. Nat Rev Drug Discov. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert E, Van Damme P, Poesen K, Dhondt J, Hersmus N, Kiraly D, Scheveneels W, Robberecht W, Van Den Bosch L. VEGF Protects Motor Neurons Against Excitotoxicity by Upregulation of GluR2. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2008.12.007. [Epub ahead of print] Pubmed ID: 19185395. [DOI] [PubMed] [Google Scholar]
- Brown MD, Hudlicka O. Modulation of Physiological Angiogenesis in Skeletal Muscle by Mechanical Forces: Involvement of VEGF and Metalloproteinases. Angiogenesis. 2003;6:1–14. doi: 10.1023/a:1025809808697. [DOI] [PubMed] [Google Scholar]
- Cao Y. Positive and Negative Modulation of Angiogenesis by VEGFR1 Ligands. Sci Signal. 2009;2:re1. doi: 10.1126/scisignal.259re1. [DOI] [PubMed] [Google Scholar]
- Collinson DJ, Donnelly R. Therapeutic Angiogenesis in Peripheral Arterial Disease: Can Biotechnology Produce an Effective Collateral Circulation? European Journal of Vascular and Endovascular Surgery. 2004;28:9–23. doi: 10.1016/j.ejvs.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Ebos JM, Bocci G, Man S, Thorpe PE, Hicklin DJ, Zhou D, Jia X, Kerbel RS. A Naturally Occurring Soluble Form of Vascular Endothelial Growth Factor Receptor 2 Detected in Mouse and Human Plasma. Mol Cancer Res. 2004;2:315–326. [PubMed] [Google Scholar]
- Feng D, Nagy JA, Dvorak HF, Dvorak AM. Ultrastructural Studies Define Soluble Macromolecular, Particulate, and Cellular Transendothelial Cell Pathways in Venules, Lymphatic Vessels, and Tumor-Associated Microvessels in Man and Animals. Microsc Res Tech. 2002;57:289–326. doi: 10.1002/jemt.10087. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T. The Biology of Vascular Endothelial Growth Factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Filion RJ, Popel AS. Intracoronary Administration of FGF-2: A Computational Model of Myocardial Deposition and Retention. Am J Physiol Heart Circ Physiol. 2005;288:263–279. doi: 10.1152/ajpheart.00205.2004. [DOI] [PubMed] [Google Scholar]
- Filion RJ, Popel AS. A Reaction-Diffusion Model of Basic Fibroblast Growth Factor Interactions with Cell Surface Receptors. Ann Biomed Eng. 2004;32:645–663. doi: 10.1023/b:abme.0000030231.88326.78. [DOI] [PubMed] [Google Scholar]
- Forsten KE, Fannon M, Nugent MA. Potential Mechanisms for the Regulation of Growth Factor Binding by Heparin. J Theor Biol. 2000;205:215–230. doi: 10.1006/jtbi.2000.2064. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of Vascular Endothelial Growth Factor Gene Transcription by Hypoxia-Inducible Factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu BM, Shen S. Structural Mechanisms of Acute VEGF Effect on Microvessel Permeability. Am J Physiol Heart Circ Physiol. 2003;284:2124–2135. doi: 10.1152/ajpheart.00894.2002. [DOI] [PubMed] [Google Scholar]
- Gagnon ML, Bielenberg DR, Gechtman Z, Miao HQ, Takashima S, Soker S, Klagsbrun M. Identification of a Natural Soluble Neuropilin-1 that Binds Vascular Endothelial Growth Factor: In Vivo Expression and Antitumor Activity. Proc Natl Acad Sci U S A. 2000;97:2573–2578. doi: 10.1073/pnas.040337597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlick DG, Renkin EM. Transport of Large Molecules from Plasma to Interstitial Fluid and Lymph in Dogs. Am J Physiol. 1970;219:1595–1605. doi: 10.1152/ajplegacy.1970.219.6.1595. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N. VEGF Regulates Haematopoietic Stem Cell Survival by an Internal Autocrine Loop Mechanism. Nature. 2002;417:954–958. doi: 10.1038/nature00821. [DOI] [PubMed] [Google Scholar]
- Girling JE, Rogers PA. Recent Advances in Endometrial Angiogenesis Research. Angiogenesis. 2005;8:89–99. doi: 10.1007/s10456-005-9006-9. [DOI] [PubMed] [Google Scholar]
- Goldman D, Popel AS. A Computational Study of the Effect of Capillary Network Anastomoses and Tortuosity on Oxygen Transport. J Theor Biol. 2000;206:181–194. doi: 10.1006/jtbi.2000.2113. [DOI] [PubMed] [Google Scholar]
- Gordon MS, Margolin K, Talpaz M, Sledge GW, Jr, Holmgren E, Benjamin R, Stalter S, Shak S, Adelman D. Phase I Safety and Pharmacokinetic Study of Recombinant Human Anti-Vascular Endothelial Growth Factor in Patients with Advanced Cancer. J Clin Oncol. 2001;19:843–850. doi: 10.1200/JCO.2001.19.3.843. [DOI] [PubMed] [Google Scholar]
- Gschwind A, Fischer OM, Ullrich A. The Discovery of Receptor Tyrosine Kinases: Targets for Cancer Therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Haigh JJ. Role of VEGF in Organogenesis. Organogenesis. 2008;4:247–256. doi: 10.4161/org.4.4.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SJ, Bates DO. VEGF-A Splicing: The Key to Anti-Angiogenic Therapeutics? Nat Rev Cancer. 2008;8:880–887. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JW, Mac Gabhann F, Popel AS. Skeletal Muscle VEGF Gradients in Peripheral Arterial Disease: Simulations of Rest and Exercise. Am J Physiol Heart Circ Physiol. 2007;293:H3740–H3749. doi: 10.1152/ajpheart.00009.2007. [DOI] [PubMed] [Google Scholar]
- Ji JW, Tsoukias NM, Goldman D, Popel AS. A Computational Model of Oxygen Transport in Skeletal Muscle for Sprouting and Splitting Modes of Angiogenesis. J Theor Biol. 2006;241:94–9108. doi: 10.1016/j.jtbi.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-Inducible Factor 1 Levels Vary Exponentially Over a Physiologically Relevant Range of O2 Tension. Am J Physiol. 1996;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- Kerbel RS. Tumor Angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kut C, Mac Gabhann F, Popel AS. Where is VEGF in the Body? A Meta-Analysis of VEGF Distribution in Cancer. Br J Cancer. 2007;97:978–985. doi: 10.1038/sj.bjc.6603923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffenburger DA, Linderman JL. Receptors: models for binding, trafficking, and signaling. New York: Oxford University Press; 1993. [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF Signaling is Required for Vascular Homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by Matrix Metalloproteinases Regulates Bioavailability and Vascular Patterning in Tumors. J Cell Biol. 2005;169:681–691. doi: 10.1083/jcb.200409115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, Avraham S. Vascular Endothelial Growth Factor Mediates Intracrine Survival in Human Breast Carcinoma Cells through Internally Expressed VEGFR1/FLT1. PLoS Med. 2007;4 doi: 10.1371/journal.pmed.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd PG, Prior BM, Yang HT, Terjung RL. Angiogenic Growth Factor Expression in Rat Skeletal Muscle in Response to Exercise Training. Am J Physiol Heart Circ Physiol. 2003;284:H1668–H1678. doi: 10.1152/ajpheart.00743.2002. [DOI] [PubMed] [Google Scholar]
- Lodish H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J. Molecular Cell Biology. New York: W.H. Freeman & Co.; 2004. [Google Scholar]
- Mac Gabhann F, Popel AS. Systems Biology of Vascular Endothelial Growth Factors. Microcirculation. 2008;15:715–738. doi: 10.1080/10739680802095964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Gabhann F, Ji JW, Popel AS. Multi-Scale Computational Models of Pro-Angiogenic Treatments in Peripheral Arterial Disease. Ann Biomed Eng. 2007a;35:982–994. doi: 10.1007/s10439-007-9303-0. [DOI] [PubMed] [Google Scholar]
- Mac Gabhann F, Ji JW, Popel AS. VEGF Gradients, Receptor Activation, and Sprout Guidance in Resting and Exercising Skeletal Muscle. J Appl Physiol. 2007b;102:722–734. doi: 10.1152/japplphysiol.00800.2006. [DOI] [PubMed] [Google Scholar]
- Mac Gabhann F, Ji JW, Popel AS. Computational Model of Vascular Endothelial Growth Factor Spatial Distribution in Muscle and Pro-Angiogenic Cell Therapy. PLoS Comput Biol. 2006;2:e127. doi: 10.1371/journal.pcbi.0020127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Gabhann F, Popel AS. Targeting Neuropilin-1 to Inhibit VEGF Signaling in Cancer: Comparison of Therapeutic Approaches. PLoS Comput Biol. 2006;2:e180. doi: 10.1371/journal.pcbi.0020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Gabhann F, Popel AS. Differential Binding of VEGF Isoforms to VEGF Receptor 2 in the Presence of Neuropilin-1: A Computational Model. American Journal of Physiology-Heart and Circulatory Physiology. 2005;288:H2851–H2860. doi: 10.1152/ajpheart.01218.2004. [DOI] [PubMed] [Google Scholar]
- Mac Gabhann F, Popel AS. Model of Competitive Binding of Vascular Endothelial Growth Factor and Placental Growth Factor to VEGF Receptors on Endothelial Cells. Am J Physiol Heart Circ Physiol. 2004;286:H153–H164. doi: 10.1152/ajpheart.00254.2003. [DOI] [PubMed] [Google Scholar]
- Maharaj AS, D'Amore PA. Roles for VEGF in the Adult. Microvasc Res. 2007;74:100–113. doi: 10.1016/j.mvr.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 Stimulates Vascular Endothelial Growth Factor (VEGF) Expression and the Autocrine Activation of VEGFR2 in Endothelial Cells by Activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) Complex. J Biol Chem. 2009;284:6038–6042. doi: 10.1074/jbc.C800207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazitschek R, Giannis A. Inhibitors of Angiogenesis and Cancer-Related Receptor Tyrosine Kinases. Curr Opin Chem Biol. 2004;8:432–441. doi: 10.1016/j.cbpa.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Pollak M. Insulin and Insulin-Like Growth Factor Signalling in Neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an Anti-Vascular Endothelial Growth Factor Monoclonal Antibody for the Therapy of Solid Tumors and Other Disorders. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- Pries AR, Secomb TW. Microvascular Blood Viscosity in Vivo and the Endothelial Surface Layer. Am J Physiol Heart Circ Physiol. 2005;289:H2657–H2664. doi: 10.1152/ajpheart.00297.2005. [DOI] [PubMed] [Google Scholar]
- Qutub AA, Mac Gabhann F, Karagiannis ED, Vempati P, Popel AS. Multiscale Models of Angiogenesis. IEEE Eng Med Biol Mag. 2009;28:14–31. doi: 10.1109/MEMB.2009.931791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qutub AA, Popel AS. A Computational Model of Intracellular Oxygen Sensing by Hypoxia-Inducible Factor HIF1 Alpha. J Cell Sci. 2006;119:3467–3480. doi: 10.1242/jcs.03087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy H, Bhardwaj S, Yla-Herttuala S. Biology of Vascular Endothelial Growth Factors. FEBS Lett. 2006;580:2879–2887. doi: 10.1016/j.febslet.2006.03.087. [DOI] [PubMed] [Google Scholar]
- Segerstrom L, Fuchs D, Backman U, Holmquist K, Christofferson R, Azarbayjani F. The Anti-VEGF Antibody Bevacizumab Potently Reduces the Growth Rate of High-Risk Neuroblastoma Xenografts. Pediatr Res. 2006;60:576–581. doi: 10.1203/01.pdr.0000242494.94000.52. [DOI] [PubMed] [Google Scholar]
- Sela S, Itin A, Natanson-Yaron S, Greenfield C, Goldman-Wohl D, Yagel S, Keshet E. A Novel Human-Specific Soluble Vascular Endothelial Growth Factor Receptor 1: Cell-Type-Specific Splicing and Implications to Vascular Endothelial Growth Factor Homeostasis and Preeclampsia. Circ Res. 2008;102:1566–1574. doi: 10.1161/CIRCRESAHA.108.171504. [DOI] [PubMed] [Google Scholar]
- Simons M. Integrative Signaling in Angiogenesis. Mol Cell Biochem. 2004;264:99–102. doi: 10.1023/b:mcbi.0000044379.25823.03. [DOI] [PubMed] [Google Scholar]
- Stefanini MO, Wu FTH, Mac Gabhann F, Popel AS. A Compartment Model of VEGF Distribution in Blood, Healthy and Diseased Tissues. BMC Syst Biol. 2008;2:77. doi: 10.1186/1752-0509-2-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Breen EC, Wagner H, Brutsaert TD, Gassmann M, Wagner PD. HIF and VEGF Relationships in Response to Hypoxia and Sciatic Nerve Stimulation in Rat Gastrocnemius. Respir Physiol Neurobiol. 2004;144:71–80. doi: 10.1016/j.resp.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Truskey GA, Yuan F, Katz DF. Transport Phenomena in Biological Systems. New Jersey: Pearson Prentice Hall; 2004. Porosity, tortuosity, and available volume fraction; pp. 389–398. [Google Scholar]
- Venturoli D, Rippe B. Ficoll and Dextran Vs. Globular Proteins as Probes for Testing Glomerular Permselectivity: Effects of Molecular Size, Shape, Charge, and Deformability. Am J Physiol Renal Physiol. 2005;288:605–613. doi: 10.1152/ajprenal.00171.2004. [DOI] [PubMed] [Google Scholar]
- Verheul HM, Lolkema MP, Qian DZ, Hilkes YH, Liapi E, Akkerman JW, Pili R, Voest EE. Platelets Take Up the Monoclonal Antibody Bevacizumab. Clin Cancer Res. 2007;13:5341–5347. doi: 10.1158/1078-0432.CCR-07-0847. [DOI] [PubMed] [Google Scholar]
- Wijelath ES, Rahman S, Namekata M, Murray J, Nishimura T, Mostafavi-Pour Z, Patel Y, Suda Y, Humphries MJ, Sobel M. Heparin-II Domain of Fibronectin is a Vascular Endothelial Growth Factor-Binding Domain: Enhancement of VEGF Biological Activity by a Singular Growth factor/matrix Protein Synergism. Circ Res. 2006;99:853–860. doi: 10.1161/01.RES.0000246849.17887.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley HS, Shvartsman SY, Lauffenburger DA. Computational Modeling of the EGF-Receptor System: A Paradigm for Systems Biology. Trends Cell Biol. 2003;13:43–50. doi: 10.1016/s0962-8924(02)00009-0. [DOI] [PubMed] [Google Scholar]
- Willett CG, Boucher Y, Duda DG, di Tomaso E, Munn LL, Tong RT, Kozin SV, Petit L, Jain RK, Chung DC, Sahani DV, Kalva SP, Cohen KS, Scadden DT, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Shellito PC, Mino-Kenudson M, Lauwers GY. Surrogate Markers for Antiangiogenic Therapy and Dose-Limiting Toxicities for Bevacizumab with Radiation and Chemotherapy: Continued Experience of a Phase I Trial in Rectal Cancer Patients. J Clin Oncol. 2005;23:8136–8139. doi: 10.1200/JCO.2005.02.5635. [DOI] [PubMed] [Google Scholar]
- Wu FT, Stefanini MO, Mac Gabhann F, Kontos CD, Annex BH, Popel AS. A Computational Kinetic Model of VEGF Trapping by Soluble VEGF Receptor-1: Effects of Transendothelial and Lymphatic Macromolecular Transport. Physiol Genomics. 2009a;38:29–41. doi: 10.1152/physiolgenomics.00031.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FT, Stefanini MO, Mac Gabhann F, Popel AS. A Compartment Model of VEGF Distribution in Humans in the Presence of Soluble VEGF Receptor-1 Acting as a Ligand Trap. PLoS ONE. 2009b;4:e5108. doi: 10.1371/journal.pone.0005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK. Vascular Permeability in a Human Tumor Xenograft: Molecular Size Dependence and Cutoff Size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]