

Abstract
Solar UVA exposure plays a causative role in skin photoaging and photocarcinogenesis. Here we describe the proteomic identification of novel UVA-targets in human dermal fibroblasts following a 2D-DIGE (two-dimensional-difference-gel-electrophoresis) approach. Fibroblasts were exposed to non-cytotoxic doses of UVA or left untreated, and total protein extracts underwent CyDye-labeling followed by 2D-DIGE/mass spectrometric identification of differentially expressed proteins, confirmed independently by immunodetection. The protein displaying the most pronounced UVA-induced upregulation was identified as the nucleolar protein nucleophosmin. The protein undergoing the most pronounced UVA-induced downregulation was identified as cathepsin B, a lysosomal cysteine-protease displaying loss of enzymatic activity and altered maturation after cellular UVA exposure. Extensive lysosomal accumulation of lipofuscin-like autofluorescence and osmiophilic material occurred in UVA-exposed fibroblasts as detected by confocal fluorescence microscopy and transmission electron microscopy, respectively. Array analysis indicated UVA-induced upregulation of oxidative stress response gene expression, and UVA-induced loss of cathepsin B enzymatic activity in fibroblasts was suppressed by antioxidant intervention. Pharmacological cathepsin B-inhibition using CA074Me mimicked UVA-induced accumulation of lysosomal autofluorescence and deficient cathepsin B-maturation. Taken together, these data support the hypothesis that cathepsin B is a crucial target of UVA-induced photooxidative stress causatively involved in dermal photodamage through impairment of lysosomal removal of lipofuscin.
Introduction
Most of the solar UV energy incident on human skin derives from the deeply penetrating UVA region (> 95%, 320-400 nm). In contrast to the formation of mutagenic photoproducts including epidermal cyclobutane pyrimidine dimers through direct absorption of UVB (290 - 320 nm) radiation by skin cell DNA (1), UVA radiation results in little photoexcitation of DNA directly, and cutaneous generation of reactive oxygen species (ROS) and organic free radicals has been implicated in photo-oxidative mechanisms of UVA-induced skin damage contributing to photoaging and photocarcinogenesis (reviewed in (2-7)). Various molecular sources of ROS and redox dysregulation are thought to contribute to the generation of cutaneous photooxidative stress including NAD(P)H oxidase, endogenous photosensitizers, mitochondrial electron leakage, energy crisis and glycolytic blockade, lysosomal disruption upstream of iron dysregulation, and inflammatory signaling (8-17). However, identification of novel molecular targets of UVA mechanistically involved in the causation of skin cell photodamage remains an important subject of ongoing research.
Proteomic analysis using two-dimensional difference gel electrophoresis (2D-DIGE) in combination with mass spectrometry has been used successfully for the unbiased identification of novel molecular targets involved in various human pathologies including infectious diseases, cancer, neurodegeneration, inflammatory dysregulation, metabolic disease, and general chronological aging (18,19). DIGE is a variant of two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) involving two or more separate protein samples that are covalently labeled with different fluorescent dyes and then mixed prior to PAGE analysis (20). Using advanced fluorescent gel imaging technology, 2D-DIGE enables proteomic detection of differences in protein abundance between samples, which is then followed by mass spectrometric identification of specific proteins displaying differential abundance between samples. In this study, we have used 2D-DIGE combined with mass spectrometry as a photobiological discovery tool validated by immunodetection and biochemical analysis. Here we report the proteomic identification of the lysosomal protease cathepsin B as a novel UVA-target in cultured human skin fibroblasts.
Materials and Methods
Chemicals
CA074Me was purchased from Enzo Life Sciences (Plymouth Meeting, PA), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), and LysoTracker Red™ were purchased from Invitrogen (Carlsbad, CA). All other chemicals were from Sigma Chemical Co. (St. Louis, MO).
Cell Culture
Dermal neonatal foreskin Hs27 fibroblasts from ATCC (Manassas, VA) were cultured in DMEM containing 10% bovine calf serum. Cells were maintained at 37°C in 5% CO2, 95% air in a humidified chamber.
Irradiation with solar UVA
A KW large area light source solar simulator, model 91293, from Oriel Corporation (Stratford, CT) was used, equipped with a 1000 W Xenon arc lamp power supply, model 68920, and a VIS-IR bandpass blocking filter plus UVB and C blocking filter (output 320–400 nm plus residual 650–800 nm, for UVA). The output was quantified using a dosimeter from International Light Inc. (Newburyport, MA), model IL1700, with a SED033 detector for UVA (range 315–390 nm, peak 365 nm), at a distance of 365 mm from the source, which was used for all experiments. Using UVB/C blocking filter, the dose at 365 mm from the source was 5.39 mJ cm-2 sec-1 UVA radiation with a residual UVB dose of 3.16 μJ cm-2 sec-1.
Chronic UVA exposure regimens
For chronic UVA treatment, an exposure regimen was selected that delivered a physiologically relevant dose of UVA without causing compromised cell viability or altered proliferative rate after reseeding.
‘Three week’ UVA regimen (used for DIGE- and DIGE-related experimentation): Cells were exposed to 9.9 J/cm2 UVA twice a week for a total of 18 days (59.4 J/cm2 total UVA dose). Viability was maintained throughout the duration of the experiment as confirmed by flow cytometric analysis of annexinV/ propidium iodide-stained cells performed as published earlier (21)(data not shown). Moreover, after completion of the irradiation regimen, proliferation rate based on cumulative population doublings monitored over ten days after reseeding [(control: 4.8 ± 0.1; UVA: 4.7 ± 0.3 (mean ± SD)] was unaltered. In addition, no experimental evidence of UV-induced cellular senescence was obtained using SA-β-galactosidase staining performed according to a published standard procedure (22)(data not shown).
‘One week’ UVA regimen [used for exposure that required inclusion of pharmacological modulators (NAC and CA074Me) that would display cytotoxicity over the length of the three week regimen]: Cells were exposed to 9.9 J/cm2 UVA for four consecutive days (39.6 J/cm2 total UVA dose).
In both regimens, cells were seeded at 5×105 cells/ 100 mm dish and incubated overnight prior to UVA exposure. Before each irradiation, cells were first washed with PBS and irradiated (10 ml PBS, 30 min). After irradiation, PBS was removed and fresh culture medium was added. For mock UVA treatments, cells were washed with PBS, placed in 10 ml PBS and then incubated at room temperature in the dark for 30 min. For analysis, cells were harvested one hour after last UVA exposure occurred.
2D-DIGE (two-dimensional difference gel electrophoresis) and mass spectrometry
Sample preparation
After UVA exposure according to the ‘three week’ regimen, UVA-exposed and mock treated cells (7 × 106 per group) were harvested by scraping and washed with PBS. The pellet was frozen immediately on dry ice. For sample processing, 200 ml 2-D cell lysis buffer (30 mM Tris-HCl, pH 8.8, containing 7 M urea, 2 M thiourea and 4% CHAPS) were added to the thawed samples followed by sonication at 4 °C. After centrifugation (14,000 rpm, 4 °C, 30 min) the supernatant was collected. Protein concentration was determined using the Bio-Rad assay and the lysate samples were diluted with the sample 2-D cell lysis buffer to the same protein concentration between 5 to 8 mg/ml. 2-D DIGE and mass spectrometric analysis were then performed in collaboration with Applied Biomics (Hayward, CA).
Minimal CyDye labeling
To 30 mg of cell lysate 1.0 ml of diluted CyDye (1:5 diluted with DMF; control: Cy3; UVA: Cy5; GE Health Care, Piscataway, NJ) was added and after vortexing the tube was kept on ice for 30 min. After addition of 1.0 ml of 10 mM L-lysine to each of the samples samples were incubated on ice for an additional 15 min. After mixing Cy3 and Cy5 labeled samples and addition of 2× 2-D sample buffer (8 M urea, 4% CHAPS, 20 mg/ml DTT, 2% pharmalytes and trace amount of bromophenol blue), 100 ml destreak solution (GE Healthcare) and rehydration buffer (7 M urea, 2 M thiourea, 4% CHAPS, 20 mg/ml DTT, 1% pharmalytes and trace amount of bromophenol blue) were added (250 ml final volume). After mixing and spinning the labeled samples were loaded into the strip holder [13 cm immobilized pH gradient (IPG) strip, Amersham BioSciences].
IEF and SDS-PAGE
After IEF according to the manufacturer's protocol (Amersham BioSciences), the IPG strips were incubated in the freshly prepared equilibration buffer 1 (50 mM Tris-HCl, pH 8.8, containing 6 M urea, 30% glycerol, 2% SDS, a trace amount of bromophenol blue and 10 mg/ml DTT) for 15 min. The strips were rinsed in the fresh made equilibration buffer 2 (50 mM Tris-HCl, pH 8.8, containing 6 M urea, 30% glycerol, 2% SDS, a trace amount of bromophenol blue and 45 mg/ml iodacetamide) for 10 min. The IPG strips were then rinsed once in the SDS-gel running buffer before transfer into the SDS-gel (12% SDS-gel prepared using low fluorescent glass plates) and sealed with 0.5% (w/v) agarose solution (in SDS-gel running buffer). The SDS-gel was run at 15 °C.
Protein identification by mass spectrometry
Spots of interest were picked using the Ettan Spot Picker (Amersham BioSciences) based on in-gel analysis and spot picking design performed using the DeCyder software. Gel spots were digested in-gel with modified porcine trypsin protease (Trypsin Gold, Promega). The digested tryptic peptides were desalted by Zip-tip C18 (Millipore). Peptides were eluted from the Zip-tip with 0.5 μl of matrix solution (a-cyano-4-hydroxycinnamic acid; 5 mg/ml in 50% acetonitrile, 0.1% trifluoroacetic acid, 25 mM ammonium bicarbonate) and spotted on the MALDI plate (model ABI 01-192-6-AB). MALDI-TOF MS and TOF/TOF tandem MS/MS were performed on an ABI 4700 mass spectrometer (Applied Biosystems, Framingham, MA). MALDI-TOF mass spectra were acquired in reflectron positive ion mode, averaging 4000 laser shots per spectrum. TOF/TOF tandem MS fragmentation spectra were acquired for each sample, averaging 4000 laser shots per fragmentation spectrum on each of the 10 most abundant ions present in each sample (excluding trypsin autolytic peptides and other known background ions).
Database search
Both the resulting peptide mass and the associated fragmentation spectra were submitted to a GPS Explorer workstation equipped with MASCOT search engine (Matrix science) capabilities to search the database of the National Center for Biotechnology Information non-redundant (NCBInr). Searches were performed without constraining protein molecular weight or isoelectric point, with variable carbamidomethylation of cysteine and oxidation of methionine residues, and with one missed cleavage allowed in the search parameters. Candidates with either protein score C.I.% or Ion C.I.% greater than 95 were considered significant.
Cathepsin B enzymatic activity
Cathepsin B activity was measured using the fluorimetric cathepsin B assay kit from BioVision, Inc. (Mountain View, CA) according to manufacturer's instructions. Cells (7×105) were lysed in 0.5 ml of chilled lysis buffer. After 10 min incubation on ice, lysates were centrifuged at 10,000 g at 4 °C for 5 min and supernatant was retained for analysis. 50 μL of cell lysate was incubated with 50 μL of reaction buffer and cathepsin B substrate (Ac-Arg-Arg-AFC; 200 μM final concentration; 1 h at 37 °C). As a negative control, analysis was performed in the presence of the cathepsin B inhibitor Z-Phe-Phe-FMK (200 μM final concentration). The release of free amino-4-trifluoromethylcoumarin (AFC) was measured using a fluorescence plate reader (λex 400, λem 505; SpectraMax Gemini, Molecular Devices, Sunnyvale, CA). Additionally, protein concentration of cell lysates was determined using the Pierce™ BCA Protein Assay Kit (ThermoScientific, Rockford, IL), and cathepsin B activity was normalized to protein concentration per sample.
Measurement of Cathepsin L activity
Cathepsin L activity was measured using the fluoremetric cathespin L activity assay kit from BioVision, Inc. according to manufacturer's instructions. Processing of samples and assay protocol was identical to the cathepsin B activity assay except the cathepsin L substrate (Ac-Phe-Arg-AFC; 200 μM final concentration) was used.
Glyceraldehyde dehydrogenase activity assay
UVA-induced alteration of GAPDH specific enzymatic activity was assessed in cytosolic cell extracts prepared from Hs27 fibroblasts (5 × 106 per sample) according to a published standard procedure measuring increase in absorbance at 340 nm (formation of NADH) in a reaction mixture containing 0.4 mM NAD+, 50 mM sodium arsenate, 0.1 mM DTPA, 0.9 mM D,L-glyceraldehyde-3-phosphate, and 50 mM TrisHCl, pH 8.8. (23). Reaction was started by the addition of cytosolic extract normalized for protein content using the BCA protein assay. One unit of enzyme was defined as the amount forming 1 μmol/min NADH at 25° C.
Gene expression analysis by real time RT-PCR
One hour after last UVA exposure, total cellular RNA (5×106 cells) was prepared using the RNEasy kit from Qiagen (Valencia, CA). Reverse transcription was performed using TaqMan Reverse Transcription Reagents (Roche Molecular Systems, Branchburg, NJ) and 200 ng of total RNA in a 50 μl reaction. Reverse transcription was primed with random hexamers and incubated at 25°C for 10 min followed by 48°C for 30 min, 95°C for 5 min, and a chill at 4°C. Each PCR reaction consisted of 3.75 μl of cDNA added to 12.5 μl of TaqMan Universal PCR Master Mix (Roche Molecular Systems), 1.25 μl of gene-specific primer/probe mix [Assays-by-Design; Applied Biosystems: CTSB (assay ID Hs00947433_m1), CSTA (assay ID HS00193257_m1), CSTB (assay ID Hs00164368_m1), CSTC (assay ID Hs00969174_m1), CST6 (assay ID Hs00154599_m1), NPM1 (assay ID Hs01576587_g1) or GAPDH (assay ID Hs99999905_m1)] and 7.5 μl of PCR water. PCR conditions were: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s alternating with 60°C for 1 min (Applied Biosystems 7000 SDSGene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method described in the ABI Prism 7000 sequence detection system user guide. Expression values were averaged across three independent experiments (mean ± SD).
Human Stress and Toxicity PathwayFinder™ RT2 Profiler™ PCR Expression Array
Expression array analysis was performed as published recently (24). One hour after last UVA exposure, total cellular RNA (5 × 106 cells) was prepared using the RNeasy kit. Reverse transcription was performed using the RT2 First Strand kit (SA Biosciences, Frederick, MD) and 1 μg total RNA. The Human Stress and Toxicity PathwayFinder™ RT2 Profiler™ PCR Expression Array (SA Biosciences) profiling the expression of 84 stress- and toxicity-related genes was run using the following PCR conditions: 95 °C for 10 min, followed by 40 cycles of 95°C for 15 s alternating with 60 °C for 1 min (Applied Biosystems 7000 SDS, Foster City, CA). Gene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method as described in the ABI Prism 7000 sequence detection system user guide. Expression values were averaged across three independent array experiments (mean ± SD).
Detection of intracellular oxidative stress by flow cytometric analysis
Induction of intracellular oxidative stress by photosensitization was analyzed by flow cytometry using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) as a sensitive nonfluorescent precursor dye according to a published standard procedure (15). One hour after the last irradiation, DCFH-DA (5 μg/mL final concentration) was added to the culture medium and cells were incubated for 1 h in the dark (37°C, 5% CO2). Cells were harvested, washed with PBS, resuspended in 300 μl PBS and immediately analyzed by flow cytometry. To avoid direct photooxidation of the dye probe, cells were loaded with the indicator dye under light exclusion.
Flow cytometric quantification of cellular autofluorescence
One hour after the last irradiation, cells were harvested, washed with PBS, resuspended in 300 μL PBS, and immediately analyzed by flow cytometry (λex 488nm, λem 530 ± 15 nm).
Confocal Fluorescence Microscopy
One hour after last irradiation or CA074Me treatment, cells were trypsinized, reseeded on glass bottom 35 mm dishes (MatTek Corp., Ashland, MA) at 1×105 cells per dish and cultured overnight. Prior to live imaging, cells were incubated in LysoTracker Red DND-99 (75 nM in growth medium) for 1 h at 37 °C/ 5% CO2. Medium was removed and cells were incubated in DAPI (3 μM in HBSS) for 30 min. Cells were then washed several times and kept in HBSS for fluorescence microscopy. Using a SP5 spectral confocal microscopy system equipped with a Leica DMI6000 inverted microscope (Wetzlar, Germany), DAPI was detected between 450-550 nm with excitation at 405nm (UV laser source). Autofluorescence and LysoTracker Red were visualized using an argon laser (λex 488 nm), and a spectral scan (560-700 nm with 10 nm increments) was performed. Image analyses were performed using Leica Confocal Imaging software and distinction between fluorescent signals was accomplished by spectral separation.
Transmission Electron Microscopy
One hour after last UVA irradiation, cells were trypsinized, reseeded and cultured for 4h. Cells were fixed in situ with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH7.4), postfixed in 1% osmium tetroxide in cacodylate buffer, washed, scraped and pelleted. Cells were then stained in 2% aqueous uranyl acetate, dehydrated through a graded series (50,70, 90 and 100%) of ethanol and infiltrated with Spurr's resin, then allowed to polymerize overnight at 60 °C. Sections (50 nm) were cut, mounted onto uncoated 150 mesh copper grids, and stained with 2% lead citrate. Sections were examined in a CM12 Transmission Electron Microscope (FEI, Hillsboro, OR) operated at 80 kV with digital image collection (AMT, Danvers, MA).
Immunoblot detection
One hour after last irradiation, cells were washed with PBS, lysed in 1× SDS-PAGE sample buffer and heated for 3 min at 95°C. Samples were separated by 12% SDS-PAGE followed by transfer to nitrocellulose membranes (Optitran, Whatman, Piscataway, NJ). Membranes were incubated with primary antibody in 5% milk-PBST overnight at 4°C. HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibody (Jackson Immunological Research, West Grove, PA) was used at 1:20,000 in 5% milk-PBST followed by visualization using enhanced chemiluminescence detection reagents. Equal protein loading was examined by α-actin-detection. The following primary antibodies were used: rabbit anti-cathepsin B polyclonal antibody, 1:200 (BioVision, Inc.); rabbit anti-nucleophosmin polyclonal antibody, 1:1,000 (Cell Signaling Technology, Danvers, MA); rabbit anti-HO-1 polyclonal antibody, 1:1,500 (Stressgen Bioreagents, Ann Arbor, MI); rabbit anti-Hsp70 polyclonal antibody, 1:1,000 (Stressgen Bioreagents); mouse anti-actin monoclonal antibody, 1:1,500 (Sigma).
Statistical analysis
The results are presented as means (± SEM) of at least three independent experiments. All data were analyzed employing one-way analysis of variance (ANOVA) with Tukey's post hoc test using the Prism 4.0 software unless specified otherwise. Differences were considered significant at p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001).
Results
Nucleophosmin and cathepsin B are targets of chronic UVA exposure in human skin fibroblasts as revealed by two-dimensional difference gel electrophoresis (2D-DIGE) followed by mass spectrometry
First, proteomic changes that occur in human fibroblasts in response to chronic UVA exposure were examined using 2D-DIGE technology (Fig. 1).
Figure 1. UVA-induced proteome changes in human skin fibroblasts as analyzed by two-dimensional difference gel electrophoresis (2D-DIGE).
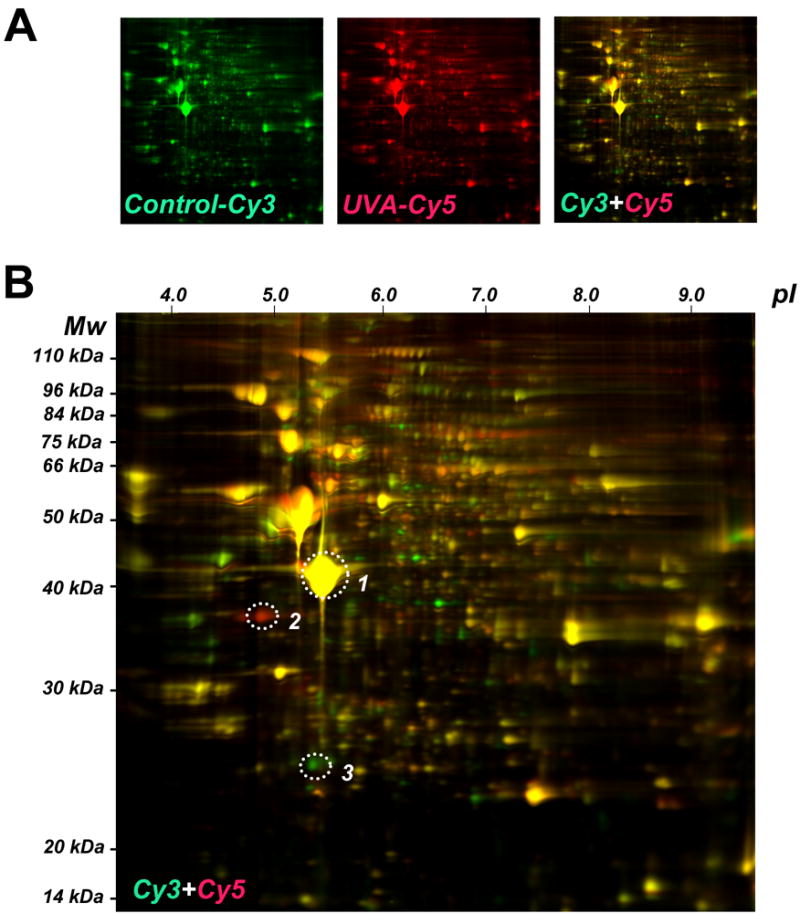
(A) After chronic UVA exposure (9.9 J/cm2, twice a week, three weeks, termed ‘three week regimen’) or mock treatment, total cell protein extracts were prepared followed by Cy3- (untreated control) and Cy5-labeling (chronic UVA exposure). After gel electrophoresis differential fluorescence image analysis was performed: Cy3-image (left panel) Cy5-image (middle panel), Cy3/Cy5-overlay (right panel). (B) Cy3/Cy5-overlay with molecular weight (Mw) and isolectric point (pI) scales. Fluorescent spots displaying the highest UVA-induced expression differential [upregulation: spot #2 (red); downregulation: spot #3 (green)] were then excised for subsequent mass spectrometric analysis. A yellow reference spot displaying equal abundance in both protein samples is also indicated (spot #1).
A regimen of chronic UVA exposure was selected that delivered a physiologically relevant dose without induction of cell death or inhibition of cellular proliferation (as detailed in Materials and methods). To this end, after chronic UVA exposure (9.9 J/cm2, twice a week, over three weeks; ‘three week UVA regimen’) or mock treatment, total cell protein extracts were prepared followed by Cy3- (untreated control) and Cy5-labeling (chronic UVA exposure) and subjected to gel electrophoresis followed by differential fluorescence image analysis (Cy3/Cy5-overlay shown in Fig. 1A, right panel, enlarged in Fig. 1B) using the DeCyder software (Fig. 2A and Fig. 3A).
Figure 2. Nucleophosmin upregulation in UVA-exposed human skin fibroblasts as detected by 2D-DIGE mass spectrometry and immunoblot analysis.
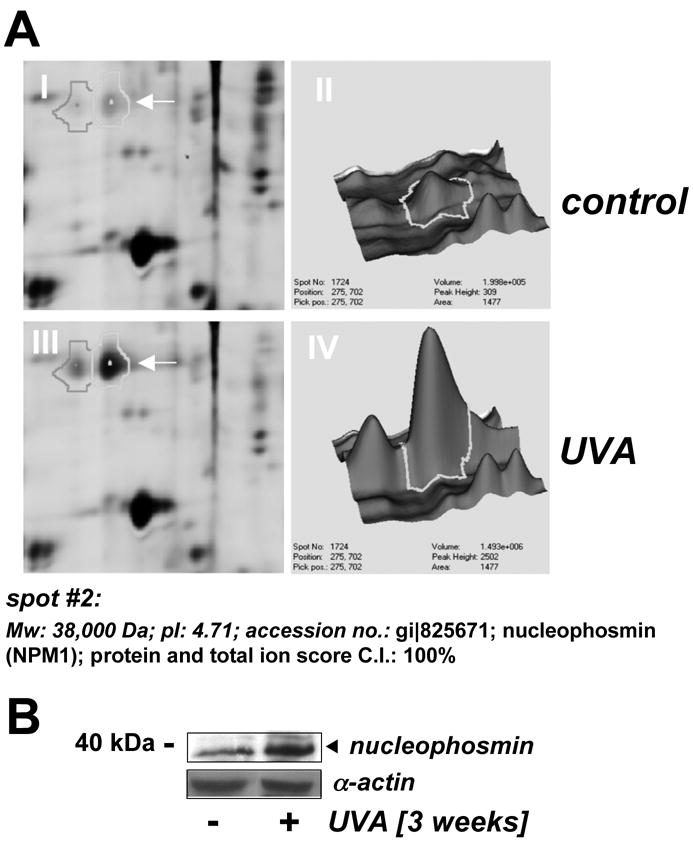
(A) Differential in-gel analysis (gel spot view: panels I and III; 3D-view: panels II and IV) was performed using the DeCyder software. The arrow indicates position of protein spot #2 from Fig. 1B (untreated: panels I and II; UVA-exposed: panels III and IV). Spot #2 was picked and identified as nucleophosmin using mass spectrometric analysis (see Materials and methods). (B) Upregulation of cellular nucleophosmin protein levels in response to UVA exposure was confirmed by Western blotting as described in Materials and methods.
Figure 3. Cathepsin B downregulation in UVA-exposed human skin fibroblasts as detected by 2D-DIGE mass spectrometry and immunoblot analysis occurs with loss of specific enzymatic activity.
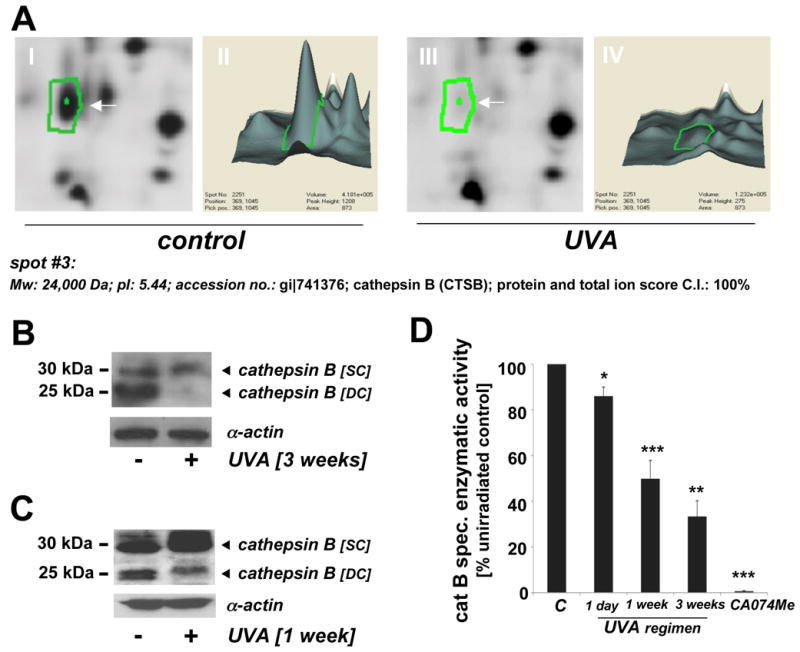
(A) Differential in-gel analysis (gel spot view: panels I and III; 3D-view: panels II and IV) was performed using the DeCyder software. The arrow indicates position of protein spot #3 from Fig. 1B (untreated: panels I and II; UVA-exposed: panels III and IV). Spot #3 was picked and identified as cathepsin B using mass spectrometric analysis (see Materials and Methods). (B) Immunoblot detection of cellular cathepsin B protein levels [double chain (DC) and single chain (SC) form] in UVA-treated (59.4 J/cm2 total dose; ‘3 week’ UVA regimen) versus untreated control cells was performed as described in Experimental Procedures. (C) Immunoblot detection of cellular cathepsin B protein levels in UVA-treated (‘1 week’ UVA regimen) and control cells. (D) Loss of cathepsin B specific enzymatic activity in human skin fibroblasts exposed to UVA exposure (1 day to 3 weeks) was detected using a fluorigenic enzyme substrate as described in Experimental Procedures. Treatment with the cathepsin B inhibitor CA074Me (1μM, q.d., four consecutive days) served as a positive control (n=3, mean ± SEM; p<0.05).
Fluorescent spots displaying the highest UVA-induced expression differential [upregulation: spot #2 (red); downregulation: spot #3 (green); Fig. 1B] were excised for subsequent mass spectrometric analysis. Additionally, spot #1 displaying equal abundance in both protein samples was identified as β-actin [actin, cytoplasmic 1 (Mw: 42,000 Da; pI: 5.4; acc. no: gi∣46397333; Fig. 1B]. Spot #2 displaying upregulation by 3.6 fold according to DeCyder analysis was identified as nucleophosmin (Mw: 38,000 Da; pI: 4.71; accession number: gi∣825671) with protein score and total ion score confidence intervals (C.I.) being 100% (Fig. 2A). Independent confirmation of UVA-induced nucleophosmin upregulation was obtained by immunodetection that indicated approximately 3.1 fold upregulation based on densitometric analysis of band intensity (Fig. 2B).
Spot #3 displaying pronounced downregulation by almost 7.1 fold according to DeCyder quantitative analysis was identified as cathepsin B (Mw: 24,000 Da; pI: 5.44; accession number: gi∣741376) with protein score and total ion score confidence intervals (C.I.) being 100% (Fig. 3A). Independent confirmation of UVA-induced cathepsin B downregulation was obtained by immunodetection [cathepsin B double chain (DC; 24 kDa) and single chain (SC; 29 kDa) forms] (Fig. 3B and C). Densitometric analysis indicated downregulation of the fully processed mature DC form in UVA-treated cells (59.4 J/cm2 total dose; ‘three week’ UVA regimen) by approximately 10.7 fold, a number in good agreement with DIGE analysis (Fig. 3A). Significant downregulation was also detectable in cells that received UVA exposure according to a shorter UVA regimen (39.6 J/cm2 total dose; ‘one week’ UVA regimen). The ‘one week’ UVA regimen was therefore chosen in subsequent experiments (displayed in Figs. 4C-D and 5D) that required co-administration of pharmacological agents displaying cytotoxic effects over the course of the ‘3 week’ UVA regimen.
Figure 4. Gene expression changes and oxidative stress in UVA-exposed human skin fibroblasts that display impaired cathepsin B activity.
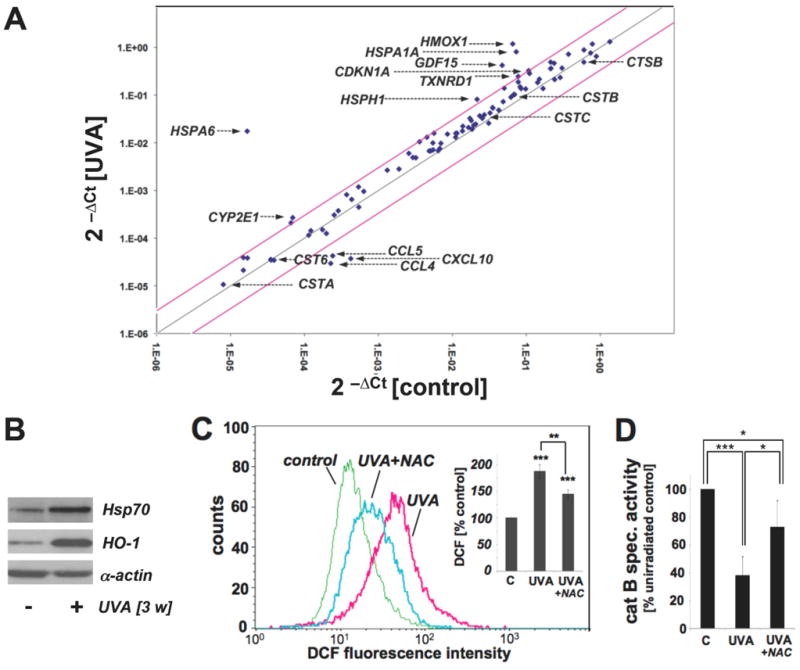
(A) Differential gene expression in response to chronic UVA exposure (conditions as in Fig. 1; ‘three week’ UVA regimen) or mock treatment was analyzed using the RT2 Human Stress and Toxicity Pathway Finder™ PCR Expression Array. Three independent repeat experiments were analyzed using the two-sided Student's t test as summarized in table 1. Changes in cycle threshold (Ct) for genes of interest relative to ACTB for untreated control (x-axis) versus UVA-exposed (y-axis) cells are displayed as scatter blot. Upper and lower lines represent the cut-off indicating three fold up- or down-regulated expression, respectively. Arrows mark genes of specific interest. (B) Induction of Hsp70 and HO-1 protein expression in UVA-exposed fibroblasts from (A) was determined by Western blot analysis using α-actin detection as a loading control. (C) Modulation of cellular oxidative stress was examined in UVA-exposed cells irradiated (‘one week’ UVA regimen) in the presence or absence of NAC (10 mM) by flow cytometric detection of DCF fluorescence. One representative histogram is shown. Bar graph depicts summarized data of three independent repeats (n=3, mean ± SEM; p<0.05). (D) UVA-induced changes in cathepsin B specific enzymatic activity were assessed in human skin fibroblasts (‘one week’ UVA regimen) irradiated in the presence or absence of NAC (10 mM) using a fluorigenic enzyme substrate as described in Experimental Procedures (n=3, mean ± SEM; p<0.05).
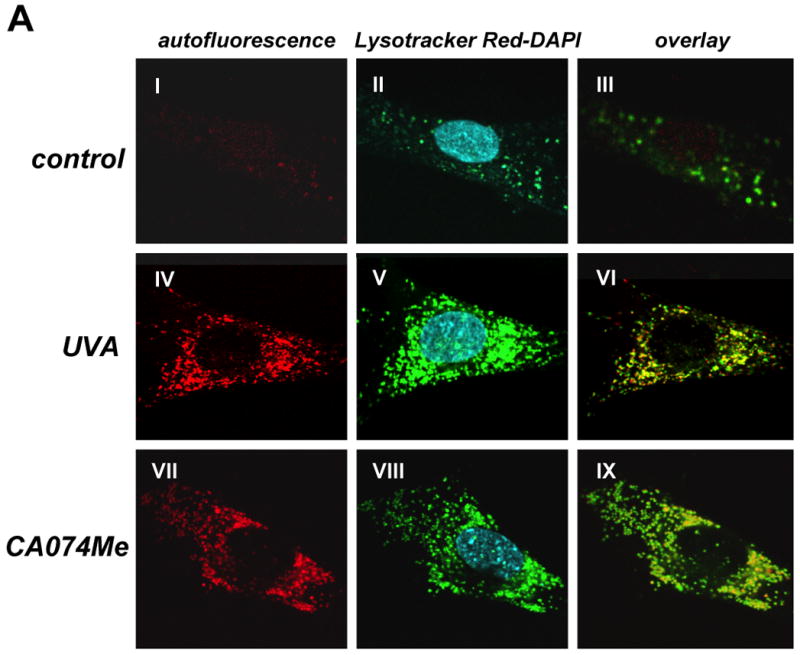
Figure 5. Pharmacological inhibition of cathepsin B mimics UVA-induced accumulation of lysosomal autofluorescence and deficient cathepsin B maturation in human skin fibroblasts.
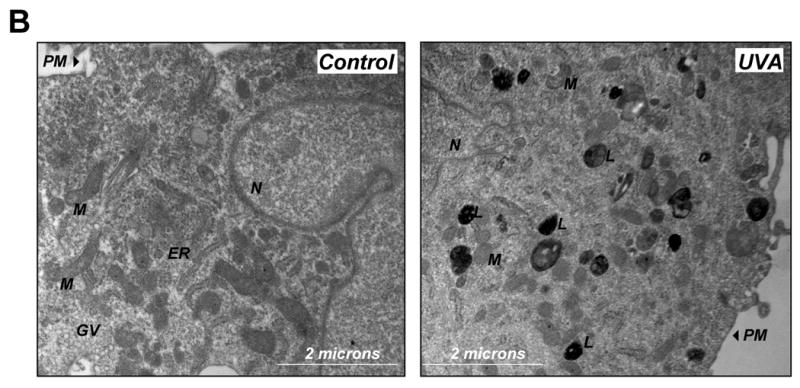
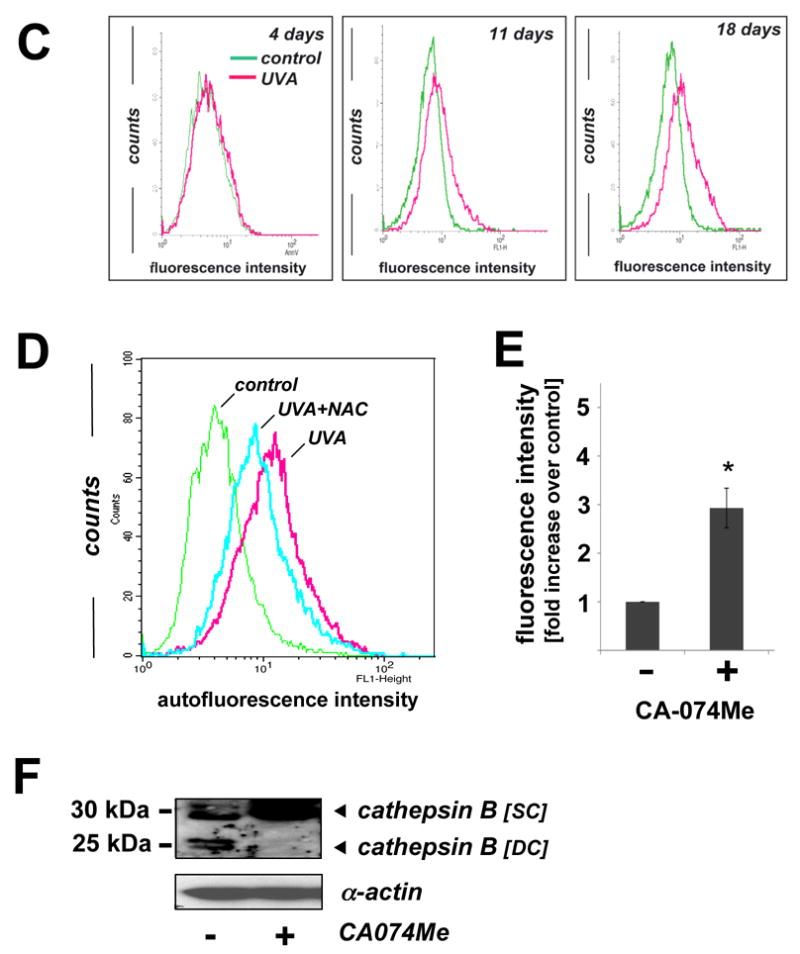
(A) After exposure to chronic UVA (59.4 J/cm2 total dose; ‘three week’ UVA regimen), cathepsin B inhibitor CA074Me (1 μM, q.d., 4 subsequent days), or mock treatment, cellular autofluorescence (panels I, IV, VII) and lysosomal staining (Lysotracker Red; panels II, V, VIII) were visualized by confocal microscopy as specified in Materials and methods. In addition, nuclei were stained using DAPI (panels II, V, VIII). Overlays of lysosomal staining and autofluorescence demonstrating colocalization are depicted in panels III, VI, and IX. (B) Cells [control and chronic UVA treated (59.4 J/cm2 total dose; ‘three week’ UVA regimen)] were examined by transmission electron microscopy (direct magnification: 8,800 x): L (Osmiophilic vesicles indicative of lysosomal lipofuscin accumulation; M (mitochondrion); N (nucleus); ER (endoplasmic reticulum); GV (Golgi vesicles). (C) The increase in UVA-induced autofluorescence intensity of cells treated as in panel (A) was examined over the course of the three week irradiation period by flow cytometric analysis. The histograms depict fluorescence intensity of UVA- and mock-treated cells after treatment for four (19.8 J/cm2 total dose; left panel), eleven (39.6 J/cm2 total dose; middle panel), and eighteen days (59.4 J/cm2 total dose; right panel) as specified in Materials and methods. One representative set of histograms out of three similar repeats is shown. (D) Exposure to UVA (39.6 J/cm2 total dose; ‘one week UVA regimen’) was performed in the presence or absence of NAC (10 mM, as specified in Fig. 4D). Cellular autofluorescence intensity was then quantified by flow cytometric analysis. One representative histogram out of three similar repeats is shown. (E) After exposure to the cathepsin B inhibitor CA074Me (treatment as for Fig. 5A, panels VII-IX), cellular autofluorescence intensity was quantified by flow cytometric analysis (n=3, mean ± SEM; p<0.05). (F) Deficient cathepsin B maturation as a result of pharmacological inhibition of cathepsin B. Immunoblot detection of cellular cathepsin B protein levels [double chain (DC) and single chain (SC) form] in CA074Me-treated (treatment as for Fig. 5A, panels VII-IX) versus untreated control cells was performed as described in Materials and methods.
Based on the detection of UVA-induced changes in cathepsin B protein levels (Fig. 3A-C), we examined the possibility that chronic UVA-exposure impairs cathepsin B specific enzymatic activity in human skin fibroblasts (Fig. 3D). As a positive control, the cathepsin B inhibitor CA074Me (1μM, q.d., four consecutive days) was used causing complete loss of cathepsin B specific enzymatic activity. After chronic UVA exposure of fibroblasts (‘three week’ UVA regimen, identical to treatment used for 2D-DIGE analysis), specific enzymatic activity of cathepsin B in total cellular extracts was reduced by more than 60% as compared to mock-treated control. A significant loss of cathepsin B activity by approximately 10% was already detectable after a single exposure to UVA (9.9 J/cm2), and inhibition by approximately 50% was observed when UVA exposure occurred over the course of four consecutive days (‘one week’ UVA regimen). In contrast, enzymatic activity of GAPDH, an active site cysteine-containing glycolytic enzyme, known to be a common target of cytotoxic oxidative insult including Rose Bengal-dependent photosensitization (25), was not changed significantly in response to UVA exposure (‘1 week’ regimen) [control: 3.3 ± 0.5 versus UVA: 3.4 ± 0.4 specific enzymatic activity (u/mg cytosolic protein; n=3; mean ± SD]. These data suggest that UVA-induced changes affecting cathepsin B structure and enzymatic activity may occur as a result of specific molecular mechanisms that do not cause the indiscriminate inactivation of other cysteine-dependent enzymes such as GAPDH.
Gene expression profiling reveals an oxidative stress response in UVA-exposed human skin fibroblasts
Modulation of gene expression in response to chronic UVA exposure [treatment conditions identical to 2D-DIGE experimentation shown in Fig. 1; ‘three week’ UVA regimen] versus mock treatment was assessed using the RT2 Human Stress and Toxicity Pathway Finder™ PCR Expression Array platform (Fig. 4A and table 1).
Table 1. Gene expression changes in human skin fibroblasts exposed to chronic UV.
(A) Genes displaying UVA-induced (‘three week’ UVA regimen) up- or downregulation of expression by at least threefold (p < 0.05) as detected using the RT2 Human Stress and Toxicity Pathway Finder™ PCR Expression Array shown in Fig. 4A. (B) Selected genes without significant expression changes in response to UVA exposure (p > 0.05).
| A | ||
|---|---|---|
| gene symbol | fold change | p-value |
| HSPA6 (NM_002155) | 1041.61 | 0.0002 |
| HMOX1 (NM_002133) | 18.05 | 0.0048 |
| HSPA1A (NM_005345) | 11.04 | 0.0026 |
| GDF15 (NM_004864) | 8.93 | 0.0011 |
| CYP2E1 (NM_000773) | 3.87 | 0.0130 |
| HSPH1 (NM_006644) | 3.76 | 0.0012 |
| TXNRD1 (NM_003330) | 3.16 | 0.0218 |
| CDKN1A (NM_000389) | 3.02 | 0.0080 |
| CCL5 (NM_002985) | -5.66 | 0.0128 |
| CCL4 (NM_002984) | -7.65 | 0.0037 |
| CXCL10 (NM_001565) | -11.35 | 0.0199 |
UVA treatment upregulated expression levels of eight genes on the array by at least three-fold including genes encoding the heat shock proteins Hsp70B' (HSPA6; 1042-fold), Hsp70 (HSPA1A; 11-fold), Hsp105 (HSPH1; 4-fold), the heat shock protein and antioxidant enzyme hemeoxygenase-1 (HMOX1; 18-fold), the antioxidant enzyme thioredoxin reductase 1 (TXNRD1; 3-fold), the cyclin dependent kinase inhibitor p21(WAF1) (CDKN1A; 3-fold), and the inflammatory transforming growth factor beta superfamily member GDF15 (GDF15; 9-fold). Of note, significant downregulation of chemokine encoding genes (CL5, CCL4, CXCL10) was observed in response to chronic UVA exposure. UVA-induced upregulation of major stress response encoding genes (HSPA1A and HMOX1) was then confirmed at the protein level by immunoblot detection that revealed 4.7 fold (Hsp70) and 9.5 fold (HO-1) upregulation as determined by densitometric analysis (Fig. 4B).
UVA-induced loss of cathepsin B enzymatic activity in human skin fibroblasts can be antagonized by the antioxidant N-acetyl-L-cysteine
Further evidence supporting the induction of cellular oxidative stress in response to chronic UVA treatment was obtained by quantitative detection of DCF-fluorescence after subjecting Hs27 cells to UVA treatment (‘one week’ UVA regimen) performed in the presence or absence of the thiol-based antioxidant N-acetyl-L-cysteine (NAC, 10 mM) (Fig. 4C). UVA exposure upregulated cellular DCF fluorescence intensity by almost 80% indicative of increased peroxide levels, an effect significantly suppressed by inclusion of NAC during irradiation. In parallel, loss of cathepsin B enzymatic activity observed earlier in response to UVA exposure (Fig. 3D) was antagonized significantly if irradiation occurred in the presence of NAC (Fig. 4D)
Cathepsin B inhibition using CA074Me mimics UVA-induced accumulation of lysosomal autofluorescence and deficient cathepsin B maturation in human skin fibroblasts
Earlier reports have demonstrated lysosomal accumulation of autofluorescent material (lipofuscin) originating from lipid peroxidation damage in oxidatively stressed human cells (26-30). Our data demonstrating UVA-induced oxidative stress (Fig. 4) and cathepsin B dysfunction (Fig. 3 and Fig. 4D) led us to examine generation of cellular autofluorescence indicative of lipofuscin accumulation in response to UVA exposure (Fig. 5A-C).
Visualization by confocal fluorescence microscopy revealed pronounced accumulation of autofluorescent material (λex 488 nm/ λem 553-611 nm) displaying a punctate cytosolic staining pattern in human fibroblasts observed after chronic UVA exposure (‘three week’ UVA regimen), but not in untreated control cells (Fig. 5A). Further studies employing lysotracker Red and nuclear DAPI staining confirmed cellular colocalization of autofluorescent chromophores and lysosomal organelles (Fig. 5A, overlay panels III, VI, and IX). In addition, substantial accumulation of lysosomal vesicles as obvious from intense lysotracker Red staining occurred in UVA-exposed fibroblasts (Fig. 5A, panel II versus panel V). Accumulation of autofluorescent material in response to chronic UVA exposure as shown in Fig. 5A was confirmed independently by flow cytometric analysis of the same treatment groups that revealed a time-dependent increase in fluorescence intensity during the course of this three week experiment (Fig. 5C) (31). Further flow cytometric analysis showed that UVA-induced increase in cellular autofluorescence was partially antagonized if irradiation occurred in the presence of the antioxidant NAC (Fig. 5D), consistent with an involvement of photooxidative mechanisms.
Lysosomal integrity seemed to be unaffected by UVA exposure as evidenced by maintenance of punctate staining observed by confocal microscopy (Fig. 5A, panel V-VI) and confirmed independently by electron microscopy (Fig. 5B) that indicated extensive cytosolic accumulation of membraneous vesicles containing osmiophilic material commonly referred to as lipofuscin (26). Interestingly, other ultrastructural changes beyond vesicular accumulation of osmiophilic lipofuscin material were observed as a result of UVA exposure including mitochondrial shrinkage and loss of functional endoplasmatic reticulum, structural changes with important implications for skin cell photodamage not addressed further in this study.
Next, we tested the hypothesis that UVA-induced inhibition of cathepsin B enzymatic activity (as observed in Fig. 3D) may contribute to the generation of autofluorescent lipofuscin pigments in these cells (Fig. 5A and E). To this end, Hs27 cells were exposed to the specific cathepsin B inhibitor CA074Me (1 μM, q.d., 4 subsequent days), and autofluorescence was examined by confocal fluorescence microscopy (Fig. 5A, panel VII) and flow cytometric detection (Fig. 5E). CA074Me-treatment caused a marked increase in cellular autofluorescence displaying colocalization with lysosomes (lysotracker Red; Fig. 5A, panels VII-IX) with striking similarities to the UVA-induced autofluorescence pattern observed earlier (Fig.5A, panels IV-VI).
After demonstrating CA074Me-induced accumulation of autofluorescent material that mimics UVA-associated changes, we tested the hypothesis that pharmacological inhibition of cathepsin B might also impact cathepsin B protein maturation in response to UVA as detected earlier by 2D-DIGE mass spectrometric analysis and immunoblot analysis (Fig. 3A-C). Indeed, immunoblot analysis revealed the complete loss of the mature double chain (DC) form and a pronounced increase in the single chain (SC) form of cathepsin B in cells exposed to CA074Me (1 μM, q.d., 4 subsequent days; Fig. 5F).
Taken together, these data demonstrate that pharmacological inhibition of cathepsin B mimics UVA-induced accumulation of lysosomal autofluorescence and deficient cathepsin B maturation in human skin fibroblasts suggesting that UVA-induced cathepsin B dysfunction plays a role in photodamage of skin fibroblasts.
Discussion
2D-DIGE is a powerful analytical tool for the unbiased identification of protein targets that display altered expression levels in cutaneous cells exposed to cytotoxic stimuli and environmental stressors including UVB (32-34). Here, using a proteomic approach that involved 2D-DIGE analysis followed by mass spectrometric identification and follow up confirmation by immunodetection, we have identified novel target proteins that display a pronounced expression differential in human dermal fibroblasts exposed to chronic UVA irradiation.
The fibroblast protein displaying the highest differential upregulation in response to chronic UVA exposure was identified as nucleophosmin (nucleolar phosphoprotein B23, NPM1) (Figs. 1 and 2). Nucleophosmin is a nucleolar protein that undergoes stress-induced nucleoplasmic relocalization displaying regulatory protein-protein interactions with important nuclear factors including cell cycle regulator checkpoint kinase 1 (Chk1), the tumor suppressor proteins p53, Rb, BRCA1, and the p53-antagonist MDM2 (35,36). Recently, it has been demonstrated that nucleophosmin is an UVC-damage response protein involved in attenuation of p53-dependent apoptosis that occurs in response to genotoxic stress in cultured human fetal WS-1 fibroblasts and U2OS osteosarcoma cells (35,37). Our data document for the first time the significant upregulation of nucleophosmin protein levels in human dermal fibroblasts exposed to chronic UVA, but no further functional implications of this UVA-induced expression differential were explored at this point.
The protein displaying the highest differential downregulation in UVA-exposed fibroblasts was identified as the 24 kDa fragment of the mature cathepsin B double chain form (Figs. 1 and 3). The lysosomal papain-like cysteine protease cathepsin B is known to undergo proteolytic maturation that encompasses cysteine protease-dependent processing of procathepsin B (41 kDa) to the proteolytically active single chain cathepsin B form (29 kDa) followed by further maturation to the cystine-linked double chain form (5 kDa and 24 kDa fragments) in the lysosomal compartment (38). Loss of the cathepsin B 24 kDa fragment occurred as a result of both UVA treatment regimens (‘one week’ and ‘three week’ exposure) employed in this study, but a more pronounced effect was observed as a result of exposure according to the ‘three week’ regimen (Fig. 3B and C).
Our data indicate for the first time that chronic UVA exposure causes alteration of cathepsin B maturation and dramatic loss of cathepsin B specific enzymatic activity in human dermal fibroblasts. Loss of cathepsin B activity was already detectable even after exposure to a single dose of UVA (9.9 J/cm2) (Fig. 3D). Importantly, fibroblasts exposed to chronic UVA (‘one week’ and ‘three week’ regimens) displayed strong cellular autofluorescence, indicating the accumulation of lysosomal fluorescent pigments as confirmed by confocal microscopy employing costaining with Lysotracker Red (Fig. 5A and C).
Consistent with a causative involvement of UVA-induced impairment of cathepsin B structure and function in the accumulation of lysosomal autofluorescence, the pharmacological cathepsin B inhibitor CA074Me that completely abolished cellular cathepsin B activity (fig. 3D) mimicked UVA-induced autofluorescence characteristics, specifically pronounced punctate cytosolic staining displaying lysosomal colocalization (Fig. 5A) and quantitative detection by flow cytometry (Fig. 5 E). Importantly, CA074Me treatment altered cathepsin B maturation causing the loss of the 24 kDa mature form with accumulation of the 29 kDa single chain form (Fig. 5F), a posttranslational change of protein structure observed earlier in response to chronic UVA exposure (Fig. 1 and Fig. 3A-C). These data are consistent with the hypothesis that loss of cathepsin B activity from UVA exposure (or as a result of pharmacological inhibition) may impair cathepsin B maturation, possibly in the context of impaired autoproteolytic cleavage.
Earlier reports have demonstrated accumulation of autofluorescent lysosomal pigment (lipofuscin) originating from lipid peroxidation damage that was observed in oxidatively stressed human cells, including retinal pigment epithelial cells that accumulate the bisretinoid lipofuscin chromophore A2E under conditions of photooxidative stress (39), skin fibroblasts exposed to oxidative stress, and fibroblasts undergoing replicative senescence (26-30). The pronounced accumulation of autofluorescent material in lysosomal organelles that was observed as a consequence of either chronic UVA exposure (Fig. 5A, panels IV-VI)) or pharmacological inhibition of cathepsin B (Fig. 5A, panels VII-IX) strongly suggests a causative role of cathepsin B structural and functional alteration in UVA-induced accumulation of autofluorescent pigments. This is consistent with the established role of cathepsin B and other lysosomal proteases in lysosomal maintenance and clearance of damaged proteins, further supported by recent experimental evidence that demonstrates the involvement of cathepsin B and D in removal of glycated AGE-modified proteins (40).
The detailed mechanism underlying UVA-induced alteration of cathepsin B structure and function remains largely unresolved at this point and may result from complex molecular changes, since it is well established that cathepsin B is regulated by posttranslational modification through enzymatic glycosylation or nonenzymatic singlet oxygen-dependent oxidation, proteolytic processing and maturation, organelle trafficking, cellular and extracellular localization, and endogenous cysteine proteinase inhibitors (cystatins), all of which could be the subject of modulation by UVA exposure (38,41-43).
Our data do not suggest the involvement of changes that occur at the transcriptional level affecting expression of genes encoding either cathepsin B itself (CTSB) or established peptide inhibitors of cathepsin B including cystatin A (CSTA), cystatin B (CSTB), cystatin C (CSTC), and cystatin 6 (cystatin M/E; CST6) (Fig. 4A and table 1) (41,44). In contrast, our experiments indicate a causative involvement of photooxidative mechanisms in UVA-induced cathepsin B alterations as evidenced by detection of UVA-dependent ROS formation (Fig. 4C), oxidative stress response gene expression profiling (Fig. 4A and table 1), and antioxidant suppression of cathepsin B inactivation (Fig. 4D) and accumulation of autofluorescence (Fig. 5D).
In earlier studies photooxidative inactivation of active site cysteine residue-containing enzymes including protein tyrosine phosphatase 1B has been demonstrated (23,45,46), and oxidative inactivation of cellular cathepsin B by Rose Bengal-dependent photosensitization and singlet oxygen generated from naphthalene endoperoxides has been reported (25,43). Further experimentation involving mass spectrometric analysis of cathepsin B isolated from fibroblasts after chronic UVA exposure will therefore address the question if UVA-induced cathepsin B alterations result from direct photooxidation. Moreover, the possibility that UVA photooxidative stress targets additional cathepsins, forming a complex network involved in protein maturation and lysosomal function, will be examined in more detail (25,47,48). Interestingly, our preliminary experimentation using Hs27 fibroblasts exposed according to the ‘1 week’ UVA regimen indicated a significant loss of specific enzymatic activity of an additional lysosomal cysteine protease, cathepsin L, by almost 40 % compared with untreated controls [60.7 % ± 13.0 residual protease activity after UVA exposure (n=3; mean ± SD; p < 0.05)]. In contrast, no UVA-induced changes affecting activity of the cysteine-dependent enzyme GAPDH were observed (as reported in the results section), consistent with UVA-induced changes that specifically target lysosomal structure and function.
It is important to note that earlier work has demonstrated the involvement of lysosomal disruption in UVA-induced fibroblast photodamage, involving photooxidative rupture of lysosomal membranes followed by cytoplasmic release of proteases including cathepsin B that degrade ferritin with mobilization of redox-active iron (16,17). However, typical UVA fluence (dose) and irradiance (intensity) employed in these studies were significantly higher than the ones used in our model of chronic UVA exposure. In our experiments, fibroblast viability, proliferative rate after reseeding, and integrity of lysosomal membranes as monitored by direct imaging involving confocal fluorescence (Fig. 5A) and transmission electron microscopy (Fig. 5B) were not impaired as a result of chronic UVA exposure, and other mechanisms different from lysosomal disintegration must therefore be involved in UVA-induced cathepsin B alterations to be addressed by future experimentation.
In the context of cathepsins as potential molecular targets involved in skin photodamage, it should be mentioned that in another recent study performed in cultured presenescent fibroblasts downregulation of cathepsin B has been observed at the transcriptional and protein level in response to psoralen-UVA (PUVA) co-treatment (8-methoxypsoralen in combination with UVA) used as a model of photoaging (49,50). Moreover, using a mixed UVA/B light source (30% UVA + 70 % UVB spectral output), loss of cathepsin B immunohistochemical staining was observed in human skin specimens after undergoing a six week exposure regimen, but no assessment of altered specific enzymatic activity was performed (50). Moreover, a role of cathepsin K in cutaneous solar elastosis has been substantiated recently in human skin fibroblasts where an age-related decline in cathepsin K maturation was shown to compromise the process of orderly intracellular elastin degradation leading to subsequent accumulation of elastin in the extracellular space (51).
Irrespective of the molecular mechanism involved in UVA-induced impairment of cathepsin B structure and enzymatic activity, our findings may be of significant relevance to cutaneous photobiology given the emerging role of cathepsin B in lysosomal removal of damaged and AGE-modified cellular proteins observed in dermal fibroblasts (40,52). In the specific context of fibroblast photoaging, it is therefore tempting to speculate that in addition to the established role of UVA as a source of cellular oxidative stress leading to ROS-dependent structural damage and formation of autofluorescent lipofuscin-like material, UVA may contribute to cutaneous photodamage through an additional, heretofore unrecognized mechanism: Our data suggest that chronic UVA exposure of dermal fibroblasts compromises lysosomal clearance of damaged cellular material through cathepsin B inactivation thereby enhancing lipofuscin accumulation (summarized in Fig. 6), a hypothesis to be substantiated by future experimentation.
Figure 6. A model of UVA-induced accumulation of lipofuscin-like material in human dermal fibroblasts.

Photooxidative stress that results from UVA-induced formation of reactive oxygen species (ROS) and intracellular release of redox-active transition metal ions (Men+) causes lipid peroxidation and glycation reactions involved in accumulation of lipofuscin-like lysosomal material including proteins with autofluorescent and crosslinking AGE-epitopes (advanced glycatione endproducts). In parallel, lysosomal clearance of lipofuscin-precursor material is impaired by UVA-induced alterations of cathepsin B protein structure and enzymatic activity, thereby further enhancing skin fibroblast photodamage.
Apart from substantiating the emerging role of cathepsin B as a novel cellular target in skin photodamage involved in lipofuscin accumulation, it will be fascinating to elucidate the functional implications of UVA-induced lipofuscin accumulation in the context of skin photocarcinogenesis and photoaging, particularly in the light of earlier studies that have documented the emerging functional involvement of lipofuscin in transition metal ion-dependent redox dysregulation and alteration of lysosomal cell death pathways in lipofuscin-loaded fibroblasts (53,54).
Acknowledgments
Preliminary data from this research were part of an oral presentation at the 35th Meeting of the American Society for Photobiology, June 12-16, 2010, in Providence, RI. Flow cytometric analysis was performed at the Arizona Cancer Center flow cytometry core facility. Supported in part by grants from the National Institutes of Health [R01CA122484, ES007091, ES006694, Arizona Cancer Center Support Grant CA023074].
References
- 1.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scharffetter-Kochanek K, Wlaschek M, Brenneisen P, Schauen M, Blaudschun R, Wenk J. UV-induced reactive oxygen species in photocarcinogenesis and photoaging. Biol Chem. 1997;378:1247–1257. [PubMed] [Google Scholar]
- 3.Gasparro FP. Sunscreens, skin photobiology, and skin cancer: the need for UVA protection and evaluation of efficacy. Environ Health Perspect. 2000;108 1:71–78. doi: 10.1289/ehp.00108s171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wondrak GT, Jacobson MK, Jacobson EL. Endogenous UVA-photosensitizers: mediators of skin photodamage and novel targets for skin photoprotection. Photochem Photobiol Sci. 2006;5:215–237. doi: 10.1039/b504573h. [DOI] [PubMed] [Google Scholar]
- 5.Zastrow L, Groth N, Klein F, Kockott D, Lademann J, Renneberg R, Ferrero L. The missing link--light-induced (280-1,600 nm) free radical formation in human skin. Skin Pharmacol Physiol. 2009;22:31–44. doi: 10.1159/000188083. [DOI] [PubMed] [Google Scholar]
- 6.Cadet J, Douki T, Ravanat JL, Di Mascio P. Sensitized formation of oxidatively generated damage to cellular DNA by UVA radiation. Photochem Photobiol Sci. 2009;8:903–911. doi: 10.1039/b905343n. [DOI] [PubMed] [Google Scholar]
- 7.Girotti AW. Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J Photochem Photobiol B. 2001;63:103–113. doi: 10.1016/s1011-1344(01)00207-x. [DOI] [PubMed] [Google Scholar]
- 8.Wlaschek M, W J, Brenneisen P, Briviba K, Schwarz A, Sies H, Scharffetter-Kochanek Singlet oxygen is an early intermediate in cytokine-dependent ultraviolet-A induction of interstitial collagenase in human fibroblasts in vitro. FEBS Lett. 1997;413:239–242. doi: 10.1016/s0014-5793(97)00919-8. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson EL, Giacomoni PU, Roberts MJ, Wondrak GT, Jacobson MK. Optimizing the energy status of skin cells during solar radiation. J Photochem Photobiol B. 2001;63:141–147. doi: 10.1016/s1011-1344(01)00211-1. [DOI] [PubMed] [Google Scholar]
- 10.Valencia A, Kochevar IE. Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes. J Invest Dermatol. 2008;128:214–222. doi: 10.1038/sj.jid.5700960. [DOI] [PubMed] [Google Scholar]
- 11.Salet C, Moreno G. Photodynamic action increases leakage of the mitochondrial electron transport chain. Int J Radiat Biol. 1995;67:477–480. doi: 10.1080/09553009514550551. [DOI] [PubMed] [Google Scholar]
- 12.Park J, Halliday GM, Surjana D, Damian DL. Nicotinamide prevents ultraviolet radiation-induced cellular energy loss. Photochem Photobiol. 2010;86:942–948. doi: 10.1111/j.1751-1097.2010.00746.x. [DOI] [PubMed] [Google Scholar]
- 13.Halliday GM. Common links among the pathways leading to UV-induced immunosuppression. J Invest Dermatol. 2010;130:1209–1212. doi: 10.1038/jid.2009.374. [DOI] [PubMed] [Google Scholar]
- 14.Sreevidya CS, Fukunaga A, Khaskhely NM, Masaki T, Ono R, Nishigori C, Ullrich SE. Agents that reverse UV-Induced immune suppression and photocarcinogenesis affect DNA repair. J Invest Dermatol. 2010;130:1428–1437. doi: 10.1038/jid.2009.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamore SD, Azimian S, Horn D, Anglin BL, Uchida K, Cabello CM, Wondrak GT. The malondialdehyde-derived fluorophore DHP-lysine is a potent sensitizer of UVA-induced photooxidative stress in human skin cells. J Photochem Photobiol B. 2010 doi: 10.1016/j.jphotobiol.2010.07.010. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pourzand C, Watkin RD, Brown JE, Tyrrell RM. Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: the role of ferritin. Proc Natl Acad Sci U S A. 1999;96:6751–6756. doi: 10.1073/pnas.96.12.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basu-Modak S, Ali D, Gordon M, Polte T, Yiakouvaki A, Pourzand C, Rice-Evans C, Tyrrell RM. Suppression of UVA-mediated release of labile iron by epicatechin--a link to lysosomal protection. Free Radic Biol Med. 2006;41:1197–1204. doi: 10.1016/j.freeradbiomed.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Issaq HJ, Veenstra TD. The role of electrophoresis in disease biomarker discovery. Electrophoresis. 2007;28:1980–1988. doi: 10.1002/elps.200600834. [DOI] [PubMed] [Google Scholar]
- 19.Hariharan D, Weeks ME, Crnogorac-Jurcevic T. Application of proteomics in cancer gene profiling: two-dimensional difference in gel electrophoresis (2D-DIGE) Methods Mol Biol. 2010;576:197–211. doi: 10.1007/978-1-59745-545-9_11. [DOI] [PubMed] [Google Scholar]
- 20.Minden JS, Dowd SR, Meyer HE, Stuhler K. Difference gel electrophoresis. Electrophoresis. 2009;30 1:S156–161. doi: 10.1002/elps.200900098. [DOI] [PubMed] [Google Scholar]
- 21.Wondrak GT, Jacobson MK, Jacobson EL. Identification of quenchers of photoexcited states as novel agents for skin photoprotection. J Pharmacol Exp Ther. 2005;312:482–491. doi: 10.1124/jpet.104.075101. [DOI] [PubMed] [Google Scholar]
- 22.Herrmann G, Brenneisen P, Wlaschek M, Wenk J, Faisst K, Quel G, Hommel C, Goerz G, Ruzicka T, Krieg T, Sies H, Scharffetter-Kochanek K. Psoralen photoactivation promotes morphological and functional changes in fibroblasts in vitro reminiscent of cellular senescence. J Cell Sci. 1998;111(Pt 6):759–767. doi: 10.1242/jcs.111.6.759. [DOI] [PubMed] [Google Scholar]
- 23.Linetsky M, Chemoganskiy VG, Hu F, Ortwerth BJ. Effect of UVA light on the activity of several aged human lens enzymes. Invest Ophthalmol Vis Sci. 2003;44:264–274. doi: 10.1167/iovs.02-0597. [DOI] [PubMed] [Google Scholar]
- 24.Lamore SD, Cabello CM, Wondrak GT. The topical antimicrobial zinc pyrithione is a heat shock response inducer that causes DNA damage and PARP-dependent energy crisis in human skin cells. Cell Stress Chaperones. 2010;15:309–322. doi: 10.1007/s12192-009-0145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahmanto AS, Morgan PE, Hawkins CL, Davies MJ. Cellular effects of photo-generated oxidants and long-lived, reactive, hydroperoxide photoproducts. Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.08.006. in press 2010 Aug 12 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 26.Nilsson E, Yin D. Preparation of artificial ceroid/lipofuscin by UV-oxidation of subcellular organelles. Mech Ageing Dev. 1997;99:61–78. doi: 10.1016/s0047-6374(97)00091-2. [DOI] [PubMed] [Google Scholar]
- 27.Sitte N, Merker K, Grune T, von Zglinicki T. Lipofuscin accumulation in proliferating fibroblasts in vitro: an indicator of oxidative stress. Exp Gerontol. 2001;36:475–486. doi: 10.1016/s0531-5565(00)00253-9. [DOI] [PubMed] [Google Scholar]
- 28.Jung T, Bader N, Grune T. Lipofuscin: formation, distribution, and metabolic consequences. Ann N Y Acad Sci. 2007;1119:97–111. doi: 10.1196/annals.1404.008. [DOI] [PubMed] [Google Scholar]
- 29.Jung T, Hohn A, Catalgol B, Grune T. Age-related differences in oxidative protein-damage in young and senescent fibroblasts. Arch Biochem Biophys. 2009;483:127–135. doi: 10.1016/j.abb.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. 2010;12:503–535. doi: 10.1089/ars.2009.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jung T, Hohn A, Grune T. Lipofuscin: detection and quantification by microscopic techniques. Methods Mol Biol. 2010;594:173–193. doi: 10.1007/978-1-60761-411-1_13. [DOI] [PubMed] [Google Scholar]
- 32.Ibuki Y, Naitou H, Ohashi N, Goto R. Proteome analysis of UV-B-induced anti-apoptotic regulatory factors. Photochem Photobiol. 2005;81:823–829. doi: 10.1562/2004-09-01-RA-297. [DOI] [PubMed] [Google Scholar]
- 33.Pollins AC, Friedman DB, Nanney LB. Proteomic investigation of human burn wounds by 2D-difference gel electrophoresis and mass spectrometry. J Surg Res. 2007;142:143–152. doi: 10.1016/j.jss.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guipaud O, Holler V, Buard V, Tarlet G, Royer N, Vinh J, Benderitter M. Time-course analysis of mouse serum proteome changes following exposure of the skin to ionizing radiation. Proteomics. 2007;7:3992–4002. doi: 10.1002/pmic.200601032. [DOI] [PubMed] [Google Scholar]
- 35.Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–475. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- 36.Chen S, Maya-Mendoza A, Zeng K, Tang CW, Sims PF, Loric J, Jackson DA. Interaction with checkpoint kinase 1 modulates the recruitment of nucleophosmin to chromatin. J Proteome Res. 2009;8:4693–4704. doi: 10.1021/pr900396d. [DOI] [PubMed] [Google Scholar]
- 37.Kurki S, Peltonen K, Laiho M. Nucleophosmin, HDM2 and p53: players in UV damage incited nucleolar stress response. Cell Cycle. 2004;3:976–979. [PubMed] [Google Scholar]
- 38.Ishidoh K, Kominami E. Processing and activation of lysosomal proteinases. Biol Chem. 2002;383:1827–1831. doi: 10.1515/BC.2002.206. [DOI] [PubMed] [Google Scholar]
- 39.Lamb LE, Simon JD. A2E: a component of ocular lipofuscin. Photochem Photobiol. 2004;79:127–136. doi: 10.1562/0031-8655(2004)079<0127:aacool>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 40.Grimm S, Ernst L, Grotzinger N, Hohn A, Breusing N, Reinheckel T, Grune T. Cathepsin D is one of the major enzymes involved in intracellular degradation of AGE-modified proteins. Free Radic Res. 2010;44:1013–1026. doi: 10.3109/10715762.2010.495127. [DOI] [PubMed] [Google Scholar]
- 41.Zeeuwen PL. Epidermal differentiation: the role of proteases and their inhibitors. Eur J Cell Biol. 2004;83:761–773. doi: 10.1078/0171-9335-00388. [DOI] [PubMed] [Google Scholar]
- 42.Pillai S, Oresajo C, Hayward J. Ultraviolet radiation and skin aging: roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation - a review. Int J Cosmet Sci. 2005;27:17–34. doi: 10.1111/j.1467-2494.2004.00241.x. [DOI] [PubMed] [Google Scholar]
- 43.Nagaoka Y, Otsu K, Okada F, Sato K, Ohba Y, Kotani N, Fujii J. Specific inactivation of cysteine protease-type cathepsin by singlet oxygen generated from naphthalene endoperoxides. Biochem Biophys Res Commun. 2005;331:215–223. doi: 10.1016/j.bbrc.2005.03.146. [DOI] [PubMed] [Google Scholar]
- 44.Kartasova T, Cornelissen BJ, Belt P, van de Putte P. Effects of UV, 4-NQO and TPA on gene expression in cultured human epidermal keratinocytes. Nucleic Acids Res. 1987;15:5945–5962. doi: 10.1093/nar/15.15.5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Montfort C, Sharov VS, Metzger S, Schoneich C, Sies H, Klotz LO. Singlet oxygen inactivates protein tyrosine phosphatase-1B by oxidation of the active site cysteine. Biol Chem. 2006;387:1399–1404. doi: 10.1515/BC.2006.175. [DOI] [PubMed] [Google Scholar]
- 46.Voss P, Hajimiragha H, Engels M, Ruhwiedel C, Calles C, Schroeder P, Grune T. Irradiation of GAPDH: a model for environmentally induced protein damage. Biol Chem. 2007;388:583–592. doi: 10.1515/BC.2007.068. [DOI] [PubMed] [Google Scholar]
- 47.Yan S, Sloane BF. Molecular regulation of human cathepsin B: implication in pathologies. Biol Chem. 2003;384:845–854. doi: 10.1515/BC.2003.095. [DOI] [PubMed] [Google Scholar]
- 48.Laurent-Matha V, Derocq D, Prebois C, Katunuma N, Liaudet-Coopman E. Processing of human cathepsin D is independent of its catalytic function and auto-activation: involvement of cathepsins L and B. J Biochem. 2006;139:363–371. doi: 10.1093/jb/mvj037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai W, Zheng Y, Ye ZZ, Su XY, Wan MJ, Gong ZJ, Xie XY, Liu W. Changes of cathepsin B in human photoaging skin both in vivo and in vitro. Chin Med J (Engl) 2010;123:527–531. [PubMed] [Google Scholar]
- 50.Zheng Y, Lai W, Wan M, Maibach HI. Expression of Cathepsins in Human Skin Photoaging. Skin Pharmacol Physiol. 2010;24:10–21. doi: 10.1159/000314725. [DOI] [PubMed] [Google Scholar]
- 51.Codriansky KA, Quintanilla-Dieck MJ, Gan S, Keady M, Bhawan J, Runger TM. Intracellular degradation of elastin by cathepsin K in skin fibroblasts--a possible role in photoaging. Photochem Photobiol. 2009;85:1356–1363. doi: 10.1111/j.1751-1097.2009.00592.x. [DOI] [PubMed] [Google Scholar]
- 52.Sitte N, Merker K, Von Zglinicki T, Grune T, Davies KJ. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part I--effects of proliferative senescence. Faseb J. 2000;14:2495–2502. doi: 10.1096/fj.00-0209com. [DOI] [PubMed] [Google Scholar]
- 53.Stroikin Y, Johansson U, Asplund S, Ollinger K. Increased resistance of lipofuscin-loaded prematurely senescent fibroblasts to starvation-induced programmed cell death. Biogerontology. 2007;8:43–53. doi: 10.1007/s10522-006-9029-7. [DOI] [PubMed] [Google Scholar]
- 54.Hohn A, Jung T, Grimm S, Grune T. Lipofuscin-bound iron is a major intracellular source of oxidants: role in senescent cells. Free Radic Biol Med. 2010;48:1100–1108. doi: 10.1016/j.freeradbiomed.2010.01.030. [DOI] [PubMed] [Google Scholar]