

Abstract
Hallucinogens mediate many of their psychoactive effects by activating serotonin 2A receptors (5-HT2AR). Although serotonin is the cognate endogenous neurotransmitter and is not considered hallucinogenic, metabolites of serotonin also have high affinity at 5-HT2AR and can induce hallucinations in humans. Here we report that serotonin differs from the psychoactive N-methyltryptamines by its ability to engage a β-arrestin2-mediated signaling cascade in the frontal cortex. Serotonin and 5-hydroxy-l-tryptophan (5-HTP) induce a head-twitch response in wild-type (WT) mice that is a behavioral proxy for 5-HT2AR activation. The response in β-arrestin2 knock-out (βarr2-KO) mice is greatly attenuated until the doses are elevated, at which point, βarr2-KO mice display a head-twitch response that can exceed that of WT mice. Direct administration of N-methyltryptamines also produces a greater response in βarr2-KO mice. Moreover, the inhibition of N-methyltransferase blocks 5-HTP-induced head twitches in βarr2-KO mice, indicating that N-methyltryptamines, rather than serotonin, primarily mediate this response. Biochemical studies demonstrate that serotonin stimulates Akt phosphorylation in the frontal cortex and in primary cortical neurons through the activation of a β-arrestin2/phosphoinositide 3-kinase/Src/Akt cascade, whereas N-methyltryptamines do not. Furthermore, disruption of any of the components of this cascade prevents 5-HTP-induced, but not N-methyltryptamine-induced, head twitches. We propose that there is a bifurcation of 5-HT2AR signaling that is neurotransmitter and β-arrestin2 dependent. This demonstration of agonist-directed 5-HT2AR signaling in vivo may significantly impact drug discovery efforts for the treatment of disorders wherein hallucinations are part of the etiology, such as schizophrenia, or manifest as side effects of treatment, such as depression.
Introduction
The serotonin 2A receptor (5-HT2AR) is highly expressed on pyramidal neurons in the frontal cortex and has been implicated in several mental health disorders, including schizophrenia, anxiety, and depression (Jakab and Goldman-Rakic, 1998; Meltzer, 2002; Miner et al., 2003; Roth et al., 2004; Berg et al., 2008). The 5-HT2AR is also the target of serotonergic hallucinogenic drugs (Aghajanian and Marek, 1999; Nichols, 2004). Hallucinogens act at the 5-HT2AR to produce a distinct head-twitch response in rodents (Corne and Pickering, 1967) that is prevented by genetic ablation or pharmacological antagonism of the 5-HT2AR (Willins and Meltzer, 1997; González-Maeso et al., 2007; Keiser et al., 2009). Furthermore, systemic injections of 5-hydroxy-l-tryptophan (5-HTP), a metabolic precursor of serotonin, can elevate serotonin in the brain to levels that induce a robust head-twitch response in mice (Corne et al., 1963; Schmid et al., 2008). Moreover, the selective antagonist M100907 [R(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorophenylethyl)]-4-piperidinemethanol] [Chemical Abstract Services (CAS) registry number 139290-65-6], blocks this response, and 5-HTP-induced head twitches are lacking in 5-HT2AR knock-out (KO) mice (Schmid et al., 2008) (B. L. Roth, personal communication). The expression of 5-HT2ARs in forebrain are likely mediating this response because the response to 5-HT2AR agonists can be rescued by expression of receptors in glutamatergic neurons in the frontal cortex of 5-HT2AR-KO mice (González-Maeso et al., 2007).
The 5-HT2AR is a G-protein-coupled receptor (GPCR), and GPCRs can be activated by chemically distinct ligands to preferentially engage select signaling pathways, a characteristic referred to as “agonist-directed signaling” or “functional selectivity” (Kenakin, 1995; Kobilka and Deupi, 2007; Urban et al., 2007). β-Arrestins are intracellular scaffolding proteins that can either dampen or facilitate GPCR signaling and therefore may represent a key point at which receptor signaling may diverge in response to particular ligands (Luttrell and Lefkowitz, 2002; Lefkowitz et al., 2006; DeWire et al., 2007; Rajagopal et al., 2010). Diverse ligands to the 5-HT2AR have been shown to differentially activate downstream signaling cascades both in vitro and in vivo (Berg et al., 1998; Kurrasch-Orbaugh et al., 2003; González-Maeso et al., 2007; Schmid et al., 2008). We showed previously that serotonin and a synthetic hallucinogen, 2,5-dimethoxy-4-iodoamphetamine (DOI), can differentially activate the 5-HT2AR in cellular models as well as in vivo, wherein 5-HTP requires β-arrestin2 for the expression of the head-twitch response yet DOI does not (Schmid et al., 2008).
In the brain, serotonin is a substrate for N-methyltransferases, enzymes that act to convert serotonin to N-methyltryptamines; these metabolites possess psychoactive properties and can be hallucinogenic in humans (Axelrod, 1962; Glennon and Gessner, 1979; Shulgin and Shulgin, 1997; McBride, 2000). It has previously been very difficult to delineate the distinct neuropharmacological effects produced by serotonin and its psychoactive metabolites in vivo because many of the biogenic amines have higher affinities than serotonin at the 5-HT2AR (Glennon and Gessner, 1979; Blair et al., 2000). In this study, we demonstrate functional selectivity at the 5-HT2AR by serotonin and N-methyltryptamines in vivo, as well as in the mouse frontal cortex and in primary cortical neurons in which we show that the actions of these neurotransmitters are functionally distinct.
Materials and Methods
Drugs.
5-HTP, serotonin (5-HT), N-methylserotonin oxalate salt (CAS registry number 1134-01-6), 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT) (CAS registry number 1019-45-0), and clorgyline were purchased from Sigma-Aldrich. MTZ [N,N′bis-(3-methyl-2-thiazolidinylidene)succinamide] (CAS registry number 65400-75-1) was purchased from Life Chemicals. LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one], Akt inhibitor VIII (Akti; 1,3-dihydro-1-((4-(6-phenyl-1H-imidazo[4,5-g]quinoxalin-7-yl)phenyl)methyl)-4-piperidnyl)-2H-benzimidazol-2-one) and Src tyrosine kinase inhibitor (PP2; 4-amino-5-(4-chlorophenyl)-7-(t-butyl)-pyrazolo[3,4-d]pyrimidine) were purchased from Cayman Chemical, Calbiochem, and Tocris Cookson, respectively. M100907 was provided by Dr. Kenner Rice (National Institute on Drug Abuse/National Institute on Alcohol Abuse and Alcoholism/National Institutes of Health, Bethesda, MD).
Primary antibodies.
Total Akt (1:2000; pan C67E7), phospho-Akt (1:1000; Thr308 C31E5E), and Src (1: 500; L4A1) antibodies were obtained from Cell Signaling Technology (Luan et al., 2009); anti-PSD-95 (1:500; K28/4) was purchased from University of California, Davis/National Institute of Neurological Disorders and Stroke/National Institute of Mental Health NeuroMab Facility (Davis, CA) (Fan et al., 2009); β-arrestin2 (1:2000; A2CT) antibody was provided by Dr. Robert Lefkowitz (Duke University, Howard Hughes Medical Institute, Durham, NC) (Bohn et al., 1999); the polyclonal antibody to the N terminus of the 5-HT2AR (1:500) was from Neuromics (Magalhaes et al., 2010); the c-Myc monoclonal antibody (1:500) was from Clontech/Takara Bio (Hu et al., 1995).
Mice.
Subjects used in the experiments include male wild-type (WT) and β-arrestin2 knock-out (βarr2-KO) mice derived via heterozygous breeding as well as male C57BL/6J mice (The Jackson Laboratory) between 2.5 and 9 months of age (Bohn et al., 1999; Schmid et al., 2008). Drugs administered intraperitoneally were prepared in 0.9% saline at a volume of 10 μl/g body weight; drugs administered intracerebroventricularly were prepared in 18 Ω purified, distilled, sterile water and injected at a volume of 5 μl, 2 mm caudal and 2 mm lateral from bregma at a depth of 3 mm. Immediately after injection with agonist, mice were placed in individual Plexiglas boxes, and the number of head twitches were counted in 5 min increments for either 30 or 60 min (Schmid et al., 2008). In all cases, mice were used only once. All experiments were performed with the approval of the Institutional Animal Care and Use Committee of The Ohio State University or The Scripps Research Institute.
Behavioral experiments.
The number of mice used in the experimental design is summarized in Table 1. For dose–response studies, WT and βarr2-KO mice were treated with 5-HTP (100, 150, and 200 mg/kg, i.p), serotonin (10, 20, and 40 μg, i.c.v.), N-methylserotonin (10, 20, and 40 μg, i.c.v.), or 5-MeO-DMT (5, 10, and 15 mg/kg, i.p.). To determine the contribution of monoamine oxidase A (MAO-A) metabolism to 5-HTP-induced head twitches, mice were pretreated (intraperitoneally) with either vehicle (0.9% saline) or clorgyline (1 mg/kg) 60 min before treatment with 5-HTP (50 and 100 mg/kg, i.p.). The 5-HT2AR antagonist M100907 (0.05 mg/kg, i.p.) was administered 10 min before 5-HTP (200 mg/kg, i.p.) or 5-HT (40 μg, i.c.v.) in Figure 1, B and D, or 50 min after treatment with clorgyline (1 mg/kg, i.p.) and 10 min before treatment with 5-HTP (100 mg/kg, i.p.) in Figure 1F. In Figure 3, M100907 was administered 10 min before treatment with N-methylserotonin (20 μg, i.c.v.) or 5-MeO-DMT (10 mg/kg, i.p.). The M100907 vehicle was 0.02% Tween 80 in water. The N-methyltransferase inhibitor MTZ (125 ng, i.c.v.) or vehicle (dH2O for studies in Figs. 2, 3 and in 1% DMSO for studies in Fig. 8) was injected 10 min before treatment with 5-HTP (200 mg/kg, i.p.), N-methylserotonin (20 μg, i.c.v.), or 5-MeO-DMT (10 mg/kg, i.p.). Vehicle (1% DMSO), LY294002 (125 ng, i.c.v.), PP2 (300 ng, i.c.v.), or AKTi (55 ng, i.c.v.) was injected 10 min before treatment with 5-HTP (200 mg/kg, i.p.). For the studies detailed in Figure 8, WT mice were pretreated with vehicle (1% DMSO), AKTi (55 ng, i.c.v.), MTZ (125 ng, i.c.v.), or both ATKi and MTZ for 10 min before treatment with 5-HTP (200 mg/kg, i.p.). Dosing of the inhibitors was based on the literature (clorgyline: Tadano et al., 1989; MTZ: Mandel et al., 1978, Rokach et al., 1980; LY294002: Beaulieu et al., 2005; PP2: Narita et al., 2006; AKTi: Xu et al., 2008).
Table 1.
Numbers of mice used in behavioral studies
| Figure | Treatment in mice | Serotonergic dose and n values | ||||
|---|---|---|---|---|---|---|
| 1A,B | 5-HTP (mg/kg, i.p.) ±M100 | 100 | 150 | 200 | M100 + 200 | |
| WT, βarr2-KO | n = 13, 8 | n = 7, 5 | n = 8, 7 | n = 6, 6 | ||
| 1C,D | 5-HT (μg, i.c.v.) ±M100 | 10 | 20 | 40 | M100 + 40 | |
| WT, βarr2-KO | n = 4, 5 | n = 7, 5 | n = 5, 6 | n = 4, 4 | ||
| 1E,F | Veh/Clor (±M100) + 5-HTP | Veh +50 | Veh +100 | Clor +50 | Clor +100 | Clor + M100 +100 |
| WT, βarr2-KO | n = 6, 5 | n = 6, 5 | n = 5, 6 | n = 9, 8 | n = 4, 4 | |
| 2A,B | Veh/MTZ + 5-HTP | Veh + 200 | MTZ + 200 | |||
| WT, βarr2-KO | n = 4, 5 | n = 4, 5 | ||||
| 3A,B | N-Me-5-HT (μg, i.c.v.) | 10 | 20 | 40 | ||
| WT, βarr2-KO | n = 4, 4 | n = 5, 5 | n = 4, 4 | |||
| 3C | Veh/M100 + N-Me-5-HT | Veh + 20 | M100 + 20 | |||
| WT, βarr2-KO | n = 4, 4 | n = 4, 4 | ||||
| 3D | Veh/MTZ + N-Me-5-HT | Veh + 20 | MTZ + 20 | |||
| WT | n = 5 | n = 4 | ||||
| 3E,F | 5-MeO-DMT (mg/kg, i.p.) | 5 | 10 | 15 | ||
| WT, βarr2-KO | n = 9, 10 | n = 10, 16 | n = 5, 5 | |||
| 3G | Veh/M100 + 5-MeO-DMT | Veh + 10 | M100 + 10 | |||
| WT, βarr2-KO | n = 5, 9 | n = 5, 5 | ||||
| 3H | Veh/MTZ + 5-MeO-DMT | Veh + 10 | MTZ + 10 | |||
| C57BL/6 | n = 6 | n = 4 | ||||
| 6A,B | Veh, LY, PP2, AKTi + 5-HTP | Veh + 200 | LY + 200 | PP2 + 200 | AKTi + 200 | |
| C57BL/6 | n = 10 | n = 5 | n = 5 | n = 5 | ||
| 6C | Veh, AKTi + 5-MeO-DMT | 10 | AKTi + 200 | |||
| C57BL/6 | n = 6 | n = 4 | ||||
| 7A,B | Veh, LY, PP2, AKTi + 5-HTP | 200 | LY + 200 | PP2 + 200 | AKTi + 200 | |
| βbarr2-KO | n = 7 | n = 6 | n = 6 | n = 6 | ||
| 8A,B | Veh, AKTi, MTZ, AKTi + MTZ + 5-HTP | 200 | MTZ + 200 | AKTi + 200 | MTZ + AKTi +200 | |
| WT | n = 11 | n = 6 | n = 5 | n = 5 | ||
Veh, Vehicle; 5-HT, serotonin; M100, M100907; Clor, clorgyline; N-Me-5-HT, N-methylserotonin; LY, LY294002. Serotonergic dosing routes: 5-HTP, mg/kg, i.p.; 5-HT and N-methylserotonin, μg, i.c.v.; 5-MeO-DMT, mg/kg, i.p. For inhibitor and antagonist dosage information, see Materials and Methods.
Figure 1.
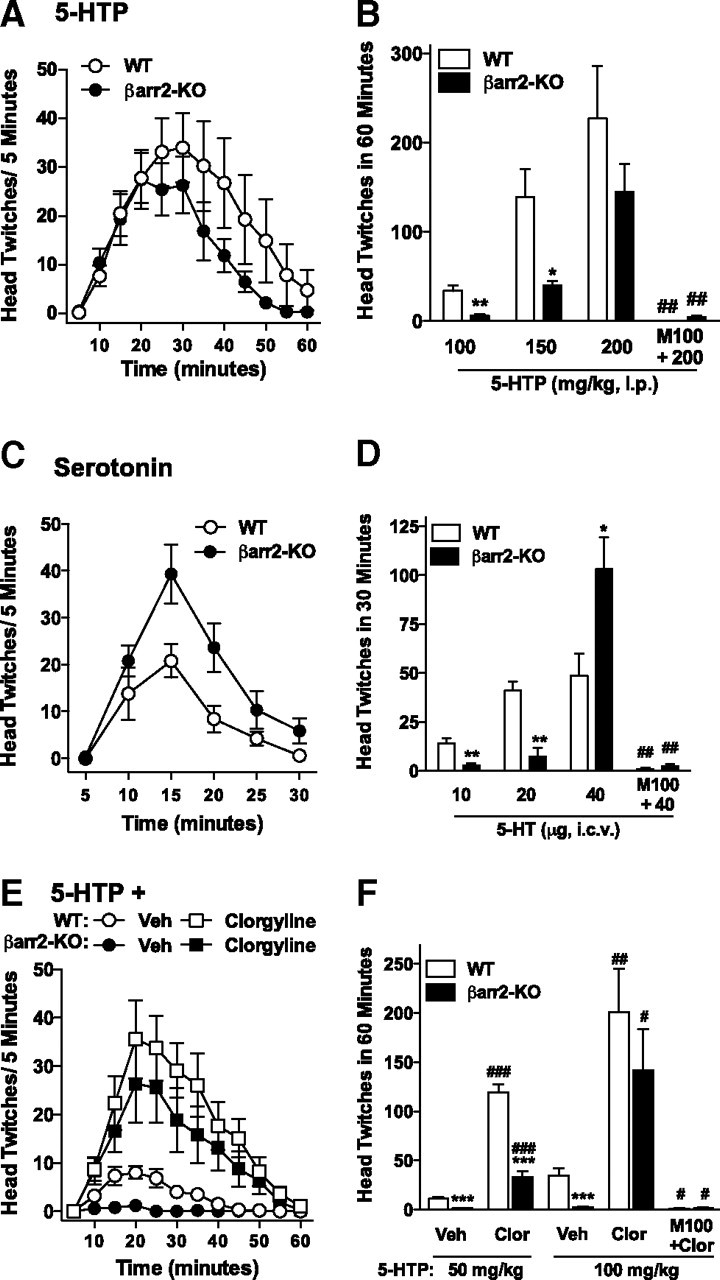
High doses of serotonin and 5-HTP can induce a head-twitch response in the βarr2-KO mice. A–D, 5-HTP and serotonin induce a head-twitch response in WT and βarr2-KO mice. Time course of 5-HTP (200 mg/kg, i.p.)-induced head-twitch response (A) and 5-HTP dose–response curves in WT and βarr2-KO mice and blockade of 5-HTP-induced (200 mg/kg) responses (B) after a 10 min pretreatment with M100907 (M100; 0.05 mg/kg, i.p.). WT versus βarr2-KO within dose: *p < 0.05; **p < 0.001; within genotype: 200 mg/kg 5-HTP versus M100 + 200 mg/kg 5-HTP, ##p < 0.001. Time course of 5-HT (40 μg, i.c.v.)-induced head-twitch response (C) and serotonin dose–response curves in WT and βarr2-KO mice and blockade of 5-HT-induced (40 μg, i.c.v.) responses (D) after a 10 min pretreatment with M100907 (0.05 mg/kg, i.p.). WT versus βarr2-KO at the same dose: *p < 0.05; **p < 0.001; within genotype: 40 μg of 5-HT versus M100 + 40 μg of 5-HT: ##p < 0.001. E, F, Inhibition of MAO-A enhances 5-HTP potency in both genotypes. Time course of 5-HTP (100 mg/kg, i.p.)-induced head-twitch responses after a 1 h pretreatment with clorgyline (Clor; 1 mg/kg, i.p.) or clorgyline vehicle (Veh; 0.9% saline, i.p.) (E) and the sum of twitches induced over the 60 min observation period at two doses of 5-HTP (50 and 100 mg/kg, i.p.) in WT and βarr2-KO mice and blockade of clorgyline-enhanced 5-HTP-induced (100 mg/kg) responses after a 10 min pretreatment with M100907 (0.05 mg/kg, i.p.) (F). WT versus βarr2-KO at the same dose: ***p < 0.0001. Vehicle pretreatment versus clorgyline pretreatment within genotype: #p < 0.05, ##p < 0.001, ###p < 0.0001; within genotype: clorgyline + 100 mg/kg 5-HTP versus M100 + clorgyline + 100 mg/kg 5-HTP: #p < 0.05. Mean ± SEM are shown.
Figure 3.

N-Methyltryptamines induce more head twitches in βarr2-KO mice than their WT littermates. βarr2-KO mice treated with either N-methylserotonin (N-Me-5-HT) (A–D) or 5-MeO-DMT (E–H) display significantly more head twitches than WT mice. Time course of head twitches induced by N-methylserotonin (20 μg, i.c.v.) (A) and dose–response curve for the total number of head twitches observed over 30 min (B). WT versus βarr2-KO: *p < 0.05, **p < 0.01. C, Pretreatment (10 min) with the 5-HT2AR antagonist M100907 (M100; 0.05 mg/kg, i.p.), but not vehicle (0.02% Tween 80), blocks the N-methylserotonin (20 μg, i.c.v.)-induced head-twitch response in both genotypes. WT versus βarr2-KO: *p < 0.05; values for M100907 treatment were 0 ± 0. D, Pretreatment (10 min) of WT mice with the N-methyltransferase inhibitor MTZ (125 ng, i.c.v.) or vehicle (5 μl of dH2O, i.c.v.) has no effect on the N-methylserotonin (20 μg, i.c.v.)-induced head-twitch response. E, F, Time course of head twitches induced by 5-MeO-DMT (10 mg/kg, i.p.) (E) and dose–response curve for the total number of head twitches observed over 30 min (F). WT versus βarr2-KO: *p < 0.05, ***p < 0.001. G, Pretreatment (10 min) with M100907 (0.05 mg/kg, i.p.), but not vehicle, blocks the 5-MeO-DMT (10 mg/kg, i.p.)-induced head-twitch response in both genotypes. WT versus βarr2-KO: *p < 0.05; vehicle versus M100 within genotype: ##p < 0.01, ###p < 0.0001. H, Pretreatment (10 min) of C57BL/6J mice with MTZ (125 ng, i.c.v.) or vehicle (5 μl of dH2O, i.c.v.) has no effect on the 5-MeO-DMT-induced (10 mg/kg, i.p.) head-twitch response. Mean ± SEM are shown.
Figure 2.

An N-methyltransferase inhibitor eliminates 5-HTP-induced head twitches in βarr2-KO mice. Pretreatment with an N-methyltransferase inhibitor, MTZ, blocks 5-HTP-induced head-twitch response in the βarr2-KO mice and attenuates the response in WT mice. Time course analysis of head twitches induced by 200 mg/kg 5-HTP (intraperitoneally) (A) and the total number of twitches observed over 60 min after a 10 min pretreatment with MTZ (125 ng, i.c.v.) or vehicle (5 μl of dH2O, i.c.v.) (B). WT versus βarr2-KO with the same treatment: ***p < 0.001. Vehicle pretreatment versus MTZ pretreatment within genotype: #p < 0.05, ##p < 0.01. Mean ± SEM are shown.
Figure 8.

5-HTP-induced head twitches in WT mice are blocked by combined inhibition of N-methyltransferase and Akt. Pretreatment with the Akt inhibitor (AKTi) or the N-methyltransferase inhibitor (MTZ) individually reduce 5-HTP-induced twitches in WT mice; coadministration of the inhibitors is additive. Time course (A) and total numbers (B) of head twitches observed in WT mice after 10 min pretreatment with either vehicle (Veh; 0.1% DMSO in dH2O, i.c.v.), AKTi (55 ng, i.c.v.), or MTZ (125 ng, i.c.v.) alone, or AKTi and MTZ concurrently before 5-HTP administration (200 mg/kg, i.p.) Vehicle versus inhibitor: ##p < 0.01, ###p < 0.001. AKTi versus AKTi + MTZ: **p < 0.01; MTZ versus AKTi + MTZ: ***p < 0.001. Mean ± SEM are shown.
Biochemistry.
Signaling in the mouse frontal cortex was determined after treatment of both WT and βarr2-KO mice with vehicle (0.9% saline, n = 23 per genotype), 5-HTP (100 mg/kg, i.p., n = 16 per genotype), or 5-MeO-DMT (10 mg/kg, i.p., n = 5 WT and 6 βarr2-KO). Mice were killed by cervical dislocation, and tissue was frozen immediately in liquid nitrogen. Tissue was homogenized by polytronic tissue grinder, followed by glass-on-glass dounce homogenization and then solubilized overnight at 4°C in immunoprecipitation lysis buffer (20 mm NaF, 10% glycerol, 50 mm Tris, 150 mm NaCl, 0.5% NP-40, 1 mm PMSF, and 1 mm EDTA) containing a Mini EDTA-free protease inhibitor cocktail tablet (Roche Diagnostics) (Luan et al., 2009). Protein levels were determined by a protein assay kit compatible with detergent-based buffers (Bio-Rad) and 25–30 μg of protein per lane were resolved on 10% Bis-Tris gels (Invitrogen). Proteins were transferred to polyvinylidene fluoride membranes (Millipore Corporation) and immunoblotted for total-Akt or phospho-Akt. Chemiluminescence was detected using a Kodak 2000R imaging system (Eastman Kodak), and fluorescence was detected using an Odyssey Infrared imaging system (LI-COR Biosciences). Blots were quantified using NIH ImageJ analysis software. For each sample, phospho-Akt levels were normalized to corresponding total-Akt levels, and fold stimulation was determined by normalizing to the average vehicle levels for each experiment.
The 5-HT2AR was immunoprecipitated from equivalent aliquots of remaining lysates by overnight incubation with a polyclonal rabbit antibody against the N terminus of the 5-HT2AR (1:133; Neuromics) at 4°C; complexes were immobilized on Protein A Sepharose 4 Fast Flow (GE Healthcare) beads for 3 h at room temperature. Beads were washed three times in lysis buffer, and proteins were eluted by heating in SDS sample buffer containing 5% β-mercaptoethanol at 55°C for 20 min. Immunoblot analysis was performed as described above, and representative Western blots are shown (vehicle, n = 11–24 WT and 19–24 βarr2-KO; 5-HTP, n = 4–18 WT and 13–18 βarr2-KO; 5-MeO-DMT, n = 5–7 WT and 6–7 βarr2-KO). Any alterations to enhance brightness or contrast were applied to the entire gel image. For quantification of the immunoprecipitation studies, the protein levels were first normalized to their corresponding 5-HT2AR immunoblots, and then drug treatment was normalized to vehicle-treated controls.
Primary neuronal cultures were generated from postnatal day 1 mouse neonates obtained from homozygous breeding of βarr2-KO or WT mice (Askwith et al., 2004; Schmid et al., 2008). Frontal cortex was removed and digested in Leibovitz's L-15 (Invitrogen) solution containing 0.025% bovine serum albumin and 0.0375% papain for 15 min at 37°C with 95% oxygen/5% carbon dioxide gently agitating the media. Tissue was washed in complete Neurobasal-A media (with 2% B-27 serum supplement, 0.5 mm l-glutamine, 1.675 μg/L sodium selenite, 2.5 mg/L insulin, 1.375 mg/L transferring, and 50 μg/ml gentamycin; Invitrogen). A single-cell suspension was generated by triturating the tissue with glass pipettes of decreasing diameter. Neurons were then plated in Neurobasal-A media in 12-well dishes (one neonate = two wells) coated with poly-l-lysine (Sigma-Aldrich).
Activation of Akt in neuronal cultures was determined on the fifth day after plating. Unless stated otherwise, neurons were incubated in serum-free MEM (Invitrogen) for 1 h before a 10 min drug treatment with 1 μm agonist. For each assay, the number of wells of neurons treated over at least four separate experiments is provided. For agonist-induced Akt studies, neurons were treated with “agonist vehicle” (2 μm ascorbate in water; n = 56 WT and 35 βarr2-KO) or serotonin (n = 35 WT and 23 βarr2-KO), N-methylserotonin (n = 19 WT and 12 βarr2-KO), or 5-MeO-DMT (n = 13 WT and 12 βarr2-KO). For Figure 5B, WT neurons were pretreated with M100907 (10 nm) (Johnson-Farley et al., 2005) or M100907 vehicle (0.0001% DMSO) during the last 15 min of the serum starvation and before agonist stimulation. Neurons were then treated with either agonist vehicle or serotonin (n = 8 for each of the 4 conditions). For time course studies, neurons were lysed 0, 1, 3, 5, 10, 20, and 30 min after treatment with serotonin (n = 4–8 WT and 4–6 βarr2-KO), N-methylserotonin (n = 3–6 per genotype), or 5-MeO-DMT (n = 4–6 per genotype). A concentration curve was completed for serotonin stimulation of Akt, wherein WT neurons were treated with agonist vehicle or 0.001–10 μm serotonin (n = 8 per dose) (data not shown). To compare basal phospho-Akt to total-Akt ratios between genotypes, WT and βarr2-KO neurons were plated concurrently and lysed after treatment with agonist vehicle (n = 9 per genotype) (data not shown). For Figure 5E, inhibitor vehicle (0.1% DMSO) or 1 μm LY294002 or PP2 was added to the serum-free media during the 1 h incubation. Neurons were then treated with either agonist vehicle or serotonin (Gingerich and Krukoff, 2008) (inhibitor vehicle + agonist vehicle, n = 17; LY294002 + agonist vehicle, n = 9; PP2 + agonist vehicle, n = 10; inhibitor vehicle + serotonin, n = 19; LY294002 + serotonin, n = 10; PP2 + serotonin, n = 11).
Figure 5.
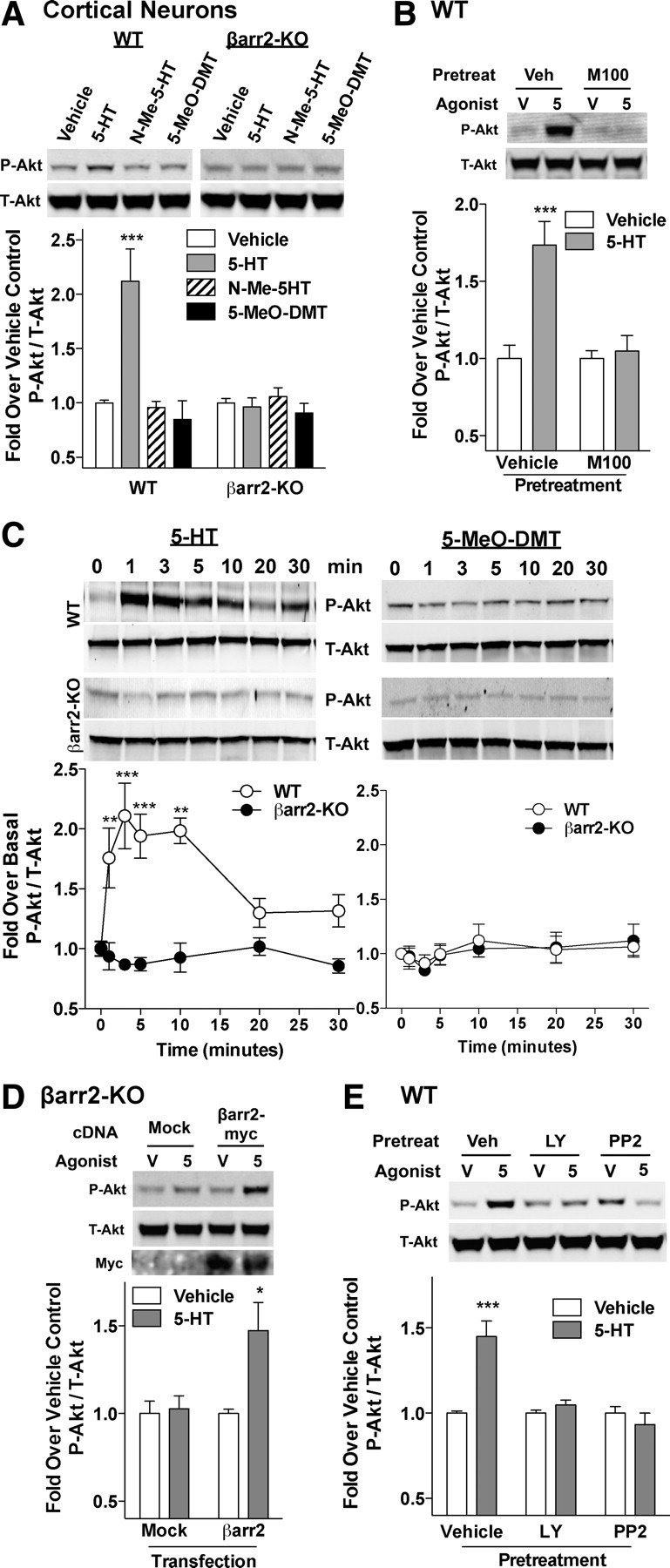
Serotonin stimulates Akt phosphorylation in mouse cortical neurons. A, A 10 min 5-HT (1 μm) treatment induces Akt phosphorylation in WT but not βarr2-KO primary cortical cultures. N-Methylserotonin (N-Me-5-HT) and 5-MeO-DMT do not stimulate Akt phosphorylation in either genotype. Vehicle versus 5-HT: ***p < 0.001. B, M100907 pretreatment (M100; 10 nm for 15 min) inhibits Akt phosphorylation by serotonin (5; 1 μm for 10 min) compared with neurons that were pretreated with vehicle [M100 vehicle (Veh; Vehicle on abscissa) = 0.0001% DMSO; serotonin vehicle (V; Vehicle in the legend) = 2 μm ascorbate] for the same time period. Vehicle versus 5-HT: ***p < 0.001, Bonferroni's post hoc analysis. C, Time course studies reveal Akt phosphorylation after treatment with 1 μm serotonin (left) but not 5-MeO-DMT (right) in primary cortical cultures from WT mice, whereas βarr2-KO neurons do not show Akt activation with either agonist. WT versus βarr2-KO: **p < 0.01, ***p < 0.001, Bonferroni's post hoc analysis. D, Serotonin induces (5; 1 μm for 10 min) Akt phosphorylation compared with vehicle (V; 2 μm ascorbate) in βarr2-KO neurons transfected with Myc-tagged β-arrestin2 (βarr2-myc), whereas those neurons transfected with empty vector (Mock) do not. Vehicle versus 5-HT: *p < 0.05. E, Pretreatment (10 μm for 1 h) with the PI3K inhibitor [LY294002 (LY)] or the Src inhibitor (PP2) blocks Akt phosphorylation induced by serotonin (5; 1 μm for 10 min) in WT primary cortical neurons [inhibitor vehicle (Veh; Vehicle on abscissa) = 0.1% DMSO; serotonin vehicle (V; Vehicle in the legend) = 2 μm ascorbate). Vehicle versus 5-HT: ***p < 0.001. Representative blots and densitometric analysis are provided. Mean ± SEM are shown.
Rescue of serotonin-mediated Akt phosphorylation was accomplished by transfecting βarr2-KO neurons with 4 μg of N-terminal Myc-tagged β-arrestin2 or empty vector (pcDNA3.1) via the calcium phosphate method (Jiang et al., 2004) (Clontech/BD Bioscience). After incubation in the DNA/calcium phosphate suspension for 2 h at 37°C, neurons were washed twice with complete Neurobasal-A medium. Forty-eight hours after transfection, neurons were treated with either agonist vehicle or serotonin (n = 9 for each condition).
After treatment with agonist, primary neurons were washed once in PBS, and lysates were prepared in lysis buffer (20 mm Tris, 150 mm NaCl, 2 mm EDTA, 0.1% SDS, 1% NP-40, 0.25% deoxycholate, 1 mm sodium orthovanadate, 1 mm PMSF, 1 mm NaF, and protease inhibitors) (Groer et al., 2007; Schmid et al., 2008). Lysates were prepared and Western blots were performed as described above to assess phospho-Akt and total-Akt levels. For β-arrestin2-rescue experiments, immunoblots were also probed with c-Myc antibody to confirm transfection. For each sample, phospho-Akt levels were normalized to corresponding total-Akt levels, and fold stimulation was determined by normalizing to the average vehicle levels for each experiment (or untreated controls for time course experiments).
Statistical analysis.
Two-way ANOVA was used to examine group differences in head-twitch responses, comparing two main effects (such as genotype and dose); three-way ANOVA was used to compare three main effects, such as pretreatment, genotype, and dose. Post hoc comparisons between the total number of twitches between drug doses or across genotypes, as well as comparisons of the biochemical data were completed with a Student's unpaired, two-tailed t test. All statistics were conducted with a confidence interval of 95% and were performed using GraphPad Prism 5.0 software (GraphPad Software) with the exception of the three-way ANOVAs, which were performed using PASW/SSPS 18.0 software (IBM). Three-way ANOVAs were followed by two-way ANOVA to further examine main effects within groups if visualization prompted the hypothesis that effects may differ within the groups. The use of three-way and one-way ANOVAs are noted in the text; otherwise, the analyses represent two-way ANOVA. Unless stated otherwise, Student's t tests are presented in the figure legends.
Results
High doses of serotonin induce head twitches in βarr2-KO mice
Previously, we showed that mice lacking β-arrestin2 do not display a head-twitch response to a moderate dose of 5-HTP (100 mg/kg, i.p.) (Fig. 1B), suggesting that serotonin requires β-arrestin2 to mediate this effect (Schmid et al., 2008). However, as the dose of 5-HTP increases, the number of twitches observed in both the WT and the βarr2-KO mice also increases (Fig. 1B: for genotype, F(1,42) = 7.99, p = 0.0072; for dose, F(2,42) = 15.30, p < 0.0001). In effect, when the dose of 5-HTP is doubled, the response in the βarr2-KO mice approaches that observed in WT mice (Fig. 1A: for genotype, F(1,156) = 8.15, p = 0.0049; for time, F(11,156) = 7.15, p < 0.0001), indicating that high doses of 5-HTP can ultimately induce a head-twitch response in mice lacking β-arrestin2.
To ensure that this response was not attributable to unanticipated differences in the conversion of 5-HTP to serotonin in the βarr2-KO animals, mice were also treated directly with serotonin. After intracerebroventricular injection, lower doses of serotonin produce robust head twitches in WT mice that greatly exceed those observed in βarr2-KO mice (Fig. 1D: interaction genotype × dose, F(2,22) = 9.27, p = 0.0012), which seems to be in agreement with the previous observations. Again, when the dose is escalated, the βarr2-KO mice display more twitches than their WT littermates (Fig. 1C: for genotype, F(1,54) = 16.61, p = 0.0002; for time, F(5,54) = 17.62, p < 0.0001). Importantly, the effects of the highest doses of 5-HTP and serotonin were blocked by pretreatment with the 5-HT2AR antagonist M100907 (Fig. 1B: effect of M100907, F(3,23) = 8.37, p = 0.0006, one-way ANOVA; Fig. 1D: effect of M100907, F(3,15) = 16.42, p < 0.0001, one-way ANOVA), demonstrating that these actions are indeed mediated via the 5-HT2AR. Therefore, a question arises as to whether the deletion of β-arrestin2 simply shifts the relative potency of serotonin at the 5-HT2AR or if serotonin really requires β-arrestin2 for signaling, as initially proposed (Schmid et al., 2008).
To further explore this question, we asked whether serotonin metabolites might contribute to the behavioral response. When serotonin levels are elevated, the neurotransmitter is primarily metabolized by MAO-A into 5-hydroxyindoleacetic acid (5-HIAA). However, serotonin can also be metabolized by N-methyltransferases into N-methyltryptamines, including N-methylserotonin, N,N-dimethylserotonin (bufotenine), and indirectly into dimethyltryptamine (Axelrod, 1962; Saavedra and Axelrod, 1972; Rosengarten and Friedhoff, 1976; Crooks et al., 1979). Whereas 5-HIAA is psychoactively inert, N-methyltryptamines are active neurotransmitters at the 5-HT2AR and have been reported to induce hallucinations in humans when taken directly (Glennon and Gessner, 1979; Shulgin and Shulgin, 1997; McBride, 2000). To determine whether the metabolism of serotonin could affect the number of head twitches observed in the WT and βarr2-KO mice, the MAO-A inhibitor clorgyline was administered before low and moderate doses of 5-HTP treatment. As anticipated, inhibition of MAO-A shifts the dose–response curve leftward such that βarr2-KO mice now respond to doses of 5-HTP that are ineffective when given alone (Fig. 1E: three-way ANOVA, multivariate analysis for the main effect of time, F(11,14) = 4.87, p = 0.003; two-way ANOVA for WT, interaction of pretreatment × time, F(11,156) = 2.89, p = 0.0017; βarr2-KO, interaction of pretreatment × time, F(11,132) = 2.18, p = 0.019; vehicle pretreatment, interaction of genotype × time, F(11,108) = 6.98, p < 0.0001; clorgyline pretreatment, genotype, F(1,180) = 6.48, p = 0.0118; Fig. 1F: three-way ANOVA, for dose, F(1,42) = 6.07, p = 0.0180; for pretreatment, F(1,42) = 26.00, p < 0.001; for genotype, F(1,42) = 4.59, p = 0.0380). Again, these effects are mediated through activation of the 5-HT2AR as the enhanced 5-HTP responses in both WT and βarr2-KO mice are completely blocked by the 5-HT2AR antagonist (Fig. 1F: effect of M100907 on 100 mg/kg clorgyline treatment, F(3,21) = 5.21, p = 0.0076, one-way ANOVA). Because clorgyline blocks the primary metabolic pathway for serotonin degradation, the increase in head twitches observed in the clorgyline pretreated βarr2-KO mice may reflect an increase in N-methyltryptamine synthesis resulting from high levels of serotonin.
Inhibitors of N-methyltransferase prevent the head-twitch response in βarr2-KO mice
High doses of 5-HTP promote high levels of serotonin in brain and may thereby facilitate N-methyltransferase metabolism of serotonin to N-methyltryptamines (Axelrod, 1962). To determine the role of N-methyltryptamines in mediating the head-twitch response, we attempted to suppress N-methyltryptamine synthesis using an N-methyltransferase inhibitor, MTZ before injection with a dose of 5-HTP (200 mg/kg), which induces the greatest number of head twitches in the βarr2-KO mice (Fig. 1A,B) (Borchardt and Wu, 1976; Domino, 1976; Mandel et al., 1978; Rokach et al., 1980). The N-methyltransferase inhibitor decreases the number of 5-HTP-induced head twitches in both WT and βarr2-KO mice, although the inhibitory effect is more pronounced in the βarr2-KO mice (Fig. 2A: three-way ANOVA for interactions of genotype × time, F(11,4) = 5.99, p = 0.0492; two-way ANOVA for WT, pretreatment, F(1,72) = 34.84, p < 0.0001; time, F(11,72) = 12.11, p < 0.001; for βarr2-KO, interaction of pretreatment × time, F(11,96) = 6.51, p < 0.0001; for vehicle pretreatment, genotype, F(1,84) = 22.30, p < 0.0001; time, F(11,84) = 18.18, p < 0.0001; MTZ pretreatment, interaction of genotype × time, F(11,84) = 6.48, p < 0.0001; Fig. 2B: for genotype, F(1,14) = 16.25, p = 0.0012; for pretreatment, F(1,14) = 21.95, p = 0.0004). These findings suggest that the head-twitch response induced in the βarr2-KO mice may predominantly be attributed to the actions of the N-methyltryptamines at the 5-HT2AR rather than those of serotonin per se.
N-methyltryptamine-induced head-twitch responses
The actions of N-methyltryptamines at the 5-HT2AR were directly evaluated in vivo using the endogenous metabolite N-methylserotonin, as well as a psychoactive yet hydrolysis-resistant dimethyltryptamine, 5-MeO-DMT. Direct intracerebroventricular injection of increasing doses of N-methylserotonin induces a greater number of head twitches in the βarr2-KO mice than their WT littermates, a response similar to that observed for the highest dose of serotonin tested (Fig. 3A: for genotype, F(1,48) = 5.23, p = 0.0002; for time, F(5,48) = 14.74, p < 0.0001; Fig. 3B: interaction genotype × dose, F(2,20) = 6.45, p = 0.0069). N-Methylserotonin induces head twitches through activation of the 5-HT2AR as the antagonist M100907 significantly inhibits the response in both genotypes (Fig. 3C: interaction genotype × pretreatment, F(1,12) = 6.60, p = 0.0246). Importantly, inhibition of N-methyltransferase by MTZ has no effect on N-methylserotonin-induced head twitches in WT mice, arguing against a nonspecific blockade of the behavior by the enzyme inhibitor (Fig. 3D: for pretreatment, p > 0.05).
A similar response profile is observed after systemic treatment with 5-MeO-DMT wherein the βarr2-KO mice display an enhanced head-twitch response compared with their WT littermates (Fig. 3E: for genotype, F(1,144) = 32.70, p < 0.0001; for time, F(5,144) = 54.67, p < 0.0001; Fig. 3F: interaction genotype × dose, F(2,49) = 3.79, p = 0.0294). In addition, 5-HT2AR blockade with M100907 abrogates 5-MeO-DMT-induced head twitches in both genotypes (Fig. 3G: interaction genotype × pretreatment, F(1,20) = 5.85, p = 0.0253), whereas MTZ pretreatment has no effect on 5-MeO-DMT-induced responses in normal mice (Fig. 3H: for pretreatment, p > 0.05). These findings suggest that N-methyltryptamines do not require β-arrestin2 to produce the head-twitch response in vivo and that β-arrestin2 may play a negative regulatory role in this cascade because the mice consistently display greater responses to the N-methyltryptamines when the protein is deleted.
Phosphorylation of Akt in the frontal cortex
The findings presented thus far suggest that the behavioral response to serotonin in the βarr2-KO mice may be fundamentally different from that observed in the WT mice. To elucidate the signaling mechanisms underlying the differences in the head-twitch response between the two genotypes, the effects of serotonin on the 5-HT2AR were evaluated at the biochemical level. Therefore, we assessed agonist-induced activation of the serine–threonine kinase Akt in WT and βarr2-KO frontal cortex after treatment with 5-HTP and 5-MeO-DMT. To attempt to exclude the contribution of the metabolites after the 5-HTP treatment, we used the dose of 5-HTP (100 mg/kg, i.p.) that only produced a head-twitch response in the WT mice, presuming that the head twitches observed in the βarr2-KO mice at higher doses are indicative of the presence of serotonin metabolites. The dose of 5-MeO-DMT (10 mg/kg, i.p.) was chosen because it induces the greatest number of head twitches in both genotypes and therefore should elicit the most robust activation of the receptor signaling cascade. Treatment of WT mice with 5-HTP induces phosphorylation of Akt at threonine 308 (Fig. 4A: WT, vehicle vs serotonin, p = 0.0018, Student's t test). However, Akt is not activated in the frontal cortex of βarr2-KO mice after 5-HTP treatment (βarr2-KO, vehicle vs 5-HTP, p > 0.05, Student's t test). Treatment with 5-MeO-DMT does not induce Akt phosphorylation in either genotype (vehicle vs 5-MeO-DMT, p > 0.05, Student's t test). Importantly, comparison of vehicle-treated animals reveals no differences in Akt phosphorylation levels between genotypes, suggesting that the lack of serotonin-induced Akt stimulation in the βarr2-KO mice is not attributable to an elevated basal state of Akt activation in the frontal cortex (data not shown). Together, these findings suggest that serotonin and 5-Meo-DMT differ in their ability to activate Akt, such that serotonin requires β-arrestin2, whereas 5-MeO-DMT does not activate this kinase in frontal cortex.
Figure 4.

5-HTP stimulates Akt phosphorylation and the association of β-arrestin2/Akt/Src complex with the 5-HT2AR in the frontal cortex. A, Treatment with 5-HTP (100 mg/kg, i.p.) for 10 min induces Akt phosphorylation (P-Akt) in the frontal cortex of WT but not βarr2-KO mice. 5-MeO-DMT (10 mg/kg, i.p.) does not induce Akt phosphorylation in either genotype [total Akt (T-Akt)]. Vehicle versus 5-HTP: **p < 0.01. B, 5-HT2AR immunoprecipitation reveals that 5-HTP treatment (100 mg/kg, i.p.) for 10 min decreases 5-HT2AR association with PSD-95 but increases 5-HT2AR associations with β-arrestin2, Src, and Akt. 5-HTP treatment has no effect on PSD-95, Src, or Akt coimmunoprecipitation with 5-HT2AR in βarr2-KO mice. C, 5-MeO-DMT treatment (10 mg/kg, i.p. for 10 min) does not displace PSD-95 from the immunoprecipitated 5-HT2AR, nor does it cause associations with β-arrestin2, Src, or Akt in either WT or βarr2-KO mice. A “no protein” control (NP; antibody + beads) is shown for each representative immunoblot. The mean ± SEM of the densitometric analysis is shown. IB, Immunoblot.
Identification of a β-arrestin2-dependent 5-HT2AR signaling scaffold in frontal cortex
To directly implicate the 5-HT2AR in the agonist-induced signaling in vivo, the receptor was immunoprecipitated from the frontal cortex of both WT and βarr2-KO mice after drug treatment. We then examined the contribution of β-arrestin2 in assembling an Akt signaling scaffold by probing for proteins that associate with the receptor in response to 5-HTP or 5-MeO-DMT. Interestingly, treatment of WT mice with 5-HTP at the same dose that stimulates Akt phosphorylation in the frontal cortex reveals a depletion of PSD-95 from the receptor complex and a recruitment of β-arrestin2, Src, and Akt (Fig. 4B: WT, vehicle vs 5-HTP: PSD-95, p = 0.0004; β-arrestin2, p = 0.0024; Src, p = 0.0017; Akt, p = 0.0002, Student's t test). In contrast, in the absence of β-arrestin2, there is no depletion of PSD-95 or recruitment of Src or Akt to the receptor in response to 5-HTP (βarr2-KO, vehicle vs 5-HTP, p > 0.05, Student's t test). In agreement with the studies presented in Figure 4A, treatment with 5-MeO-DMT does not lead to the recruitment of β-arrestin2, Src, or Akt to the 5-HT2AR in either genotype (Fig. 4C: vehicle vs 5-MeO-DMT, p > 0.05, Student's t test). These findings demonstrate that β-arrestin2 is integral to mediating serotonin-induced assembly of a 5-HT2AR/Src/Akt signaling complex and that 5-MeO-DMT does not recruit this complex in the frontal cortex.
Phosphorylation of Akt in primary cortical neurons
To further characterize 5-HT2AR-mediated serotonin-induced Akt activation, we used primary neuronal cultures generated from the frontal cortex of WT and βarr2-KO neonates. Serotonin induces robust Akt phosphorylation in WT cortical neurons, which is maximal at 1 μm (Fig. 5A: WT, vehicle vs serotonin, p < 0.0001, Student's t test; dose–response not shown). However, N-methylserotonin and 5-MeO-DMT do not activate the kinase (Fig. 5A: WT, vehicle vs N-methylserotonin or 5-MeO-DMT, p > 0.05, Student's t test). In agreement with the observations made in the adult frontal cortex in Figure 4A, none of the agonists induce Akt phosphorylation in neuronal cultures derived from βarr2-KO neonates (Fig. 5A: βarr2-KO, vehicle vs agonist, p > 0.05, Student's t test). Furthermore, pretreatment with M100907 prevents serotonin-induced Akt phosphorylation in WT neurons, demonstrating that the actions of serotonin are mediated via the 5-HT2AR (Fig. 5B: vehicle vs serotonin: vehicle-pretreated, p = 0.0009; M100907-pretreated, p > 0.05, Student's t test).
To be certain that the lack of Akt phosphorylation observed in the above studies was not attributable to temporal shifts in kinase activation profiles, we performed several time-dependent studies. Serotonin treatment over time reveals that Akt is phosphorylated within 1 min and peaks at 5–10 min in the WT neurons. The absence of Akt phosphorylation at all time points in the βarr2-KO neurons demonstrates that there is not a β-arrestin2-dependent temporal shift in the activation profile, but rather that β-arrestin2 is essential for serotonin activation of Akt (Fig. 5C, left: for genotype, F(1,69) = 55.47, p < 0.0001; for time, F(6,69) = 2.40, p = 0.0367). In addition, 5-MeO-DMT (Fig. 5C, right) and N-methylserotonin (data not shown) fail to induce Akt phosphorylation in both genotypes at any time point tested, thereby demonstrating that the lack of Akt activation is not attributable to a temporal shift in activation profiles for these agonists (p > 0.05).
To determine whether the lack of serotonin-induced Akt phosphorylation in the βarr2-KO neurons is attributable to higher relative basal Akt activation levels, we directly compared phosphorylated Akt levels from vehicle-treated neurons of both genotypes run in parallel on the same gels. The data reveal that there are no differences between genotypes, suggesting that the inability of serotonin to stimulate Akt phosphorylation in the βarr2-KO neurons is not attributable to increased basal phosphorylation of the kinase in these preparations (data not shown). In addition, treatment with fetal bovine serum stimulates Akt phosphorylation in βarr2-KO neurons, demonstrating that Akt can be activated in this system (data not shown). Finally, transfection of Myc-tagged β-arrestin2 into βarr2-KO neuronal cultures rescues serotonin-induced Akt phosphorylation, further demonstrating the necessity of β-arrestin2 in the activation of this pathway (Fig. 5D: vehicle vs serotonin: mock transfected, p > 0.05; Myc-βarr2 transfected, p = 0.0100, Student's t test).
Inhibition of serotonin-mediated Akt phosphorylation in neurons
To evaluate the interplay of the signaling elements within the assembled complex, Akt phosphorylation was assessed after inhibiting individual components of the potential signaling cascade in primary cortical neuronal cultures. Phosphoinositide 3-kinase (PI3K) is an activator of Akt downstream of GPCRs, and pretreatment with a PI3K inhibitor (LY294002) prevents serotonin-induced Akt phosphorylation in the WT neurons. The Src inhibitor PP2 also blocks Akt phosphorylation, further implicating these signaling elements in mediating the activation of Akt by serotonin (Fig. 5E: vehicle vs serotonin: vehicle-pretreated, p < 0.0001; inhibitor-pretreated, p > 0.05, Student's t test).
β-Arrestin2/PI3K/Src/Akt mediation of the head-twitch response
To test whether the β-arrestin2/PI3K/Src/Akt complex contributes to the serotonin-mediated head-twitch response, we used kinase inhibitors to disrupt the function of the complex in vivo. Normal C57BL/6J mice were injected (intracerebroventricularly) with inhibitors of PI3K [LY294002 (Beaulieu et al., 2005)], Src [PP2 (Narita et al., 2006)], and Akt [AKTi (Xu et al., 2008)] before treatment with 5-HTP (200 mg/kg, i.p.). At this dose of 5-HTP, we would predict that both serotonin and N-methyltryptamine levels would be elevated. Because Akt only appears to play a role in the actions of serotonin, we might predict that the inhibitors of the Akt signaling cascade would inhibit the serotonin effects but maintain the N-methyltryptamine effects. Accordingly, we find that each of the individual kinase inhibitors blocks ∼50% of the head-twitch response produced in the C57BL/6J mice (Fig. 6A: vehicle vs LY294002: for pretreatment, F(1,156) = 38.52, p < 0.0001; for time, F(11,156) = 5.51, p < 0.0001; PP2, for pretreatment, F(1,156) = 43.02, p < 0.0001; for time, F(11,156) = 6.17, p < 0.0001; AKTi, for pretreatment, F(1,156) = 35.94, p < 0.0001; for time, F(11,156) = 6.49, p < 0.0001; Fig. 6B: F(3,21) = 5.54, p = 0.0058, one-way ANOVA). Interestingly, the Akt inhibitor has no effect on the number of head twitches induced by 5-MeO-DMT (Fig. 6C: p > 0.05). These data suggest that the head twitches induced by a high dose of 5-HTP can be partially blocked by inhibition of the Akt signaling cascade, suggesting that the remaining activity is either attributable to 5-HT2AR activity that is independent of this cascade or, alternatively, that metabolites of serotonin are signaling in an Akt-independent manner to mediate the head-twitch response.
Figure 6.
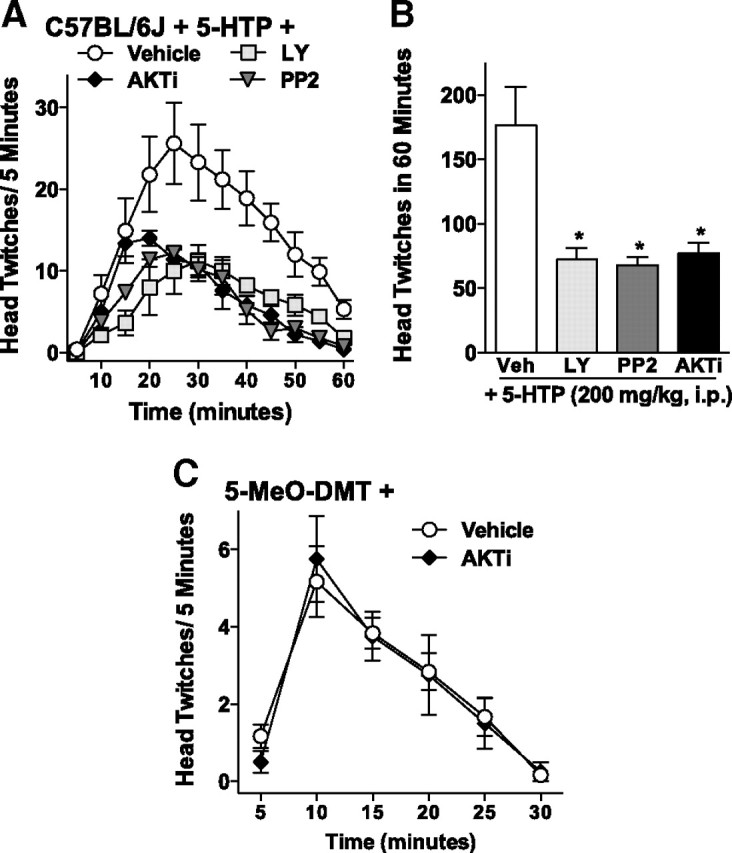
Inhibition of PI3K, Src, or Akt attenuates 5-HTP-mediated but not N-methyltryptamine-mediated head twitches in mice. A, B, Inhibitors of PI3K (LY294002), Src (PP2), or Akt (AKTi) attenuated 5-HTP-induced head twitches in normal mice. Time course of head-twitch responses (A) and the total number of head twitches (B) observed in C57BL/6J mice that were pretreated for 10 min with either vehicle (Veh; 1% DMSO in dH2O, i.c.v.) or LY294002 (LY; 125 ng, i.c.v.), PP2 (300 ng, i.c.v.), or AKTi (55 ng, i.c.v.) before 5-HTP administration (200 mg/kg, i.p.). Vehicle versus inhibitor: *p < 0.05. C, Pretreatment with AKTi (55 ng, i.c.v. for 10 min) has no effect on 5-MeO-DMT-induced (10 mg/kg, i.p.) head twitches in C57BL/6J mice compared with vehicle-pretreated (1% DMSO in dH2O, i.c.v.) mice. Mean ± SEM are shown.
In contrast, treatment of βarr2-KO mice with PI3K, Src, and Akt inhibitors before treatment with 5-HTP (200 mg/kg, i.p.) does not lead to suppression of the head-twitch response (Fig. 7A: LY294002, PP2, and AKTi, for pretreatment, p > 0.05; Fig. 7B: p > 0.05, one-way ANOVA). This may not be unanticipated because the behavioral data suggest that the highest dose of 5-HTP produces the head-twitch response in the βarr2-KO mice primarily as a result of the actions of the N-methyltryptamine metabolites (Fig. 2) and these metabolites do not activate Akt in vivo or in neuronal cultures (Figs. 4, 5). These findings further support a model wherein the head twitches that can be observed in the βarr2-KO mice occur independent of a PI3K, Src, and Akt signaling mechanism.
Figure 7.

Inhibition of PI3K, Src, or Akt has no affect on 5-HTP-induced head twitches in βarr2-KO mice. Inhibitors of PI3K (LY294002), Src (PP2), or Akt (AKTi) have no effect on 5-HTP-induced head twitches in βarr2-KO mice. Time course of head-twitch responses (A) and the total number of head twitches (B) observed in βarr2-KO mice that were pretreated for 10 min with either vehicle (Veh; 1% DMSO in dH2O, i.c.v.) or LY294002 (LY; 125 ng, i.c.v.), PP2 (300 ng, i.c.v.), or AKTi (55 ng, i.c.v.) before 5-HTP administration (200 mg/kg, i.p.). Mean ± SEM are shown.
Separation of serotonin and N-methyltryptamine responses in vivo
Together, the biochemical and behavioral data suggest that the head-twitch response induced by high doses of 5-HTP or serotonin in WT mice may reflect the activity of both serotonin and N-methyltryptamines. To directly test this hypothesis, WT mice were treated with the Akt inhibitor (to block serotonin-mediated signaling) and the N-methyltransferase inhibitor (to prevent N-methyltryptamine synthesis) before treatment with a dose of 5-HTP (200 mg/kg, i.p.) that produces the greatest response when given alone (Fig. 1A). Remarkably, this double blockade nearly abolishes the 5-HTP-induced head-twitch response in WT mice (Fig. 8: compared with vehicle: AKTi, for pretreatment, F(1,180) = 96.55, p < 0.0001; for time, F(11,180) = 19.77, p < 0.0001; MTZ, for pretreatment, F(1,168) = 53.73, p < 0.0001; for time, F(11,168) = 18.82, p < 0.0001; AKTi + MTZ, for pretreatment, F(1,168) = 172.24, p < 0.0001; for time, F(11,168) = 10.77, p < 0.0001).
Discussion
In this study, we demonstrate functional selectivity at the 5-HT2AR whereby serotonin and N-methyltryptamines differentially induce the head-twitch response in mice and promote differential signaling in mouse frontal cortex and in primary cortical neurons. We show that serotonin activates the formation of a receptor signaling complex that includes β-arrestin2, Src, and Akt and that blockade of any individual component, as well as PI3K, prevents the full expression of the head-twitch response, thus demonstrating the physiological relevance of this complex formation. Furthermore, we also show that, although N-methyltryptamines mediate the head-twitch response through activation of the 5-HT2AR, the mechanism is independent of β-arrestin2 and does not require Akt activation (summarized in Fig. 9). Together, these findings point to a signaling pathway that may represent a signature of the actions of serotonin at the 5-HT2AR in cortex that is distinct from those activated by N-methyltryptamines.
Figure 9.

Schematic representing 5-HT2AR signaling in vivo. When serotonin levels are increased (by direct administration or by blocking the MAO-A route of metabolism to the inactive 5-HIAA using clorgyline), an elevation of psychoactive N-methyltryptamine metabolites can occur via N-methyltransferase (NMT) metabolism of serotonin and tryptamines. Serotonin and these psychoactive metabolites then differentially activate the 5-HT2AR to produce the head-twitch response in mice. Left, Serotonin promotes disengagement of PSD-95 and recruitment of β-arrestin2, PI3K, Src, and Akt to the 5-HT2AR. This leads to Akt phosphorylation, which can be prevented when any member of this scaffold is disrupted. Inhibition of the kinases or disruption of the scaffold by removal of β-arrestin2 reduces serotonin-mediated head twitches in WT mice by ∼50%. Right, N-Methyltryptamines at the 5-HT2AR do not induce Akt phosphorylation and mediate head twitches in mice independent of β-arrestin2 and Akt, because neither kinase inhibition nor β-arrestin2 deletion blocks N-methyltryptamine-induced head twitches. Furthermore, β-arrestin2 appears to dampen the effect of N-methyltryptamines in the head-twitch response as the response to the N-methyltryptamines is enhanced in the βarr2-KO mice. All of these effects are mediated by the 5-HT2AR as the antagonist M100907 blocks the response in its entirety. Finally, an inhibitor of N-methyltransferase (MTZ) prevents the β-arrestin2-independent head-twitch response that occurs after administration of high doses of serotonin, further indicating that this response is mediated by the N-methyltryptamines.
The differences between these neurotransmitters, observed in both behavioral and biochemical responses, fit into the conceptual framework of functional selectivity, an idea based on the understanding that a ligand can selectively stabilize a receptor conformation, causing the receptor to preferentially interact with particular intracellular signaling components (Kenakin, 1995; Mailman, 2007; Urban et al., 2007; Violin and Lefkowitz, 2007; Rajagopal et al., 2010). Agonist-directed signaling at the 5-HT2AR has been demonstrated previously in vitro wherein certain agonists have been shown to preferentially stimulate different degrees of phospholipase C or phospholipase A2 activity (Berg et al., 1998; Kurrasch-Orbaugh et al., 2003; Moya et al., 2007). Moreover, functional selectivity at the 5-HT2AR has long been speculated in vivo, because not all agonists at the receptor induce hallucinations in humans (Pieri et al., 1978). Studies addressing differences between hallucinogenic and non-hallucinogenic agonists have shown that LSD (hallucinogen) and lisuride (non-hallucinogen) differ in their ability to induce the head-twitch response and activate distinct patterns of gene expression in the frontal cortex (González-Maeso et al., 2003, 2007). Our findings further support the physiological relevance of agonist-directed signaling at the 5-HT2AR in vivo, because two endogenous neurotransmitters (hallucinogenic N-methyltryptamines and non-hallucinogenic serotonin) differentially use β-arrestin2 to signal and induce the head-twitch response.
The fact that functional selectivity can be observed between two endogenous neurotransmitters may have a significant impact on how modulation of 5-HT2AR activation is targeted from a drug discovery perspective. This may be particularly important for the treatment of depression wherein traditional therapies focus on elevating endogenous serotonin levels that can produce adverse side effects such as a “serotonin syndrome.” This condition can be accompanied by hallucinations and is particularly problematic when selective serotonin reuptake inhibitors are taken with monoamine oxidase inhibitors (Insel et al., 1982; Sternbach, 1991; Boyer and Shannon, 2005) when serotonin, and presumably N-methyltryptamine, levels would be greatly elevated. If we surmise that the actions of serotonin are beneficial as opposed to the actions of the N-methyltryptamines (which can be hallucinogenic), then, from a therapeutic perspective, it would be beneficial to directly mimic the actions of serotonin at the receptor. This would involve the identification of agonists that preferentially engage the β-arrestin2-dependent pathway as opposed to a G-protein-mediated, β-arrestin2-independent signaling pathway.
The divergence in neurotransmitter-directed 5-HT2AR signaling may also have implications for the treatment of schizophrenia, because there has been a long-standing hypothesis that the hallucinations associated with this disease may be attributed to N-methyltryptamine actions (Kety, 1959; Snyder et al., 1974; Domino, 1976). Moreover, schizophrenic patients that experienced hallucinations as part of the pathology were found to have elevated N-methyltryptamine levels in urine, which may indicate higher endogenous levels of these amines (Bidder et al., 1974; Lipinski et al., 1974; Angrist et al., 1975; Takeda et al., 1995; Emanuele et al., 2010). Because both serotonin and the N-methyltryptamines produce the same behavior in mice (the head-twitch response), it is not clear whether the two agonists will promote the same physiological responses in humans or whether the N-methyltryptamines contribute to the hallucinations experienced in schizophrenia. However, the endogenous neurotransmitter serotonin is not considered to be a hallucinogen, and the direct administration of N-methyltryptamines can produce hallucinations, further supporting the idea that these two agonists likely have divergent signaling mechanisms in humans. Our studies in mice demonstrate a mechanistic divergence of these neurotransmitters and indicate that it may be possible to pharmacologically inhibit the actions of the N-methyltryptamines while simultaneously permitting signaling that mimics that promoted by serotonin.
The critical role scaffolding molecules play in neurotransmitter receptor signaling may become more apparent when these receptors are studied in neuronal systems. For example, the activation of D2 dopamine receptors leads to β-arrestin2-dependent inhibition of Akt by forming a PP2A/Akt/β-arrestin2 complex, suggesting that β-arrestin2 plays a facilitatory role toward deactivating Akt in this system (Beaulieu et al., 2005, 2008). However, interactions between Akt and Src are necessary for full activation of Akt in some systems (Chen et al., 2001; Jiang and Qiu, 2003), and our findings suggest that this β-arrestin2-facilitated interaction is of functional consequence for serotonin signaling at the 5-HT2AR. In addition to β-arrestin2, the postsynaptic density scaffolding protein PSD-95 has also been shown to interact with the 5-HT2AR and to play an essential role in regulating signal transduction for hallucinogenic drugs at this receptor; PSD-95 promotes clustering of the 5-HT2AR on the plasma membrane in HEK-293 cells (Xia et al., 2003) and is integral for the dendritic targeting of the receptor in cortical pyramidal neurons (Abbas et al., 2009). Our previous work demonstrates that the 5-HT2AR is more highly localized to the plasma membrane of cortical neurons in βarr2-KO mice, although it is constitutively internalized in the WT neurons (Schmid et al., 2008). Our findings here show that, in the absence of β-arrestin2, PSD-95 is not displaced from the 5-HT2AR in response to 5-HTP (Fig. 4B). Altogether, the interplay between β-arrestin2 and PSD-95 may determine whether the receptor is internalized or remains on the cell surface. Moreover, an attractive hypothesis is that β-arrestin2, in concert with PSD-95, represents a fulcrum in the ligand-directed signaling of this receptor in vivo.
The question remains as to how the N-methyltryptamines induce the head-twitch response. Because this effect is independent of β-arrestin2, it is likely that G-protein-dependent signaling mediates the effects of these metabolites. It is well established that the 5-HT2AR couples primarily to Gαq and can also couple to Gαi/o- and Gα12/13-proteins. Interestingly, the genetic deletion of Gαq decreases responsiveness to DOI-induced head twitches, suggesting that this G-protein plays an important role in mediating the effects of DOI in vivo (Garcia et al., 2007). Furthermore, DOI induces head twitches in the βarr2-KO mice, suggesting that its effects are not mediated via a β-arrestin2-dependent pathway (Schmid et al., 2008). Together, these findings suggest that at least part of the β-arrestin2-independent head-twitch response may be mediated through a G-protein pathway.
Interestingly, an enhanced head-twitch response is observed in the βarr2-KO mice after treatment with both 5-MeO-DMT and N-methylserotonin, suggesting that β-arrestin2 may play a negative regulatory role on 5-HT2AR function when activated by these agonists, a phenomenon observed for other GPCRs in vivo (Bohn et al., 1999; Breivogel et al., 2008; Deshpande et al., 2008; Schmid and Bohn, 2009). However, if β-arrestin2 was negatively regulating the receptor, then one might expect to find β-arrestin2 coimmunoprecipitated with the 5-HT2AR after 5-MeO-DMT treatment. This does not occur (Fig. 4C), and it is not clear whether this lack of coimmunoprecipitation is a result of a lack of interaction or a difference in the agonist-induced nature of the interaction that renders the complex less stable during the immunoprecipitation process. Behaviorally, the N-methyltryptamines (either by direct administration or metabolically derived from high doses of serotonin) consistently induce more head twitches in the βarr2-KO mice than in the WT mice, whereas lower doses of serotonin only induce twitches in the WT mice. Therefore, β-arrestin2 appears to be both facilitating (for serotonin) and dampening (for N-methyltryptamines) a particular receptor-mediated response, wherein the agonist determines the regulatory role that β-arrestin2 plays during interaction with the 5-HT2AR (Fig. 9).
Our studies have focused primarily on the 5-HT2AR because this receptor has been most highly correlated with the agonist-induced display of the head-twitch response in mice, although other serotonin receptor subtypes, including the 5-HT2CR and the 5-HT1AR, can modulate the extent of this behavioral response (Arnt and Hyttel, 1989; Berendsen and Broekkamp, 1991; Schreiber et al., 1995; Canal et al., 2010). As with any pharmacological studies performed in vivo, drug effects may be influenced by actions at nontarget receptors, and the variations in the head-twitch response profiles observed between the agonists could reflect differential contributions of other serotonin receptors. Nevertheless, the immunoprecipitation studies reported herein, which use an N-terminal-directed antibody that does not detect the protein in the 5-HT2AR-KO mice (Magalhaes et al., 2010), support the conclusion that the lack of Akt activation by serotonin in the βarr2-KO mice is attributable to disruption of 5-HT2AR signaling.
Overall, these studies demonstrate that, although both serotonin and its metabolic products act at the 5-HT2AR, the functionality of the receptor diverges in vivo at the level of the ligand-directed receptosome formation, which dictates signaling that underlies the display of the head-twitch response in mice. Future drug discovery efforts to identify serotonin mimetics should consider agonist-dependent signal bifurcation at β-arrestin2 because this may have particular relevance to the development of antipsychotic and antidepressant therapies.
Footnotes
This work is funded by National Institute on Drug Abuse Grant DA025158 (L.M.B.). We acknowledge Dr. Robert J. Lefkowitz for providing the initial breeders for the β-arrestin2-KO mouse line and for the A2CT antibody and Dr. Kenner Rice for providing the M100907 compound.
References
- Abbas AI, Yadav PN, Yao WD, Arbuckle MI, Grant SG, Caron MG, Roth BL. PSD-95 is essential for hallucinogen and atypical antipsychotic drug actions at serotonin receptors. J Neurosci. 2009;29:7124–7136. doi: 10.1523/JNEUROSCI.1090-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanian GK, Marek GJ. Serotonin and hallucinogens. Neuropsychopharmacology. 1999;21:16S–23S. doi: 10.1016/S0893-133X(98)00135-3. [DOI] [PubMed] [Google Scholar]
- Angrist B, Thompson H, Shopsin B, Gershon S. Clinical studies with dopamine-receptor stimulants. Psychopharmacologia. 1975;44:273–280. doi: 10.1007/BF00428906. [DOI] [PubMed] [Google Scholar]
- Arnt J, Hyttel J. Facilitation of 8-OHDPAT-induced forepaw treading of rats by the 5-HT2 agonist DOI. Eur J Pharmacol. 1989;161:45–51. doi: 10.1016/0014-2999(89)90178-7. [DOI] [PubMed] [Google Scholar]
- Askwith CC, Wemmie JA, Price MP, Rokhlina T, Welsh MJ. Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem. 2004;279:18296–18305. doi: 10.1074/jbc.M312145200. [DOI] [PubMed] [Google Scholar]
- Axelrod J. The enzymatic N-methylation of serotonin and other amines. J Pharmacol Exp Ther. 1962;138:28–33. [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, Wetsel WC, Lefkowitz RJ, Gainetdinov RR, Caron MG. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- Berendsen HH, Broekkamp CL. Attenuation of 5-HT1A and 5-HT2 but not 5-HT1C receptor mediated behaviour in rats following chronic treatment with 5-HT receptor agonists, antagonists or anti-depressants. Psychopharmacology (Berl) 1991;105:219–224. doi: 10.1007/BF02244313. [DOI] [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- Berg KA, Harvey JA, Spampinato U, Clarke WP. Physiological and therapeutic relevance of constitutive activity of 5-HT 2A and 5-HT 2C receptors for the treatment of depression. Prog Brain Res. 2008;172:287–305. doi: 10.1016/S0079-6123(08)00914-X. [DOI] [PubMed] [Google Scholar]
- Bidder TG, Mandel LR, Ahn HS, VandenHeuvel WJ, Walker RW. Letter: blood and urinary dimethyltryptamine in acute psychotic disorders. Lancet. 1974;1:165. doi: 10.1016/s0140-6736(74)92455-6. [DOI] [PubMed] [Google Scholar]
- Blair JB, Kurrasch-Orbaugh D, Marona-Lewicka D, Cumbay MG, Watts VJ, Barker EL, Nichols DE. Effect of ring fluorination on the pharmacology of hallucinogenic tryptamines. J Med Chem. 2000;43:4701–4710. doi: 10.1021/jm000339w. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Borchardt RT, Wu YS. Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 5. Role of the asymmetric sulfonium pole in the enzymatic binding of S-adenosyl-L-methionine. J Med Chem. 1976;19:1099–1103. doi: 10.1021/jm00231a004. [DOI] [PubMed] [Google Scholar]
- Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112–1120. doi: 10.1056/NEJMra041867. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Lambert JM, Gerfin S, Huffman JW, Razdan RK. Sensitivity to delta9-tetrahydrocannabinol is selectively enhanced in beta-arrestin2−/− mice. Behav Pharmacol. 2008;19:298–307. doi: 10.1097/FBP.0b013e328308f1e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canal CE, Olaghere da Silva UB, Gresch PJ, Watt EE, Sanders-Bush E, Airey DC. The serotonin 2C receptor potently modulates the head-twitch response in mice induced by a phenethylamine hallucinogen. Psychopharmacology (Berl) 2010;209:163–174. doi: 10.1007/s00213-010-1784-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Kim O, Yang J, Sato K, Eisenmann KM, McCarthy J, Chen H, Qiu Y. Regulation of Akt/PKB activation by tyrosine phosphorylation. J Biol Chem. 2001;276:31858–31862. doi: 10.1074/jbc.C100271200. [DOI] [PubMed] [Google Scholar]
- Corne SJ, Pickering RW. A possible correlation between drug-induced hallucinations in man and a behavioural response in mice. Psychopharmacologia. 1967;11:65–78. doi: 10.1007/BF00401509. [DOI] [PubMed] [Google Scholar]
- Corne SJ, Pickering RW, Warner BT. A method for assessing the effects of drugs on the central actions of 5-hydroxytryptamine. Br J Pharmacol Chemother. 1963;20:106–120. doi: 10.1111/j.1476-5381.1963.tb01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks PA, Dreyer RN, Sulens CH, Gillis CN, Coward JK. Determination of 5-hydroxytryptamine metabolites in isolated perfused rabbit lung by high-performance liquid chromatography. Anal Biochem. 1979;93:143–152. [PubMed] [Google Scholar]
- Deshpande DA, Theriot BS, Penn RB, Walker JK. Beta-arrestins specifically constrain beta2-adrenergic receptor signaling and function in airway smooth muscle. FASEB J. 2008;22:2134–2141. doi: 10.1096/fj.07-102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Domino EF. Search for new treatment approaches in schizophrenia: in vitro studies of potential N-methyltransferase inhibitors. Arch Int Pharmacodyn Ther. 1976;221:75–86. [PubMed] [Google Scholar]
- Emanuele E, Colombo R, Martinelli V, Brondino N, Marini M, Boso M, Barale F, Politi P. Elevated urine levels of bufotenine in patients with autistic spectrum disorders and schizophrenia. Neuro Endocrinol Lett. 2010;31:117–121. [PubMed] [Google Scholar]
- Fan J, Cowan CM, Zhang LY, Hayden MR, Raymond LA. Interaction of postsynaptic density protein-95 with NMDA receptors influences excitotoxicity in the yeast artificial chromosome mouse model of Huntington's disease. J Neurosci. 2009;29:10928–10938. doi: 10.1523/JNEUROSCI.2491-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia EE, Smith RL, Sanders-Bush E. Role of G(q) protein in behavioral effects of the hallucinogenic drug 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane. Neuropharmacology. 2007;52:1671–1677. doi: 10.1016/j.neuropharm.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingerich S, Krukoff TL. Activation of ERbeta increases levels of phosphorylated nNOS and NO production through a Src/PI3K/Akt-dependent pathway in hypothalamic neurons. Neuropharmacology. 2008;55:878–885. doi: 10.1016/j.neuropharm.2008.06.058. [DOI] [PubMed] [Google Scholar]
- Glennon RA, Gessner PK. Serotonin receptor binding affinities of tryptamine analogues. J Med Chem. 1979;22:428–432. doi: 10.1021/jm00190a014. [DOI] [PubMed] [Google Scholar]
- González-Maeso J, Yuen T, Ebersole BJ, Wurmbach E, Lira A, Zhou M, Weisstaub N, Hen R, Gingrich JA, Sealfon SC. Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5-hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J Neurosci. 2003;23:8836–8843. doi: 10.1523/JNEUROSCI.23-26-08836.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, Lira A, Bradley-Moore M, Ge Y, Zhou Q, Sealfon SC, Gingrich JA. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Groer CE, Tidgewell K, Moyer RA, Harding WW, Rothman RB, Prisinzano TE, Bohn LM. An opioid agonist that does not induce mu-opioid receptor-beta-arrestin interactions or receptor internalization. Mol Pharmacol. 2007;71:549–557. doi: 10.1124/mol.106.028258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Klippel A, Muslin AJ, Fantl WJ, Williams LT. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science. 1995;268:100–102. doi: 10.1126/science.7701328. [DOI] [PubMed] [Google Scholar]
- Insel TR, Roy BF, Cohen RM, Murphy DL. Possible development of the serotonin syndrome in man. Am J Psychiatry. 1982;139:954–955. doi: 10.1176/ajp.139.7.954. [DOI] [PubMed] [Google Scholar]
- Jakab RL, Goldman-Rakic PS. 5-Hydroxytryptamine2A serotonin receptors in the primate cerebral cortex: possible site of action of hallucinogenic and antipsychotic drugs in pyramidal cell apical dendrites. Proc Natl Acad Sci U S A. 1998;95:735–740. doi: 10.1073/pnas.95.2.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Deng L, Chen G. High Ca2+-phosphate transfection efficiency enables single neuron gene analysis. Gene Ther. 2004;11:1303–1311. doi: 10.1038/sj.gt.3302305. [DOI] [PubMed] [Google Scholar]
- Jiang T, Qiu Y. Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation. J Biol Chem. 2003;278:15789–15793. doi: 10.1074/jbc.M212525200. [DOI] [PubMed] [Google Scholar]
- Johnson-Farley NN, Kertesy SB, Dubyak GR, Cowen DS. Enhanced activation of Akt and extracellular-regulated kinase pathways by simultaneous occupancy of Gq-coupled 5-HT2A receptors and Gs-coupled 5-HT7A receptors in PC12 cells. J Neurochem. 2005;92:72–82. doi: 10.1111/j.1471-4159.2004.02832.x. [DOI] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KL, Edwards DD, Shoichet BK, Roth BL. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kety SS. Biochemical theories of schizophrenia. I. Science. 1959;129:1528–1532. doi: 10.1126/science.129.3362.1528. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Deupi X. Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:397–406. doi: 10.1016/j.tips.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Kurrasch-Orbaugh DM, Parrish JC, Watts VJ, Nichols DE. A complex signaling cascade links the serotonin2A receptor to phospholipase A2 activation: the involvement of MAP kinases. J Neurochem. 2003;86:980–991. doi: 10.1046/j.1471-4159.2003.01921.x. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for beta-arrestins in cell signaling: not just for seven-transmembrane receptors. Mol Cell. 2006;24:643–652. doi: 10.1016/j.molcel.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Lipinski JF, Mandel LR, Ahn HS, Vanden Heuvel WJ, Walker RW. Blood dimethyltryptamine concentrations in psychotic disorders. Biol Psychiatry. 1974;9:89–91. [PubMed] [Google Scholar]
- Luan B, Zhao J, Wu H, Duan B, Shu G, Wang X, Li D, Jia W, Kang J, Pei G. Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature. 2009;457:1146–1149. doi: 10.1038/nature07617. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- Magalhaes AC, Holmes KD, Dale LB, Comps-Agrar L, Lee D, Yadav PN, Drysdale L, Poulter MO, Roth BL, Pin JP, Anisman H, Ferguson SS. CRF receptor 1 regulates anxiety behavior via sensitization of 5-HT2 receptor signaling. Nat Neurosci. 2010;13:622–629. doi: 10.1038/nn.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel LR, Rokach J, Rooney CS, Cragoe EJ., Jr Inhibition of dimethyltryptamine biosynthesis by N,N′-bis-(3-methyl-2-thiazolidinylidene) succinamide (I) and 2-imino-3-methylthiazolidine (II) Mol Pharmacol. 1978;14:930–939. [PubMed] [Google Scholar]
- McBride MC. Bufotenine: toward an understanding of possible psychoactive mechanisms. J Psychoactive Drugs. 2000;32:321–331. doi: 10.1080/02791072.2000.10400456. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. Mechanism of action of atypical antipsychotic drugs. In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: the fifth generation of progress. New York: Raven; 2002. pp. 819–832. [Google Scholar]
- Miner LA, Backstrom JR, Sanders-Bush E, Sesack SR. Ultrastructural localization of serotonin2A receptors in the middle layers of the rat prelimbic prefrontal cortex. Neuroscience. 2003;116:107–117. doi: 10.1016/s0306-4522(02)00580-8. [DOI] [PubMed] [Google Scholar]
- Moya PR, Berg KA, Gutiérrez-Hernandez MA, Sáez-Briones P, Reyes-Parada M, Cassels BK, Clarke WP. Functional selectivity of hallucinogenic phenethylamine and phenylisopropylamine derivatives at human 5-hydroxytryptamine (5-HT)2A and 5-HT2C receptors. J Pharmacol Exp Ther. 2007;321:1054–1061. doi: 10.1124/jpet.106.117507. [DOI] [PubMed] [Google Scholar]
- Narita M, Kato H, Kasukawa A, Narita M, Suzuki M, Takeuchi T, Suzuki T. Role of Src family kinase in the rewarding effect and hyperlocomotion induced by morphine. Neuroreport. 2006;17:115–119. doi: 10.1097/01.wnr.0000198950.92820.c7. [DOI] [PubMed] [Google Scholar]
- Nichols DE. Hallucinogens. Pharmacol Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Pieri L, Keller HH, Burkard W, Da Prada M. Effects of lisuride and LSD on cerebral monoamine systems and hallucinosis. Nature. 1978;272:278–280. doi: 10.1038/272278a0. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov. 2010;9:373–386. doi: 10.1038/nrd3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokach J, Girard Y, Hamel P, Reader G, Rooney CS, Mandel LR, Cragoe EJ, Jr, Zacchei AG. Inhibitors of indoleethylamine N-methyltransferase. Derivatives of 3-methyl-2-thiazolidinimine. In vitro, in vivo, and metabolic studies. J Med Chem. 1980;23:773–780. doi: 10.1021/jm00181a014. [DOI] [PubMed] [Google Scholar]
- Rosengarten H, Friedhoff AJ. A review of recent studies of the biosynthesis and excretion of hallucinogens formed by methylation of neurotransmitters or related substances. Schizophr Bull. 1976;2:90–105. doi: 10.1093/schbul/2.1.90. [DOI] [PubMed] [Google Scholar]
- Roth BL, Hanizavareh SM, Blum AE. Serotonin receptors represent highly favorable molecular targets for cognitive enhancement in schizophrenia and other disorders. Psychopharmacology (Berl) 2004;174:17–24. doi: 10.1007/s00213-003-1683-8. [DOI] [PubMed] [Google Scholar]
- Saavedra JM, Axelrod J. Psychotomimetic N-methylated tryptamines: formation in brain in vivo and in vitro. Science. 1972;175:1365–1366. doi: 10.1126/science.175.4028.1365. [DOI] [PubMed] [Google Scholar]
- Schmid CL, Bohn LM. Physiological and pharmacological implications of beta-arrestin regulation. Pharmacol Ther. 2009;121:285–293. doi: 10.1016/j.pharmthera.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CL, Raehal KM, Bohn LM. Agonist-directed signaling of the serotonin 2A receptor depends on beta-arrestin-2 interactions in vivo. Proc Natl Acad Sci U S A. 2008;105:1079–1084. doi: 10.1073/pnas.0708862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R, Brocco M, Audinot V, Gobert A, Veiga S, Millan MJ. (1-(2,5-dimethoxy-4 iodophenyl)-2-aminopropane)-induced head-twitches in the rat are mediated by 5-hydroxytryptamine (5-HT) 2A receptors: modulation by novel 5-HT2A/2C antagonists, D1 antagonists and 5-HT1A agonists. J Pharmacol Exp Ther. 1995;273:101–112. [PubMed] [Google Scholar]
- Shulgin A, Shulgin A. TiHKAL: the continuation. Berkeley, CA: Transform; 1997. [Google Scholar]
- Snyder SH, Banerjee SP, Yamamura HI, Greenberg D. Drugs, neurotransmitters, and schizophrenia. Science. 1974;184:1243–1253. doi: 10.1126/science.184.4143.1243. [DOI] [PubMed] [Google Scholar]
- Sorensen SM, Kehne JH, Fadayel GM, Humphreys TM, Ketteler HJ, Sullivan CK, Taylor VL, Schmidt CJ. Characterization of the 5-HT2 receptor antagonist MDL 100907 as a putative atypical antipsychotic: behavioral, electrophysiological and neurochemical studies. J Pharmacol Exp Ther. 1993;266:684–691. [PubMed] [Google Scholar]
- Sternbach H. The serotonin syndrome. Am J Psychiatry. 1991;148:705–713. doi: 10.1176/ajp.148.6.705. [DOI] [PubMed] [Google Scholar]
- Tadano T, Satoh S, Satoh N, Kisara K, Arai Y, Kim SK, Kinemuchi H. Potentiation of para-hydroxyamphetamine-induced head-twitch response by inhibition of monoamine oxidase type A in the brain. J Pharmacol Exp Ther. 1989;250:254–260. [PubMed] [Google Scholar]
- Takeda N, Ikeda R, Ohba K, Kondo M. Bufotenine reconsidered as a diagnostic indicator of psychiatric disorders. Neuroreport. 1995;6:2378–2380. doi: 10.1097/00001756-199511270-00024. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Willins DL, Meltzer HY. Direct injection of 5-HT2A receptor agonists into the medial prefrontal cortex produces a head-twitch response in rats. J Pharmacol Exp Ther. 1997;282:699–706. [PubMed] [Google Scholar]
- Xia Z, Hufeisen SJ, Gray JA, Roth BL. The PDZ-binding domain is essential for the dendritic targeting of 5-HT2A serotonin receptors in cortical pyramidal neurons in vitro. Neuroscience. 2003;122:907–920. doi: 10.1016/s0306-4522(03)00589-x. [DOI] [PubMed] [Google Scholar]
- Xu X, Chua CC, Gao J, Chua KW, Wang H, Hamdy RC, Chua BH. Neuroprotective effect of humanin on cerebral ischemia/reperfusion injury is mediated by a PI3K/Akt pathway. Brain Res. 2008;1227:12–18. doi: 10.1016/j.brainres.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]