

Abstract
As a tumor marker for colorectal cancers, CEA enhances the metastatic potential of cancer cells. CEA functions as an intercellular adhesion molecule and is up-regulated in a wide variety of human cancers. However, the molecular mechanisms by which CEA mediate metastasis remain to be understood. TGF-β signaling regulates both tumor suppression and metastasis, and also contributes to the stimulation of CEA transcription and secretion in colorectal cancer cells. However, it remains unknown whether CEA, in-turn, influences TGF-β functions and if a regulatory cross-talk exists between CEA and TGF-β signaling pathway. Here we report that CEA directly interacts with TGF-β receptor and inhibits TGF-β signaling. Targeting CEA with either CEA specific antibody or siRNA rescues TGF-β response in colorectal cancer cell lines with elevated CEA, thereby restoring the inhibitory effects of TGF-β signaling on proliferation. CEA also enhances the survival of colorectal cancer cells in both local colonization and liver metastasis in animal study. Our study provides novel insights into the interaction between CEA and TGF-β signaling pathway and establishes a negative feed-back loop in amplifying the progression of colon cancer cells to more invasive phenotypes. These findings offer new therapeutic opportunities to inhibit colorectal cancer cell proliferation by co-targeting CEA in promoting tumor inhibitory action of TGF-β pathway.
Keywords: TGF-β signaling, colorectal cancer, CEA
Introduction
CEA is a member of the immunoglobulin superfamily. In humans, the carcinoembryonic antigen family consists of 29 genes. CEA and CEACAM6 belong to GPI-anchored carcinoembryonic antigen family, and are normally predominantly expressed in the gastrointestinal tract, but overexpressed in as many as 70% of all human cancers (1, 2). It has been demonstrated that all CEA family members function as homotypic intercellular adhesion molecules (3–5). Currently, CEA is used as a tumor marker for the clinical management of colorectal cancer (CRC). Elevated blood levels of CEA indicate metastasis and poor prognosis (6, 7). There is mounting evidence that CEA has been involved in multiple biological aspects of neoplasia such as cell adhesion, metastasis, suppression of cellular immune mechanisms, and inhibition of apoptosis (8–13). For instance, CEA increases the ability of weakly metastatic CRC to colonize in the liver and develop spontaneous hematogeneous liver and lung metastasis (14–16). CEA expression also correlates well with resistance to cytotoxic chemotherapy (12) and to anoikis (10, 17). The inhibitory role of CEA in cell differentiation (9, 13, 18) and anoikis (17, 19) has been extensively documented. However, the molecular mechanism by which CEA enhances tumor metastasis continues to be poorly understood.
The TGF-β signaling pathway is involved in the control of multiple biological processes, including cell proliferation, differentiation, migration and apoptosis (20, 21). It is one of the most commonly altered cellular signaling pathways in human cancers (22). Three TGF-β isoforms (TGFB1, TGFB2 and TGFB3) are expressed in mammalian epithelium, each encoded by a unique gene and expressed in both a tissue specific and developmentally regulated manner. Among these TGFB1 is the most abundant and ubiquitously expressed isoform. TGF-β signaling is initiated by the binding of TGF-β ligand to the type II TGF-β receptors (TBRII). The ligand binds tightly to the ectodomain of the type II receptor first; this binding allows the subsequent incorporation of the TGF-β type I receptor (TBRI), forming a large ligand-receptor complex involving a ligand dimer and four receptor molecules. Binding to the extracellular domains of both types of the receptors by the dimeric ligand induces a close proximity and a productive conformation for the intracellular kinase domains of the receptors, facilitating the phosphorylation and subsequent activation of the type I receptor (23).
With the help of adaptor proteins such as SARA and β2SP (β2 spectrin), activated TBRI then recruits and phosphorylates two downstream transcription factors, Smad2 and Smad3, allowing them to bind to Smad4 (20, 21, 24, 25). The resulting Smad complexes translocate into the nucleus and interact with other transcription factors in a cell-specific manner to regulate the transcription of a multitude of TGF-β responsive genes (26). Some of the downstream targets of TGF-β signaling are important cell-cycle checkpoint genes, including p21, p27 and p15, and their activation leads to growth arrest (20). In normal and premalignant cells, TGF-β enforces homeostasis and suppresses tumor progression through cell-autonomous tumor-suppressive effects (cytostasis, differentiation, and apoptosis) or through effects on the stroma (suppression of inflammation and stroma-derived mitogens). However, when cancer cells lose TGF-β tumor-suppressive responses, they can use TGF-β to their advantage to initiate differentiation into an invasive phenotype and metastatic dissemination (24). Current data strongly support the notion that TGF-β signaling suppresses colorectal cancers. Many colorectal cancers escape the tumor-suppressor effects of TGF-β signaling and are resistant to TGF-β induced growth inhibition (27).
Aberrant upregulation of CEA and alteration of TGF-β signaling are common features of colorectal cancers. Since both CEA and TGF-β signaling are involved in the development and progression of colorectal tumors, the possible interaction between them has been investigated by several groups. It is known that TGF-β induces CEA secretion in a dose dependent manner (28). Also, CEA and CEACAM6 are identified as target genes for Smad3-mediated TGF-β signaling (29). However, little is known about the effects of CEA on TGF-β signaling pathway. Our data demonstrate the interaction of CEA with TBRI, indicating the possible influence of CEA on TGF-β signaling.
In the current study, we focus on the effects of CEA on the TGF-β signaling in both normal cells and in colorectal cancer cells. Our studies demonstrate that CEA directly binds to TBRI. Furthermore, overexpression of CEA inhibits TGF-β signaling. In colorectal cancer cells with elevated CEA, targeting CEA with specific antibody or siRNA rescues their response to TGF-β stimulation, thereby restoring the inhibitory effects of TGF-β on the proliferation of these cancer cells.
Materials and Methods
Reagents
TGF-β1 (Sigma T 1654), purified CEA protein (abcam Ab742), and HA peptide (Sigma 12149) were purchased.
DNA constructs
The constructs for the expression of HA-TGFBR1, HA-TGFBR2, V5-Smad3, HA-Smad4, and V5-β2SP were previously described(25). Plasmid that expresses wt CEA was as described previously (17).
Tissue culture, transfections and lentivirus infection
Human colorectal carcinoma cell lines (microsatellite instable cell lines: Lovo, HCT116, DLD-1, HCT-15, LS174T LS180 and HCT-6; microsatellite stable cell lines: HT-29, Caco-2, SW480, SK-CO-1 and Colo205) and 293T cells were obtained and characterized by ATCC with PCR within 6 months. Clone A, a human metastatic CRC cells(17), was provided and characterized by Dr. Jessup’s lab two years ago, and tested again by Radil Research Animal Diagnostic Laboratory with PCR one year ago. All colorectal cancer cell lines were maintained in DMEM or RPMI 1640 supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C, 5% CO2 in a humidifier chamber. 293T cells were grown in DMEM with 10% FBS and antibiotics at 37°C and at 5% CO2. Transfections were performed with Lipofectamine 2000 (Invitrogen). Generation of stable Clone A transfectants was described previously(17). GIPZ lentiviral shRNA particles targeting human CEA (ThermoFisher RHS4348) and control lentiviral shRNA particles were from Openbiosystems. Cells were infected with lentiviral shRNA particles according to manufacturer’s instructions.
Antibodies and immunotechniques
Antibodies against CEA (Thermo MS-613-P0, MS-613-P1), Smad3 (Invitrogen 51–1500), phosphorylated Smad3 (Santa Cruze Sc-130218), TGFβRI (Santa Cruz Sc-398), TGFβRII (upstate 06-318), V5 (Invitrogen R960-25), and HA (Sigma-aldrich H 3663) were purchased. Secondary antibodies conjugated with horseradish peroxidase (Chemicon), were purchased. Immunoprecipitation and immunoblotting procedures were described elsewhere (30). To avoid signal noise from IgG chains, Trueblot IP beads (00-8800, 00-8811) and Trueblot western Blot Kit (88-8887, 88-8886) were utilized according to manufacturer’s instruction.
RT-PCR for TGF-β regulated gene expression
The primers used for amplifications were as follows: c-Myc F: 5’TCAAGAGGCGAACACACAAC-3’, R: 5’-GGCCTTTTCATTGTTTTCCA-3’; GAPDH F: 5’-CATTGACCTTCACTACATGGT-3’, R: 5’-ACCCTTCAAGTGAGCCCCAG-3’. RT-PCR was performed as previously described (25).
Confocal and fluorescence microscopy
Cells were fixed in 4% formaldehyde and permeabilized in 0.1% triton X-100. Then cells were incubated with primary antibodies overnight at 4°C, followed by incubating with secondary antibodies conjugated with TR (Santa Cruz) and FITC (Santa Cruz) for 2 h at RT. Confocal microscopy was carried out using an Olympus Fluoview confocal microscope in the Lombardi Comprehensive Cancer Center.
Small-scale biochemical fractionation
Small-scale biochemical fractionation was performed as described previously (31).
In vitro binding assay
Recombinant HA-TBRI and HA-TBRII proteins expressed in 293T cells were purified with Flag HA Tandem Affinity Purification Kit (Sigma) according to the manufacturer’s recommendations. Purified HA-TBRI (2 μg) or HA-TBRII (2 μg) proteins were incubated with purified CEA (2 μg) protein in binding buffer (50 mM Tris HCl [pH 7.5], 100 mM NaCl, 50 mM NaF, 10 nM okadaic acid, 0.1% Nonidet P-40) for 60 min at 4°C. The reactions were then incubated with monoclonal anti-HA-Agrose beads (sigma A2095) for another 1 h. The beads were extensively washed with binding buffer, and associated proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Reporter assay
C-myc-luciferase assay was performed according to the manufacturer's instructions (Promega), and the results were standardized against the β-galactosidase activity.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as described previously (32). Briefly, cells were cross-linked with formaldehyde and sonicated on ice to fragment the chromatin into an average length of 500 bp to 1 kb. Antibodies were used to immunoprecipitate the respective antigens at 4°C overnight. Protein A Sepharose beads saturated with bovine serum albumin and single-strand DNA were added to the lysate to isolate the antibody-bound complexes. The beads were washed to remove nonspecific binding, and the antibody-bound chromatin was eluted. The eluate was “de-crosslinked” by heating at 65°C for 6 h, and then treated with RNase and proteinase K. DNA was extracted using the phenol chloroform method. PCR was performed by using the following primers for human c-myc promoter: Region −5 to −233, F: TTTATAATGCGAGGGTCTGGACGGC, R: ACAGCGAGTTAGATAAAGCCCCGAAAA; Region −607 to −751 F: ATCATTCTAGGCATCGTTTTCCTC, R: GGGAAAGGGC-CGCGCTTTGATCAA.
EMSA
Nuclear extracts were prepared using a Nonidet P-40 lysis method. EMSA for Smad3 DNA binding was performed using the annealed and [γ-32P] ATP end-labeled PCR product of human c-myc promoter region (−5 to −233 ) for 15 min at 20°C. Samples were run on a nondenaturing 5% polyacrylamide gel and imaged by autoradiography. Specific competitions were performed by adding a 100-molar excess of competitor to the incubation mixture, and supershift EMSAs were performed by adding 200 ng of the Smad3 antibody.
Cell proliferation assay
10,000 of the trypan blue-negative live cells per well were seeded into 96-well plates. Cell proliferation was assessed by using a colorimetric WST-1 cell proliferation kit (Roche) according to the manufacturer’s recommendations.
Results
CEA interacts with TGF-β receptor I
It has been reported that TGF-β regulated CEA expression and secretion (28, 29). However, little is known about the influence of CEA on TGF-β signaling. We first examined whether there was a direct interaction between CEA and components of TGF-β signaling pathway by coimmunoprecipitation assays. Wild type CEA was co-transfected with one of the five elements of TGF-β signaling pathway as indicated in Figure 1A. Reciprocal coimmunoprecipitation experiments were performed to examine the interaction between CEA and these five elements. We found that, indeed, CEA could be effectively co-immunoprecipitates with the TBRI but not with other elements. We also noticed the endogenous interaction between CEA and TBRI but not TBRII in two CRC cell lines (Figure 1B). To evaluate whether the noticed interaction between CEA and TBRI was direct, we performed in vitro binding assay. Recombinant HA-TBRI and HA-TBRII proteins purified from 293T cells were incubated with CEA protein. Association between CEA with TBRI but not TBRII was observed (Figure 1C). These findings demonstrated that the interaction between CEA and TBRI was direct and did not require the presence of other proteins. We next assessed where these two proteins interacted with each other in-situ by immunofluorescence staining. Cells were transfected with or without CEA. 24 h later, cells were fixed and stained for CEA and TBRI. The results demonstrated the colocalization of CEA and TBRI in the cell membrane (Figure 1D). These data indicate that CEA may regulate TGF-β signaling by binding to TBRI at the membrane. Together, for the first time these findings establish physical interaction of CEA and TBRI in physiological relevant settings.
Figure 1. Direct association of CEA with TBRI.

A) 293T cells were co-transfected with wt CEA and one of the five elements of TGF-β signaling pathway, namely β2SP, Smad3, TBRI, TBRII, and Smad4 as indicated. Coimmunoprecipitation experiments were performed to examine the interaction between CEA and these five elements. B) LS174T and LS180 cells lysate were used for the coimmunoprecipitation assay to determine the association of endogenous CEA with endogenous TBRI. C) In vitro binding assay. Purified CEA protein was incubated with purified HA-TBRI or HA-TBRII. Binding of CEA to TBRI or TBRII was assessed by immunoprecipitation followed by immunoblotting. D) 293T Cells were transfected with wt CEA plasmid or vector pcDNA. Cells were then fixed and stained for CEA and TBRI 24 h after transfection.
CEA inhibits TGF-β signaling
Next we investigated whether the noticed association of CEA and TBRI modulates TGF-β signaling. TGF-β signals through a hetero-dimeric receptor complex consisting of both TBRI and TBRII. Activated TBRI recruits and phosphorylates R-Smads, and enables the resulting complex to bind to Smad4. Following binding to Smad4, the complex translocates into nuclear to activate transcription of various target genes. First, we examined the impact of CEA overexpression on the recruitment of Smad3 to TBRI. 293T cells were co-transfected with TBRI, Smad3 and CEA, treated with TGF-β for 1 h., and total cell lysates were subjected to coimmunoprecipitation assays. As shown in Figure 2A, association of TBRI with Smad3 was attenuated in the presence of CEA compared to in the absence of CEA. We then sought to determine if Smad3 phosphorylation was modified by CEA. 293T cells were transfected with or without CEA for 24 h, and then stimulated with TGF-β for different time-periods. Cells were harvested and the levels of p- Smad3 and Smad3 proteins were evaluated by Western blotting. Increase of Smad3 phosphorylation was observed in the cells without CEA expression. In contrast, the levels of phosphorylated- Smad3 were constant in the cells with overexpressed CEA (Figure 2B). We subsequently examined the influence of CEA on the nuclear translocation of Smad3. 293T cells were transfected with or without CEA, stimulated with TGF-β for I h, and fixed cells were stained for Smad3. Indeed, Smad3 nuclear translocation was reduced in the cells transfected with CEA (Figure 2C). To independently verify these results from confocal microscopy, total cell lysates were fractionated into the cytoplasmic and nuclear fractions. As expected from the preceding results, we found a substantial decrease in the levels of nuclear Smad3 in the cells with overexpressed CEA after TGFβ treatment (Figure 2D). To establish a modulating effect of CEA on the functionality of Smad3, we next examined the level of c-myc mRNA, one of the targets of TGF-β signaling pathway. While TGF-β induced downregulation of c-myc transcription in control cells, overexpression of CEA blocked the inhibitory effects of TGF-β on c-myc transcription (Figure 2E). The observed inhibitory effects of CEA on TGF-β-target genes was not restricted to c-myc, as the level of other target genes such as p21 also followed a similar pattern (data not shown).
Figure 2. Inhibition of TGF-β signaling by CEA.

A) 293T cells were co-transfected with HA-TBRI, V5- Smad3 and CEA as indicated. 24 h after transfection, cells were treated with TGF-β (100 pM) for 1 h. Coimmunoprecipitation was carried out to evaluate the association of TBRI with Smad3. B) 293T cells were transfected with CEA or vector plasmid for 24 h, and then stimulated with TGF-β (100 pM) for different time periods as indicated. Cells were harvested and p- Smad3 and Smad3 protein levels were evaluated by Western blotting. Histogram shows the fold increase of p- Smad3 intensity compared to that at the time point 0. C) 293T cells were transfected as in B, then stimulated with TGF-β (100 pM) for I h. Cells were then fixed and stained for Smad3 and CEA. Counterstain, DAPI). D) Cells were transfected and stimulated with TGF-β (100 pM) for I h. Cells were lysed, and cytoplasmic and nuclear fractions were separated. Smad3 levels in different fractions were evaluated by immunoblotting. Actin served as loading control. E) 293T cells were treated as in C. c-myc mRNA levels were assessed by RT-PCR. GAPDH served as loading control.
Impairment of TGF-β signaling in colorectal cancer cells with elevated CEA
To determine the functional significance of the interaction between CEA and TGF-β signaling in colorectal tumorigenesis, we first assessed the possible existence of a correlation between the levels of CEA and TGF-β-induced Smad3 phosphorylation in 12 colorectal cancer cell lines. The extent of Smad3 phosphorylation was measured by the fold increase of p- Smad3 (ratio of p-Smad3 upon TGF-β stimulation to the basal p- Smad3 levels). The cell lines were classified into 3 groups based on the levels of CEA expression. As seen in Figures 3A, there was an inverse correlation between the levels of CEA levels and the degree of Smad3 phosphorylation (p<0.05). To corroborate these findings, we treated colorectal cancer cells LS180 with anti-CEA antibody to block CEA before stimulating the cells with TGF-β. As shown in Figure 3B, nuclear translocation of Smad3 was enhanced in cells treated with anti-CEA antibody. Consistent with these results, we found that blocking CEA action with antibody also restored the inhibitory effects of TGF-β on c-myc transcription (Figure 3C). Finally, we showed that anti-CEA antibody was also able to block transcription from a c-myc-promoter-luc reporter system (Fig. 3D), presumably due to an enhanced recruitment of Smad-3 to the c-myc-gene chromatin (Fig. 3E) in TGF-β stimulated HCT116 cells. To demonstrate a potential direct binding of Smad3 to the human c-myc promoter, we next performed EMSA using a PCR product encompassing the region −5 to −233 of c-myc promoter and nuclear extracts from HCT 116 cells with or without TGF-β stimulation either in the presence of IgG or CEA antibody. As expected from the preceding results, TGF-β stimulation of HCT116 cells in the presence of CEA antibody promoted the Smad3/DNA complex formation ( Fig 3F, lanes 11–13) compared to those in the presence of IgG antibody (Fig. 3F, lanes 5–7). The specificity of the noted complex was further verified by supershift experiments using anti-Samd3 (Fig. 3F, lane 12) or control IgG (Fig. 3F lane 13). Collectively, these findings suggest that elevated levels of CEA may counteract the inhibitory activity of TGF-β, leading to a possible functional inactivation of TGF-β signaling in colorectal cancer cells.
Figure 3. TGF-β signaling is impaired in colorectal cancer cells with elevated CEA.

A) CEA expression and TGF-β induced Smad3 phosphorylation were evaluated in 12 colorectal cancer cell lines by immunoblotting. The extent of Smad3 phosphorylation was measured by the fold increase of p- Smad3 (ratio of p- Smad3 with TGF-β stimulation to p- Smad3 without TGF-β stimulation). According to CEA expression levels, the cell lines were classified into 3 groups. The scatter plot graph demonstrates an inverse correlation between CEA expression levels and the extent of Smad3 phosphorylation. Blue circles: cell lines 1,2,3 and 8; Yellow squares: cell lines 4,6,7, and 12; Red diamonds: cell lines 5,9,10 and 11. Broken lines represent average levels of p-Smad3 fold increase in each group. The bars indicate the standard error. 1. SK-CO-1; 2. LS180; 3. LS174T; 4. Caco-2; 5. HCT-6; 6. Colo205; 7. HT-29; 8. Lovo; 9. HCT116; 10. SW480; 11. HCT-15; 12. DLD-1. B) LS174T cells were treated with anti-CEA antibody (3 μg/ml) or naive IgG for 24 h to block CEA and then treated with or without TGF-β (100 pM) for 1 h. Nuclear translocation of Smad3 was determined as in Figure 2C. C). Five CRC cell lines were treated with or without anti-CEA antibody for 24 h as indicated, then treated with TGF-β (100 pM) for 1 h. Transcription levels of c-myc were assessed as in Figure 2E. The histogram shows the quantification of c-myc mRNA levels in the left graph. D–F) Anti-CEA Ab promotes Smad3-dependent repression of c-Myc expression by TGF-β. D. Effect of IgG or CEA antibody on the c-Myc-luc promoter activity (lower panel) and on the c-Myc protein in the HCT116 cells treated with or without TGF-β. *P<0.05. Western blot analysis was performed with the cell lysates obtained from the luciferase assay samples. E. ChIP analysis showing the recruitment of Smad3 but not β-spectrin onto human c-myc promoter in the HCT116 cells treated with IgG or CEA antibody in the presence or absence of TGF-β treatment. F. EMSA analysis of the Smad3 binding in the human c-Myc promoter using the PCR product encompassing the region −5 to −233 in HCT116 cells treated with TGF-β in presence of either IgG or CEA antibody.
Targeting CEA restores the inhibitory effects of TGF-β signaling on proliferation of colorectal cancer cells
TGF-β signaling plays an important role in suppressing cell proliferation and tumorigenesis (24). However, some cancer cells lose their responses to the proliferation inhibiting effects of TGF-β signaling during development. In addition to the mutations of TGF-β receptors or Smads, we assume that increased CEA may also contribute to the loss of response to TGF-β signaling as supported by data in the preceding paragraph. To test this hypothesis, we next carried out proliferation assay using colorectal cells stably transfected either with the control pcDNA vector or with plasmid encoding CEA (17). We assessed the effects of CEA on the inhibition of proliferation induced by TGF-β in the presence or absence of anti-CEA antibody. In cells transfected with vector, TGF-β treatment led to approximately 20% proliferation inhibition. Interestingly, combination of TGF-β and anti-CEA antibody had similar effects as using TGF-β alone. In contrast, in cells transfected with CEA, TGF-β treatment had little effects on cell proliferation. However, when combined with anti-CEA antibody, TGF-β remarkably reduced cell proliferation (p<0.05 Figure 4A). This result strongly suggests that targeting CEA may restore the tumor suppressing effects of TGF-β in some colorectal cancer cells. To confirm this finding, 3 colorectal cancer cell lines with augmented CEA levels were treated with TGF-β and different doses of anti-CEA antibody as indicated. Anti-CEA antibody enhanced TGF-β-mediated growth inhibition of target cells in a dose-dependant manner (Figure 4B). For further confirmation, we suppressed CEA expression with specific siRNA, and then treated cells with TGF-β. As shown in Figure 4C, treating cells with siRNA targeting CEA rescued the inhibitory effects of TGF-β on the proliferation of CRC cells.
Figure 4. Targeting CEA rescues the inhibitory effects of TGF-β signaling on proliferation of colorectal cancer cells.

A) Clone A cells were stably transfected either with pcDNA vector or with plasmids encoding wt CEA. 10,000 cells were seeded into each well of 96 well plates, and treated with TGF-β (100 pM) and/or anti-CEA antibody (3 μg/ml) as indicated for 24 h. Cell proliferation rates were assessed by WST assay. Proliferation rate was calculated as (absorbance with treatment/absorbance without treatment) × 100%. The graph depicts percentages of proliferation with data from two independent experiments (each carried out in triplicate). *, P< 0.05 for the comparison with cells treated with TGF-β alone (Student’s t test). B). Three colorectal cancer cell lines were treated with TGF-β and different doses of anti-CEA antibody as indicated. Cell proliferation assays were performed as in A. C). DLD- cells were infected with lentiviral control siRNA or siRNA targeting CEA. Western blot demonstrated that the siCEA suppressed CEA level efficiently. Stable infected cells were selected by puromycin treatment. Cells were then treated with or without TGF-β (100 pM) for different time periods. Cell proliferation assay was performed as in A. *: p<0.05 for the comparison between the TGF-β treatment and non-treatment groups at the same time point.
CEA enhances liver metastasis of colorectal cancer cells
Previous studies have shown that CEA enhances liver metastasis of colorectal cancer cells in animal experiments (16, 17, 33). To corroborate the role of CEA in liver metastasis, colorectal cancer cells stably transfected with CEA expression plasmid or pcDNA were injected intra-splenically into nude mice (2×106 viable cells/animal, n=10 animal each group). Mice were autopsied to determine spleen and liver colonization 30 days post-injection. As shown in Figure 5, CEA enhanced both liver and spleen colonization of the cancer cells. Liver metastasis was detected in 50% recipients of cells transfected with CEA. In contrast, liver colonies were found in only 10% recipients of control cells (p<0.05).
Figure 5. CEA enhances liver metastasis of colorectal cancer cells.
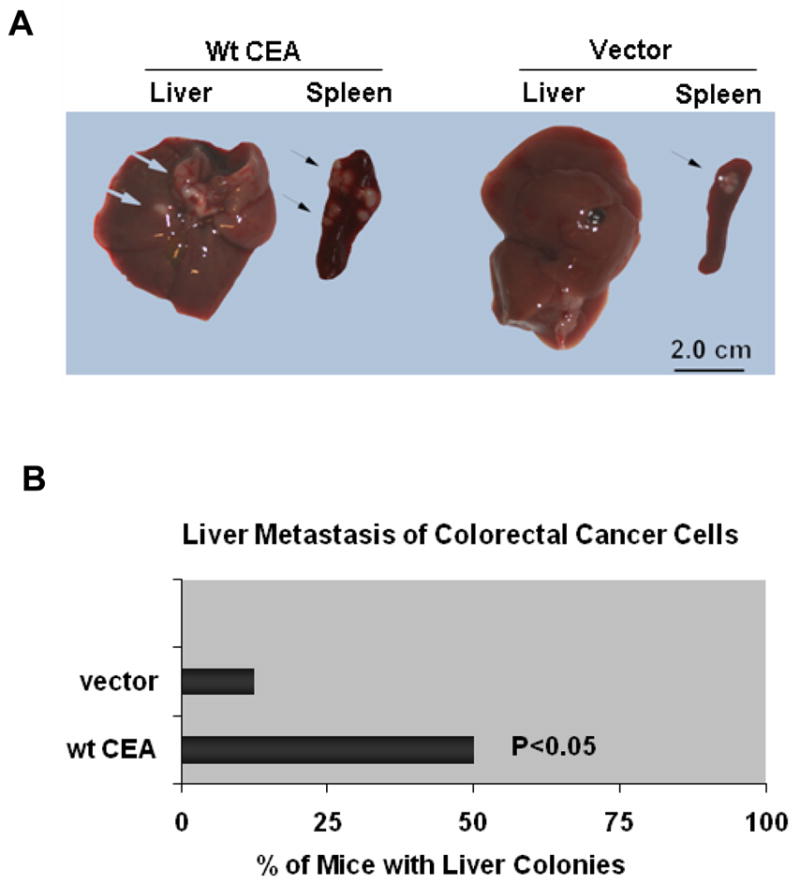
Clone A cells were stably transfected either with pcDNA vector or with plasmids encoding wt CEA, and then injected intrasplenically into nude mice. 2×106 viable cells were injected for each of 10 mice per group. Mice were autopsied to determine spleen and liver colonization 30 days after injection. A) Representative pictures show the tumor colonies in spleens and livers. Arrows indicate the colonies formed by different cell types. B) The graph shows the percentage of mice with liver colonies.
Discussion
Increased CEA levels are observed in a wide variety of human cancers such as colon, breast and lung cancers. Previous studies have demonstrated that CEA contributes to tumorigenesis by inhibiting cell differentiation and anoikis (8–13, 17, 19). Interestingly, CEA is a GPI-linked protein that lacks transmembrane and cytoplasmic domains (34–36), which is suggestive that CEA has to exert these effects by modulating other signaling pathways. It was reported that CEA mediated anoikis inhibition through integrin (37, 38) and DR5 signaling (17, 39). Here we report that CEA directly binds to TBRI, and inhibits TGF-β signaling pathway.
Our data demonstrate that CEA binds to TBRI but not TBRII, and that these two molecules colocalize on the plasma membrane (Figure 1). Our findings that CEA overexpression attenuates the TGF-β signaling (Figure 2) is significant as it implies that CEA can contribute to tumor development and progress by down-regulating TGF-β signaling pathway. Although a dual role of TGF-β in cancers has been noted, the genetic and mechanistic basis for gastrointestinal cancers has remained elusive. There is considerable genetic evidence that the TGF-β signaling pathway is a tumor suppressor in gastrointestinal epithelial cells. First, TGF-β signaling pathway has a major influence on cell lineage determination and terminal differentiation, and suppress tumorigenesis by driving precursor cells into a less proliferative state (40). Secondly, TGF-β can induce apoptosis through both Smad-dependent and -independent mechanisms (41). These mechanisms include the induction of multiple proapoptotic factors, such as the signaling factor GADD45b, the death-associated protein kinase DAPK, the death receptor FAS, and the proapoptotic effector BIM. Moreover, Smads interaction with Akt pathway and TGF-β receptor interaction with the p38 MAPK activator DAXX have also been proposed as an alternative mechanism of proapoptotic effects of TGF-β (24). Given these tumor suppressing effects of TGF-β, inhibition of TGF-β signaling by augmented CEA may be of high significance in the colorectal tumorigenesis. The role of TGF-β signaling in colorectal tumorigenesis has been recognized over the past decade. There is growing evidence that TGF-β signaling alterations mediated by Microsatellite Instability (MSI) contribute to colon cancer development and progression. MSI contributes to the TGFβ signaling resistance of CRC by resultant mutations of TGFβ receptors or Smads. Here, we provide evidence that TGF-β signaling alterations may be also mediated by enhanced CEA level. This can explain why some colorectal cancer cells can still escape the inhibitory effects of TGF-β signaling without detectable mutations or polymorphisms of TGF-β receptors or Smads. Around 80% of all microsatellite instable CRCs contain mutations in TBRII. In these cases, targeting CEA will not be able to rescue TGF-β response due to the impaired TGFβ signaling. However, for some microsatellite instable cells in which the components of TGFβ signaling pathway are not mutated, targeting CEA can also rescues TGFβ response.
Development of liver metastasis is a frequent complication in the course of gastro-intestinal malignancies. In support of a role of CEA in the process of liver metastasis, we provide experimental data supporting the notion that CEA increases the survival of cancer cells in both local colonization and distant metastasis (Figure 5). After entering the liver via the portal circulation, cancer cells are encountered by Kupffer cells (KC) in the liver sinusoids (42). Kupffer cells represent ~10% of all liver cells, and have the ability to kill tumor cells. As such, these cells may have an intrinsic role in the protection against outgrowth of hepatic metastasis. The cytotoxic function of KC is regulated by multiple cytokines. Functionally, TGF-β was found to be chemotactic for Kupffer cells and regulate Kupffer cell functions (43). In addition to inhibiting TGF-β signaling in cancer cells, it is possible that CEA produced by cancer cells may also affect TGF-β signaling in Kupffer cells and thereby enhance the metastatic potential of tumor cells. However, this possibility needs to be experimental tested, as yet.
In colorectal cancer cells with augmented CEA expression, targeting CEA with specific antibody or siRNA can rescue TGF-β response. Increase of CEA expression and alteration of TGF-β signaling are commonly observed in colorectal cancers. Our study provides insight into understanding how these two important events interact with each other during tumorigenesis. Since CEA is a frequently overexpressed tumor-associated antigen in tumors, specific antibodies targeting CEA have been developed as a novel therapeutic approach for treatment of tumors expressing CEA on their surface (44–47). In this context, results presented here offer new opportunity to combine CEA antibody and TGF-β to inhibit the proliferation and metastasis of colorectal cancer.
Acknowledgments
This study was supported by NIH grants CA106614-01A2 and CA4285718A1 to LM, and work in RK’s lab is supported by NIH CA98823.
Abbreviations
- TGF-β
transforming growth factor-β
- CEA
carcinoembryonic antigen
- TBR
TGF-β receptor
- β2SP
β2 spectrin
- CRC
colorectal cancer
References
- 1.Nollau P, Scheller H, Kona-Horstmann M, et al. Expression of CD66a (human C-CAM) and other members of the carcinoembryonic antigen gene family of adhesion molecules in human colorectal adenomas. Cancer Res. 1997;57:2354–2357. [PubMed] [Google Scholar]
- 2.Rosenberg M, Nedellec P, Jothy S, Fleiszer D, Turbide C, Beauchemin N. The expression of mouse biliary glycoprotein, a carcinoembryonic antigen-related gene, is down-regulated in malignant mouse tissues. Cancer Res. 1993;53:4938–4945. [PubMed] [Google Scholar]
- 3.Benchimol S, Fuks A, Jothy S, Beauchemin N, Shirota K, Stanners CP. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell. 1989;57:327–334. doi: 10.1016/0092-8674(89)90970-7. [DOI] [PubMed] [Google Scholar]
- 4.Oikawa S, Inuzuka C, Kuroki M, Matsuoka Y, Kosaki G, Nakazato H. Cell adhesion activity of non-specific cross-reacting antigen (NCA) and carcinoembryonic antigen (CEA) expressed on CHO cell surface: homophilic and heterophilic adhesion. Biochem Biophys Res Commun. 1989;164:39–45. doi: 10.1016/0006-291x(89)91679-3. [DOI] [PubMed] [Google Scholar]
- 5.Stanners CP. F. A. Properties of adhesion mediated by the human CEA family. Cell Adhesion and Communication Mediated by the CEA Family: Basic and Clinical Perspectives. 1998;Chapter 3:57–71. [Google Scholar]
- 6.Bast RC, Jr, Ravdin P, Hayes DF, et al. 2000 update of recommendations for the use of tumor markers in breast and colorectal cancer: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol. 2001;19:1865–1878. doi: 10.1200/JCO.2001.19.6.1865. [DOI] [PubMed] [Google Scholar]
- 7.Henson DE, Fielding LP, Grignon DJ, et al. College of American Pathologists Conference XXVI on clinical relevance of prognostic markers in solid tumors. Summary. Members of the Cancer Committee. Arch Pathol Lab Med. 1995;119:1109–1112. [PubMed] [Google Scholar]
- 8.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. CEACAM6 gene silencing impairs anoikis resistance and in vivo metastatic ability of pancreatic adenocarcinoma cells. Oncogene. 2004;23:465–473. doi: 10.1038/sj.onc.1207036. [DOI] [PubMed] [Google Scholar]
- 9.Eidelman FJ, Fuks A, DeMarte L, Taheri M, Stanners CP. Human carcinoembryonic antigen, an intercellular adhesion molecule, blocks fusion and differentiation of rat myoblasts. J Cell Biol. 1993;123:467–475. doi: 10.1083/jcb.123.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ordonez C, Screaton RA, Ilantzis C, Stanners CP. Human carcinoembryonic antigen functions as a general inhibitor of anoikis. Cancer Res. 2000;60:3419–3424. [PubMed] [Google Scholar]
- 11.Screaton RA, DeMarte L, Draber P, Stanners CP. The specificity for the differentiation blocking activity of carcinoembryonic antigen resides in its glycophosphatidyl-inositol anchor. J Cell Biol. 2000;150:613–626. doi: 10.1083/jcb.150.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soeth E, Wirth T, List HJ, et al. Controlled ribozyme targeting demonstrates an antiapoptotic effect of carcinoembryonic antigen in HT29 colon cancer cells. Clin Cancer Res. 2001;7:2022–2030. [PubMed] [Google Scholar]
- 13.Taheri M, Saragovi HU, Stanners CP. The adhesion and differentiation-inhibitory activities of the immunoglobulin superfamily member, carcinoembryonic antigen, can be independently blocked. J Biol Chem. 2003;278:14632–14639. doi: 10.1074/jbc.M212500200. [DOI] [PubMed] [Google Scholar]
- 14.Blumenthal RD, Osorio L, Hayes MK, Horak ID, Hansen HJ, Goldenberg DM. Carcinoembryonic antigen antibody inhibits lung metastasis and augments chemotherapy in a human colonic carcinoma xenograft. Cancer Immunol Immunother. 2005;54:315–327. doi: 10.1007/s00262-004-0597-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hostetter RB, Augustus LB, Mankarious R, et al. Carcinoembryonic antigen as a selective enhancer of colorectal cancer metastasis. J Natl Cancer Inst. 1990;82:380–385. doi: 10.1093/jnci/82.5.380. [DOI] [PubMed] [Google Scholar]
- 16.Thomas P, Gangopadhyay A, Steele G, Jr, et al. The effect of transfection of the CEA gene on the metastatic behavior of the human colorectal cancer cell line MIP-101. Cancer Lett. 1995;92:59–66. doi: 10.1016/0304-3835(95)03764-n. [DOI] [PubMed] [Google Scholar]
- 17.Samara RN, Laguinge LM, Jessup JM. Carcinoembryonic antigen inhibits anoikis in colorectal carcinoma cells by interfering with TRAIL-R2 (DR5) signaling. Cancer Res. 2007;67:4774–4782. doi: 10.1158/0008-5472.CAN-06-4315. [DOI] [PubMed] [Google Scholar]
- 18.Screaton RA, Penn LZ, Stanners CP. Carcinoembryonic antigen, a human tumor marker, cooperates with Myc and Bcl-2 in cellular transformation. J Cell Biol. 1997;137:939–952. doi: 10.1083/jcb.137.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camacho-Leal P, Stanners CP. The human carcinoembryonic antigen (CEA) GPI anchor mediates anoikis inhibition by inactivation of the intrinsic death pathway. Oncogene. 2008;27:1545–1553. doi: 10.1038/sj.onc.1210789. [DOI] [PubMed] [Google Scholar]
- 20.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 21.Mishra L, Shetty K, Tang Y, Stuart A, Byers SW. The role of TGF-beta and Wnt signaling in gastrointestinal stem cells and cancer. Oncogene. 2005;24:5775–5789. doi: 10.1038/sj.onc.1208924. [DOI] [PubMed] [Google Scholar]
- 22.Akhurst RJ. TGF beta signaling in health and disease. Nat Genet. 2004;36:790–792. doi: 10.1038/ng0804-790. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 24.Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 26.Elliott RL, Blobe GC. Role of transforming growth factor Beta in human cancer. J Clin Oncol. 2005;23:2078–2093. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 27.Hoosein NM, McKnight MK, Levine AE, et al. Differential sensitivity of subclasses of human colon carcinoma cell lines to the growth inhibitory effects of transforming growth factor-beta 1. Exp Cell Res. 1989;181:442–453. doi: 10.1016/0014-4827(89)90101-8. [DOI] [PubMed] [Google Scholar]
- 28.Chakrabarty S, Tobon A, Varani J, Brattain MG. Induction of carcinoembryonic antigen secretion and modulation of protein secretion/expression and fibronectin/laminin expression in human colon carcinoma cells by transforming growth factor-beta. Cancer Res. 1988;48:4059–4064. [PubMed] [Google Scholar]
- 29.Han SU, Kwak TH, Her KH, et al. CEACAM5 and CEACAM6 are major target genes for Smad3-mediated TGF-beta signaling. Oncogene. 2008;27:675–683. doi: 10.1038/sj.onc.1210686. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Kumar KG, Tang W, Spiegelman VS, Fuchs SY. Negative regulation of prolactin receptor stability and signaling mediated by SCF(beta-TrCP) E3 ubiquitin ligase. Mol Cell Biol. 2004;24:4038–4048. doi: 10.1128/MCB.24.9.4038-4048.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wysocka J, Reilly PT, Herr W. Loss of HCF-1-chromatin association precede temperature-induced growth arrest of tsBN67 cells. Mol Cell Biol. 2001;21:3820–3829. doi: 10.1128/MCB.21.11.3820-3829.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F, Adam L, Vadlamudi RK, et al. p21-activated kinase 1 interacts with and phosphorylates histone H3 in breast cancer cells. EMBO Rep. 2002;3:767–773. doi: 10.1093/embo-reports/kvf157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jessup JM, Petrick AT, Toth CA, et al. Carcinoembryonic antigen: enhancement of liver colonisation through retention of human colorectal carcinoma cells. Br J Cancer. 1993;67:464–470. doi: 10.1038/bjc.1993.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fournes B, Sadekova S, Turbide C, Letourneau S, Beauchemin N. The CEACAM1-L Ser503 residue is crucial for inhibition of colon cancer cell tumorigenicity. Oncogene. 2001;20:219–230. doi: 10.1038/sj.onc.1204058. [DOI] [PubMed] [Google Scholar]
- 35.Huber M, Izzi L, Grondin P, et al. The carboxyl-terminal region of biliary glycoprotein controls its tyrosine phosphorylation and association with protein-tyrosine phosphatases SHP-1 and SHP-2 in epithelial cells. J Biol Chem. 1999;274:335–344. doi: 10.1074/jbc.274.1.335. [DOI] [PubMed] [Google Scholar]
- 36.Luo W, Wood CG, Earley K, Hung MC, Lin SH. Suppression of tumorigenicity of breast cancer cells by an epithelial cell adhesion molecule (C-CAM1): the adhesion and growth suppression are mediated by different domains. Oncogene. 1997;14:1697–1704. doi: 10.1038/sj.onc.1200999. [DOI] [PubMed] [Google Scholar]
- 37.Camacho-Leal P, Zhai AB, Stanners CP. A co-clustering model involving alpha5beta1 integrin for the biological effects of GPI-anchored human carcinoembryonic antigen (CEA) J Cell Physiol. 2007;211:791–802. doi: 10.1002/jcp.20989. [DOI] [PubMed] [Google Scholar]
- 38.Ordonez C, Zhai AB, Camacho-Leal P, Demarte L, Fan MM, Stanners CP. GPI-anchored CEA family glycoproteins CEA and CEACAM6 mediate their biological effects through enhanced integrin alpha5beta1-fibronectin interaction. J Cell Physiol. 2007;210:757–765. doi: 10.1002/jcp.20887. [DOI] [PubMed] [Google Scholar]
- 39.Laguinge LM, Samara RN, Wang W, et al. DR5 receptor mediates anoikis in human colorectal carcinoma cell lines. Cancer Res. 2008;68:909–917. doi: 10.1158/0008-5472.CAN-06-1806. [DOI] [PubMed] [Google Scholar]
- 40.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 41.Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 42.van der Bij GJ, Oosterling SJ, Meijer S, Beelen RH, van Egmond M. Therapeutic potential of Kupffer cells in prevention of liver metastases outgrowth. Immunobiology. 2005;210:259–265. doi: 10.1016/j.imbio.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 43.Kossmann T, Manthey CL, Brandes ME, et al. Kupffer cells express type I TGF-beta receptors, migrate to TGF-beta and participate in streptococcal cell wall induced hepatic granuloma formation. Growth Factors. 1992;7:73–83. doi: 10.3109/08977199209023939. [DOI] [PubMed] [Google Scholar]
- 44.Shibata S, Raubitschek A, Leong L, et al. A phase I study of a combination of yttrium-90-labeled anti-carcinoembryonic antigen (CEA) antibody and gemcitabine in patients with CEA-producing advanced malignancies. Clin Cancer Res. 2009;15:2935–2941. doi: 10.1158/1078-0432.CCR-08-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lutterbuese R, Raum T, Kischel R, et al. Potent control of tumor growth by CEA/CD3-bispecific single-chain antibody constructs that are not competitively inhibited by soluble CEA. J Immunother. 2009;32:341–352. doi: 10.1097/CJI.0b013e31819b7c70. [DOI] [PubMed] [Google Scholar]
- 46.Blumenthal RD, Hansen HJ, Goldenberg DM. In vitro and in vivo anticancer efficacy of unconjugated humanized anti-CEA monoclonal antibodies. Br J Cancer. 2008;99:837–838. doi: 10.1038/sj.bjc.6604548. author reply 839–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Govindan SV, Cardillo TM, Moon SJ, Hansen HJ, Goldenberg DM. CEACAM5-targeted therapy of human colonic and pancreatic cancer xenografts with potent labetuzumab-SN-38 immunoconjugates. Clin Cancer Res. 2009;15:6052–6061. doi: 10.1158/1078-0432.CCR-09-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]