

Abstract
The Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling is one of the major pathways for cytokine signal transduction. However, the role of the JAK/STAT pathway in liver I/R is not clear. This study focuses on JAK2, which functions upstream of STAT-1 in JAK/STAT, and its role in the mechanism of liver IRI. Partial warm ischemia was produced in the hepatic lobes of C57BL/6 mice for 90min, followed by 6h of reperfusion. Mice were treated with JAK-2 inhibitor (Tyrphostin AG490; 40 mg/kg, i.p) or vehicle, 60min prior to ischemic insult. JAK2 blockade resulted in significant reduction of hepatocyte apoptosis and liver injury. Macrophage and neutrophil infiltration, as assessed by immunohistochemistry, was markedly decreased in AG490-treated livers, compared with controls. The expression of pro-inflammatory cytokines (TNF-α, IL-6, IL-1β) and chemokines (CXCL-10 and CXCL-2) was also significantly reduced in AG490-treated group, compared with controls. AG490-treated livers showed less TUNEL positive cells and reduced cleaved caspase-3 protein expression in parallel with increased Bcl-XL expression. We employed AG490 (75 mM) in primary bone marrow-derived macrophage (BMM) and hepatoma cell (CRL1830) cultures, both stimulated with LPS (10 ng/ml). In BMM cultures, AG490 depressed otherwise LPS-induced pro-inflammatory gene expression programs (IL-6, IL-12p40, IL-1β, CXCL-10 and iNOS). In hepatoma cells, AG490 reduced cleaved-caspase-3 expression. Moreover, JAK2 blockade inhibited STAT1 and STAT3 phosphorylation. This is the first report, which documents that JAK2 signaling is essential in the pathophysiology of liver IRI, as its selective blockage ameliorated the disease process and protected livers from inflammation and apoptosis.
Keywords: Liver, ischemia/reperfusion injury, inflammation, JAK/STAT pathway
Introduction
Ischemia/reperfusion injury (IRI), a multifactorial process that occurs in a number of clinical settings such as hepatic resection, trauma, or transplantation, significantly affects both early and late liver graft function (1–4). Although its mechanism has not been fully elucidated, IR-triggered generation of reactive oxygen species (ROS) subsequent to re-oxygenation inflicts tissue damage, which initiates a cascade of deleterious innate immunity-dominated responses leading to circulatory disturbances, local inflammation, cell death, and ultimate organ failure.
The Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway was identified through the study of transcriptional activation in response to IFNs (5–7). The cellular stress induced by IR, ROS, endotoxin, or hyperosmolarity can all activate the JAK/STAT pathway (8–10), critically important for broad immune and hematopoietic cell functions. Indeed, targeting JAK2, the most conserved isoform of JAK family, results in embryonic lethality in mice around day 12 due to erythropoiesis failure (11–12). Recently identified somatic mutations of JAK2 have stimulated interest for the development of orally bioactive small-molecule JAK2 inhibitors and their testing in preclinical studies and clinical trials, primarily in the myeloproliferative disorders (reviewed in 13). However, detailed information on their effects from animal studies or from use in human subjects is presently unavailable.
Back in 2003, we proposed that the hepatocellular damage in transplant recipients due to reperfusion following prolonged periods of ischemia should be considered as an innate immunity-dominated inflammation response (1). Despite its localized nature inevitably leading to the organ failure, the systemic host innate immune component became soon appreciated. Indeed, our group was among the first to document that TLR system was selectively involved in the development of hepatic IRI (14). Of note, activation of sentinel TLR4, but not TLR2, was required for the induction of intrahepatic inflammation and hepatocellular damage in a murine model of liver IRI. Recently, we have demonstrated that cytoprotection rendered by heme oxygenase-1 (HO-1) overexpression results from depressed activation of STAT1, which in turn via type-1 IFN pathway decreases the production CXCL-10, a chemokine indispensable for the hepatocellular damage (15).
This study focuses on JAK2, which functions upstream of STAT-1 in JAK/STAT pathway, and its role in the mechanism of liver IRI. To the best of our knowledge, this is the first report to document that JAK2 signaling is indeed essential in the disease process, as its selective blockage ameliorated IR-induced organ damage, and protected livers from otherwise fulminant inflammation and apoptosis. These results should strengthen recent efforts to develop selective JAK inhibitors, which are now coming into the clinical use, and to design novel strategies to prevent/ameliorate liver IRI cascade in the clinics.
Materials and Methods
In vivo studies
Animals
Male wild-type (WT; C57BL/6) mice (8–12 weeks old) were used (Jackson Laboratory, Bar Harbor, ME). Animals were housed in the University of California Los Angeles animal facility under specific pathogen-free conditions, and received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institute of Health (NIH publication 86-23 revised 1985).
Liver IRI model
We used an established mouse model of partial warm hepatic IRI, as originally described by Zwacka et al. (16), with modifications (17). Briefly, mice were anesthetized, injected with heparin (100 U/kg), and an atraumatic clip was used to interrupt the arterial and portal venous blood supply to the left and middle liver lobes. After 90 min of partial warm ischemia, the clamp was removed, initiating hepatic reperfusion. Mice were sacrificed at 6 h after reperfusion; liver and blood were collected. Sham WT controls underwent the same procedure, but without vascular occlusion.
Treatment protocol
Mice were treated with JAK2 inhibitor tyrphostin AG490 [(2-cyano-3- (3,4-dihydroxyphenyl)-N- (benzyl)-2-propenamide, Sigma-Aldrich, St Louis, MO); 40 mg/kg i.p.] or vehicle (45% DMSO and 55% PBS) at 1 hour prior to the ischemic insult. The AG490 dosage was chosen based on significant inhibition of JAK2 phosphorylation detected in our study (data notshown), and consistent with the published data (18).
Hepatocellular function
Serum alanine aminotransferase (sALT) and aspartate aminotransferase (sAST) levels, the enzymatic indicators of epatocellular damage, were measured in peripheral blood samples after reperfusion with an autoanalyzer (ANTECH Diagnostics, Los Angeles, CA).
Liver histology
Liver specimens were fixed in 10% buffered formalin solution and embedded in paraffin. Liver paraffin sections (5-μm thick) were stained with hematoxylin and eosin. The severity of liver IRI was graded blindly using modified Suzuki’s criteria (19), with sinusoidal congestion, hepatocyte necrosis, and ballooning degeneration graded from 0 to 4. The absence of necrosis, congestion, or centrilobular ballooning is given a score of 0; severe congestion/ballooning degeneration, as well as >60% lobular necrosis, is given a value of 4.
Immunohistochemistry
Liver specimens were embedded in optimal cutting temperature (OCT) compound (Tissue-Tec, Sakura Finetek, Inc, CA), snap frozen, and cryostat sections (5 μm) were fixed in acetone. Endogenous peroxidase activity was inhibited with 0.3% hydrogen peroxidase. Sections were then blocked with 10% normal goat serum. Primary rat Abs against mouse macrophage CD11b (Mac-1, M/70; BD Biosciences), neutrophil Ly-6G (1A8; BD Biosciences), and CD3 (17A2; BD Biosciences) were diluted (1/50; 1/200; 1/50 in 3% normal goat serum, respectively), and 100 μL was added to each section. The primary Ab was incubated for 1 h. The secondary Ab, a biotinylated goat anti-rat immunoglobulin G (Vector; diluted 1:200), was incubated for 40 min. Sections were incubated with immunoperoxidase (ABC Kit, Vector), washed, and developed with a 3,3′-diaminobenzidine kit (Vector). Slides were counterstained with hematoxylin. Negative control was prepared by omission of primary Ab. Sections were evaluated blindly by counting labeled cells in 30 high-power fields (HPF), and results expressed as average number of positive cells/HPF.
TUNEL assay
Livers tissues were removed and fixed in 10% formalin, dehydrated, and embedded in paraffin. FragEL™ DNA Fragmentation Detection kit (Calbiochem®) was used according to the manufacturer’s protocol on 5-μm paraffin sections. TUNEL-positive cells were detected under light microscopy. Terminal Deoxynucleotidyl Transferase (Tdt) was omitted as a negative control. Positive controls were generated by treatment with DNase 1 (1μg/μl in 1X TBS/1mM MgSO4 for 20 min). Sections were evaluated blindly by counting labeled cells in triplicates in 10 HPF; results are expressed as average number of positive cells/HPF
In vitro studies
Cell cultures
Bone marrow macrophages (BMM), separated from the femurs and tibias of C57BL/6 mice, were suspended in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FCS (Gemini Bio-Products, Sacramento, CA), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (Invitrogen), with the addition of 15% L929-conditioned medium. Cells (5 × 106 cells/well) were cultured with 5% CO2 and 95% air at 37°. AG 490 (75μM) was added 2 h prior to the stimulation with LPS (10 ng/mL, Sigma; St. Louis, MO). Cells were incubated for 4 h prior to harvesting for qRT-PCR. We also analyzed mouse hepatoma cells (CRL-1830 ATCC, Manassas, VA) with AG 490 (75 μM) added 2 h prior to LPS stimulation (10 ng/mL). Cells were incubated from 2 min up to 1 h, and then harvested for Western Blot analyses.
Quantitative RT-PCR
RNA was extracted from liver tissue or cultured cells using Trizol Reagent (Invitrogen, Carlsbad, CA). Total RNA (5μg) was reverse transcribed to complementary DNA using Super- ScriptTM III First-Strand Synthesis System (Invitrogen). Quantitative PCR was performed using the DNA Engine with Chromo 4Detector (MJ Research, Waltham, MA). In a final reaction of 20 μL, the following were added: 1 X SuperMix (Platinum SYBRGreen qPCR Kit, Invitrogen, Carlsbad, CA), complementary DNA, and 0.1 μM of each primer. Amplification conditions were: 50°C (2 min), 95°C (5 min), followed by 45 cycles of 95°C (15 sec), 60°C (30 sec). Primers used to amplify a specific mouse gene fragments are listed as follows: TNF α s: 5′ GCCTCTTCTCATTCCTGCTT GT 3′; TNF α as: 5′ GATGATCTGAGTGTGAGGGTCTG 3′; IL-6 s: 5′ GCTACCAAACTGGATATAATCAGGA 3′; IL-6 as: 5′ CCAGGTAGCTATGGTACTCCAGAA 3′; IL-1β s: 5′ GAGCTGAAAGCTCTCCACCTCA 3′; IL-1β as: 5′ TCGTTGCTTGGTTCTCCTTGTAC 3′; CXCL-10 s: 5′ GCTGCCGTCATTTTCTGC 3′; CXCL-10 as: 5′ TCTCACTGGCCCGTCATC 3′; CXCL-2 s: 5′ ACTTCAAGAACATCCAGAG 3′; CXCL-2 as: 5′ CTTTCCAGGTCAGTTAGC 3′; IL-12p40 s: 5′ ACAGCACCAGCTTCTTCATCAG 3′; IL-12p40 as: 5′ TCTTCAAAGGCTTCATCTGCAA 3′; INOS s: 5′ CCTTGGAGTTCACCCAGTTG 3′; INOS as: 5′ CACATCAAAGCGGCCATAG 3′; HPRTs: 5′-TCAACGGGGGACATAAAAGT- 3′; HPRTas:5′-TGCATTGTTTTACCAGTGTCAA- 3′. Target gene expressions were calculated by their ratios to the housekeeping gene HPRT.
Western blots
Whole cell proteins were extracted from liver samples or cultured cells using lysis buffer (50mM Hepes, 10mM MgCl2, 1mM EDTA, 1mM EGTA, 0.08mM sodium molybdate, 2mM sodium pyrophosphate 0.01% Triton X-100, protease and phosphatase inhibitors). Cytoplasmic and nuclear proteins were extracted from liver samples or cultured cells using lysis buffer (50mM Hepes, 10mM MgCl2, 1mMEDTA, 1mM EGTA, 0.08mM sodium molybdate, 2mM sodium pyrophosphate 0.01% Triton X-100, and protease inhibitor). The proteins were then separated by SDS - PAGE and electrophoretically transferred onto polyvinylidene fluoride membranes. The membranes were probed with Abs against phosphorylated and total JAK2, STAT3 (Tyr705) and STAT1 (Ser727), cleaved and total caspase-3, bcl-XL (Cell Signaling Technology, Danvers, MA) and β-actin (Abcam Inc., Cambridge, MA) at 4°C, and then incubated with a HPRT-conjugated secondary Ab (Cell Signaling Technology, Danvers, MA). Detection was performed using a SuperSignal West Pico Chemiluminescent Substrate system from Thermo Fisher Scientific (Rockford, IL). Relative quantities of protein were determined using a densitometer (Image J software, NIH, Bethesda, MD).
Statistical analysis
All data are expressed as means ± SD. Differences between experimental groups were analyzed using one-way analysis of variance or Student’s t test for unpaired data. All differences were considered statistically significant at the P value of <0.05.
Results
JAK2 signaling is required for IR-triggered hepatocellular damage
To study the functional significance of JAK2 in the pathophysiology of liver IRI, separate cohorts of mice were treated with AG490, a selective JAK2 inhibitor, or vehicle. One hour later, animals were subjected to 90 min of partial hepatic warm ischemia, followed by 6 h of reperfusion. Recipients given AG490 showed decreased IR-induced hepatocellular damage, as measured by sALT/AST levels (IU/L), compared with vehicle-treated controls (Fig. 1A, sALT: 24952 ± 9666 vs. 6705 ±7220; p<0.0003; sAST: 10627 ± 2657 vs. 4388 ± 3769; p<0.001. These data correlated with Suzuki’s criteria of liver damage (Fig. 1B). Indeed, those after AG 490 treatment showed well-preserved lobular architecture with minimal signs of hepatocyte necrosis (score=3.25±2.54), whereas control livers revealed severe sinusoidal/vascular congestion with marked vacuolization, focally associated with necrosis (score = 6.75 ± 0.70; p<0.05) (Fig. 1C).
Figure 1. Liver IRI.


(A) The hepatocellular function at 6 h of reperfusion after 90 min of warm ischemia. Serum alanine aminotransferase (sALT) and serum aspartate aminotransferase (sAST) levels were significantly reduced in AG490-treated livers, as compared with vehicle-treated controls (*p<0.01; n=6–8/group). Means and SD are shown. (B) Representative liver histology (Hematoxylin-eosin staining; magnification x200) of ischemic liver lobes after reperfusion (6h). (C) Suzuki’s score, *p<0.01, n=6–8/group.
JAK2 signaling is needed for liver macrophage/neutrophil infiltration
We performed immunohistochemical staining for macrophage, neutrophils and T cells in livers at 6 h of reperfusion following 90 min of warm ischemia (Fig. 2A). Disruption of JAK2 signaling diminished numbers of infiltrating macrophages (12.8 ± 10.7 vs. 47.2 ± 13.1, p< 0.05) and neutrophils (2.7 ± 1.9 vs. 17.8 ± 5.0, p<0.05), as compared with controls (Fig. 2B). The staining for liver infiltrating CD3 positive cells was relatively weak, and AG490 treatment had little effect upon local T cell sequestration (1.3 ± 0.4 vs.1.7 ± 1.1), as compared with controls (Fig. 2B).
Figure 2. Liver cell infiltration.
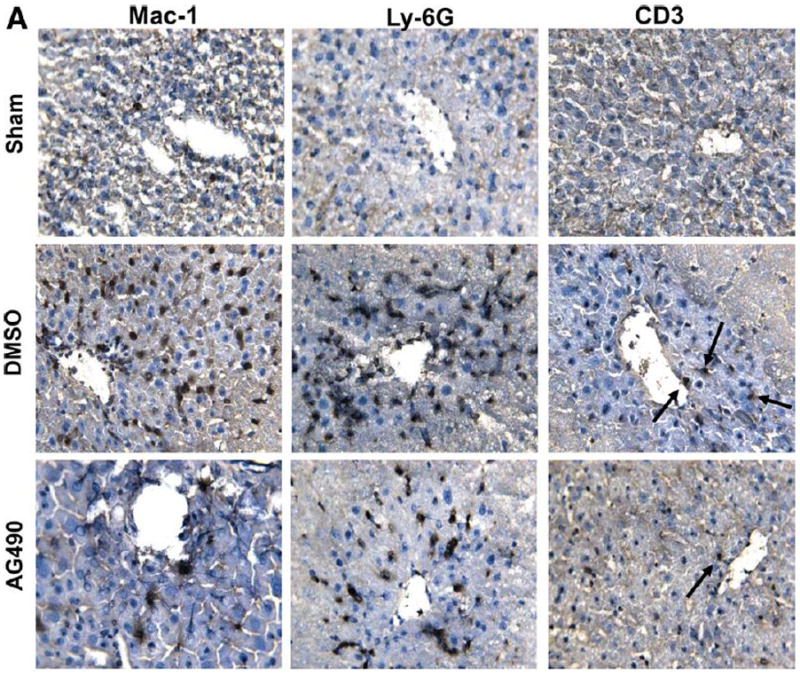

(A) Representative immunoperoxidase staining of macrophages (Mac-1), neutrophils (Ly-6G) and T cells (CD3) in ischemic liver lobes harvested at 6 h of reperfusion after 90 min of warm ischemia. Arrows denote labeled cells. (B) Quantitation of cellular infiltration by immunohistochemistry (*p < 0.01; n=4/group) (x400 magnification). Means and SD are shown.
JAK2 signaling promotes IR-triggered liver cytokine/chemokine programs
We used qRT-PCR to analyze liver expression of cytokines (TNF-α, IL-6, IL-1β), and chemokines (CXCL-2, CXCL-10), and calculated the ratio between post-IR and constitutive mRNA levels in each animal (Figure 3). Compared with controls, AG490 treatment attenuated liver expression of macrophage-associated pro-inflammatory mediators, including TNF-α (0.03 ± 0.02 vs. 0.09 ± 0.03, p < 0.01), IL-6 (0.14 ± 0.10 vs. 1.68 ± 0.82, p < 0.01), IL-1β (0.29 ± 0.21 vs. 0.97 ± 0.38, p < 0.01), and CXCL-10 (0.03 ± 0.04 vs. 0.19 ± 0.13, p < 0.05) at 6 h of reperfusion. Moreover, the expression of MIP-2 (CXCL-2), a chemokine associated with neutrophil activation, was diminished in AG490-treated livers (0.11 ± 0.11 vs. 0.37 ± 0.12, p<0.05).
Figure 3. Liver cytokine/chemokine programs.

Livers from groups of AG490-treated vs. vehicle-treated (DMSO) controls were analyzed by real-time RT-PCR for cytokine/chemokine gene expression at 6 h of reperfusion following 90 min of warm ischemia. Data were normalized to HPRT gene expression (*p < 0.05, **p < 0.01; n =6/group). Means and SD are shown.
JAK2 signaling modulates hepatocyte apoptosis
The TUNEL assay has revealed a significant decrease in a number of apoptotic positive cells at 6 hours of reperfusion in AG490-treated livers, as compared with vehicle-treated controls (8.1 ± 18.1 vs. 77.6 ± 14.6, p<0.05; Fig. 4A/B). Such a striking reduction in hepatocyte apoptosis correlated with lower cleaved caspase-3 protein expression in AG490-treated livers, as compared to controls (0.16 ± 0.02 vs. 0.40 ± 0.14; Fig. 4C). AG490-mediated JAK2 inhibition simultaneously increased the expression of anti-apoptotic Bcl-xl, as compared with controls (0.52 ± 0.14 vs. 1.08 ± 0.06, p<0.05; Fig. 4C).
Figure 4. Hepatocyte apoptosis.


(A) Representative TUNEL-assisted detection of hepatic apoptosis in ischemic liver lobes harvested at 6 h after 90 min of reperfusion (H&E stains; x400 magnification). (B) Quantitation of hepatic apoptosis by TUNEL staining. Vehicle-treated (DMSO) controls had a higher frequency of TUNEL-positive cells than the AG490-treated group. (*p < 0.05; n = 5/group). Means and SD are shown. (C) Western blot analysis of cleaved caspase-3 and Bcl-xl protein expression. Total caspase-3 and β-actin were used as internal controls (*p<0.05; n=3/group). Means and SD are shown.
JAK2 blockade decreases cytokine/chemokine expression in LPS-stimulated BMM
We further analyzed the immunomodulatory function of JAK2 signaling in well-controlled cell culture experiments, designed to mimic in vivo liver IRI model. First, to determine LPS-induced pro-inflammatory cytokine/chemokine production profile, we incubated BMM with LPS (10 ng/mL), followed by qRT-PCR assessment at 4 h. As shown in Fig. 5, addition of LPS did trigger, as expected, the expression of “signature” pro-inflammatory genes. In marked contrast, cells that were treated with AG 490 (75 μM) 2 h prior to LPS stimulation were characterized by diminished cytokine/chemokine levels: IL-6 (0.36 ± 0.24 vs. 0.79 ± 0.26, p<0.05); IL1-β (0.31 ± 0.21 vs. 1.06 ± 0.25, p<0.01); CXCL-10 (0.30 ± 0.09 vs. 0.84 ± 0.23, p<0.05); iNOS (0.06 ± 0.05 vs. 1.33 ± 0.6, p<0.05); IL-12p40 (0.06 ± 0.02 vs. 1.15 ± 0.42, p<0.05).
Figure 5. Cytokine/chemokine programs in LPS-stimulated BMM.

Quantitative RT-PCR analysis at 4 h after LPS stimulation. AG490 was added 2 h prior to LPS. Data were normalized to HPRT gene expression (*p < 0.05, **p < 0.01; n =2/group). Means and SD are shown.
JAK2 blockade depresses apoptosis in LPS-stimulated hepatoma cells
The hepatoma cells were pretreated with AG 490 (75μM) 2 h prior to LPS stimulation, followed by Western blot-assisted measurement of caspase-3. The relative expression levels were determined by densitometry, and expressed as ratios of cleaved caspase-3/Total caspase-3. LPS-stimulated cells were characterized by increased expression of cleaved caspase-3, as compared to unstimulated controls (Fig. 6). The AG490 supplement significantly reduced the expression of cleaved caspase -3 in hepatoma cell culture (0.49 ± 0.09 vs. 1.75 ± 0.11, p<0.01).
Figure 6. Cleaved caspase-3 expression in LPS-stimulated hepatoma cells.

Representative Western-blot analysis of cleaved caspase-3 protein expression in hepatoma cells (CRL1830) after LPS stimulation. Cleaved caspase-3 expression was increased within 4 h after stimulation with LPS. AG490 treatment, 2 h prior to LPS stimulation, decreased cleaved caspase-3 expression in hepatoma cells (CRL1830) (*p < 0.01, n= 2–3/group).
JAK2 blockade reduces STAT1/STAT3 and JAK2 phosphorylation in LPS-stimulated hepatoma cells
To further explore potential mechanisms, and since JAK2 is one of the major upstream activators of STAT1/STAT3, we investigated the effect of AG 490 upon their phosphorylation. First, hepatoma cells were cultured with LPS (10 ng/mL), followed by Western Blot assay at 1 h. As shown in Fig. 7, LPS readily induced phosphorylation of STAT1 serine, along with STAT3 tyrosine. However, when cells were pretreated with AG 490 (75μM) 2 hours prior to LPS, we observed decreased expression of pSTAT1 (Ser727): 0.12 ± 0.006 vs. 0.44 ± 0.02; p<0.05) and pSTAT3 (Tyr 705) (0.15 ± 0.02 vs. 0.47 ± 0.01; p < 0.05) as compared to LPS controls.
Figure 7. Activation of STAT1, STAT3 and JAK2 in LPS-stimulated hepatoma cells.

(A). Representative Western-blot analysis of phospho- and total STAT1 and phospho- and total STAT3. Within 1 h after treatment with LPS, STAT1 and STAT3 were robustly phosphorylated. Treatment with AG490, 2 h prior to LPS stimulation, significantly decreased both phospho-STAT1 and phospho-STAT3 expression in hepatoma cells (CRL1830) (*p < 0.05, n= 2/group). (B). Representative Western-blot analysis of JAK2 phosphorylation. Within 2 min after treatment with LPS, JAK2 became phosphorylated. Treatment with AG490, 2 h prior to LPS stimulation, significantly decreased phospho-JAK2 expression in hepatoma cells (CRL1830) (*p < 0.05, n= 2/group).
As shown in Figure 7B, AG490-mediated inhibition of JAK2 phosphorylation occurred as early as 2 min after LPS stimulation (0.29 ± 0.14 vs. 0.72 ± 0.02; p < 0.05). This inhibitory effect, however, was short lived. Although it was still well pronounced at 5 min, no significant differences could be readily detected between LPS vs. LPS + AG490 groups by 10 min of culture (data not shown).
Discussion
The results of this study document the essential role of JAK2 signaling in the mechanism of IR-triggered innate immunity-dominated liver damage. We used Tyrphostin AG490, a selective pharmacological inhibitor of JAK2 phosphorylation (20, 21), in a well-established mouse liver model of 90 min warm ischemia followed by 6 h of reperfusion. Unlike in vehicle-treated controls, liver function and histology, assessed by sALT/AST levels and Suzuki scores, respectively, significantly improved after AG490 treatment, consistent with the functional significance of JAK2 thyrosine kinase activation in the IRI cascade. These beneficial effects were accompanied by local reduction of macrophage and neutrophil sequestration; downregulation of pro-inflammatory cytokine/chemokine gene programs; reduced apoptosis with simultaneous sparing of anti-apoptotic cytoprotective pathway. The parallel in vitro culture data support our in vivo findings, and suggest that liver IRI proceeds via JAK2-mediated STAT-1 activation pathways. Although suppressors of cytokine signaling-1 (SOCS1) and SOCS3 have been shown to act via negative feedback on JAK/STAT in liver IRI (22), our study is the first to provide direct evidence that JAK2 inhibition ameliorates the disease process and exerts local cytoprotection. Hence, our results identify JAK2 as a novel target for putative therapeutic intervention against liver IRI.
Our interest in dissecting the complex mechanistic gears of liver IRI stems from the original observation that activation of TLR4 and its IRF-3-dependent (but MyD88-independent) downstream signaling are required to initiate the disease process (14). Then, it proceeds via type-1 (but not type-2) IFN pathway (23), to elaborate CXCL-10, which is indispensable for the inflammation response and the ultimate hepatocellular injury (24). Indeed, we have shown that cytoprotection rendered by HO-1 results from depressed activation of STAT1, and decreased CXCL-10 production (15). We have also detected selective reduction of IR-induced liver extracellular signal-regulated kinase (ERK) but intact Jun N-terminal kinase (JNK) in the absence of CXCL10, providing evidence for altered immune response in IR-resistant livers (24). In sum, our previous findings provide the rationale for, and support the notion that JAK2, an upstream element of the JAK/STAT complex, represents a focal point in the signaling events during IR-mediated liver damage.
Relatively little is known as to how the JAK/STAT pathway influences IR-induced solid organ injury. In a rat kidney IRI model, AG490-mediated JAK2 disruption improved renal function, attenuated histological lesions and reduced apoptosis of tubular epithelial cells while simultaneously inhibiting downstream STAT1/STAT3 signaling (25). This is in agreement with the protection rendered by SOCS-1 and SOCS-2, endogenous inhibitors of JAK/STAT, against the ischemic acute renal failure (26). In another study in rats, AG490 treatment prevented myocardial IRI by reducing cardiomyocyte apoptosis, and the myocardial infarct size, suggesting that JAK/STAT signaling associates with the cardiac dysfunction during ischemia (27). Finally, addition of AG490 exerted protection upon rat cardiomyocytes from oxidative stress-induced apoptosis in vitro (28), consistent with the involvement of JAK2 activation in hydrogen peroxide-induced apoptosis of vascular smooth muscle cells (29).
Our results suggest that JAK2 signaling mediates local inflammatory cell infiltration. Indeed, in the first phase of IR-mediated inflammatory response, activation of macrophages/Kupffer cells results in the release of TNF-α, IL-1β, IL-6, and CXCL-10, the “signature” markers of liver IRI (30–33). Our study also shows AG490 pretreatment significantly blunted not only the number of macrophages sequestered in the liver itself, but also their pro-inflammatory gene expression program, as compared with vehicle-treated controls. To further confirm the effect of JAK2 signaling on cytokine elaboration profiles, we employed in vitro BMM cultures. Indeed, pretreatment with AG490 efficiently diminished otherwise LPS-triggered increased induction of numerous mediators, including CXCL-10, and iNOS by BMM. This finding is consistent with the reported role of STAT1 signaling acting through JAK2 pathway to enhance TNF-α–induced CXCL-10 production in human monocytes (34). Moreover, in agreement with our data, the Geller group, using a specific JAK2 inhibitor (tyrphostin B42), has confirmed the importance of that very pathway in regulating iNOS gene transcription (35). In the second phase of IRI, neutrophils become activated, to generate ROS, and dominate the local damage cascade (36, 37). Indeed, we observed a marked increase in Ly-6G neutrophil infiltration in vehicle-treated livers subjected to IR, as compared to sham controls. Moreover, unlike those in vehicle-treated group, livers in AG490-pretreated mice revealed decreased neutrophil sequestration, along with diminished CXCL2 (MIP-2) levels, the chemokine that attracts polymorphonuclear granulocytes to the infection site (38). These results suggest that JAK2 signaling contributes to neutrophil recruitment and function in our model. This is consistent with delayed human neutrophil apoptosis, which occurs via up-regulation of its inhibitor (cIAP2), in JAK2 (and STAT3) activation-dependent and AG490-dependent fashions (39).
Although the exact mechanisms of cell death in hepatic I/R injury are multifactorial and unclear, the accumulating evidence suggests that apoptosis plays the fundamental role (40, 41). Our present in vivo TUNEL-staining findings, suggesting that the JAK/STAT pathway is involved in the pro-apoptotic process, are supported by earlier results from a rat kidney IRI model, where AG490 significantly decreased the number of apoptotic tubular epithelial cells (25). To confirm our hypothesis that JAK2 blockage did indeed protect hepatocytes from apoptotic cell death, we stimulated mouse hepatoma cells with LPS in vitro. Although LPS-stimulated cells were characterized by increased expression of cleaved caspase-3, compared with non-stimulated controls, AG490 supplement significantly reduced cleaved caspase-3 expression in culture. Both pro- and anti-apoptotic functions have been attributed to JAK2 tyrosine kinase activity in a variety of signaling systems (27, 29, 42). Although cell type and/or stimulus-specific factors may play a role, the exact factors determining the different outcomes remain unknown. Recently, in agreement with our findings, it has been reported that JAK2 inhibitor AG490 prevented H2O2-induced death of bovine aorta endothelial cells, human umbilical vein endothelial cells, as well as renal proximal tubular epithelial cells. The mechanism of cytoprotection included sparing or augmenting the anti-apoptotic intrinsic Bcl2 expression, and preventing caspase-3 activity (43). Indeed, these all are consistent with our present results, which are the first to highlight the role of JAK2 in liver IRI and in vitro hepatocyte culture systems that mimic the in vivo liver damage scenario. It is plausible JAK2 activation may change pro-apoptotic/anti-apoptotic ratio, which in turn dictates the susceptibility to the apoptotic programs executed by factors such as caspases.
Both, STAT1 and STAT3, downstream of JAK2, may produce opposing effects on biological responses, including cell proliferation, survival, differentiation and apoptosis (44). Whereas STAT-1 plays a key role in promoting apoptosis in cardiac myocytes exposed to IRI (45), STAT-3 often exerts anti-apoptotic and cytoprotective functions (46). Moreover, whilst STAT-3 promotes cell proliferation, STAT-1 appears to have an anti-proliferative effect. The role of STAT1 and STAT3 in cell apoptosis and proliferation remains controversial, with some studies associating STAT-3 activation with apoptosis (47, 48). In the liver, the induction of CD40-mediated cholangiocyte apoptosis required JAK2-mediated phosphorylation of STAT3 as well as sustained JNK1/2 and ERK1/2 activation. The latter supports STAT3 proapoptotic function in primary human liver epithelial cells (49). STAT1 is mainly activated by IFNs, and consistent with our ongoing studies in KO mice, STAT1 plays a key role in facilitating inflammation and injury in the liver (50, 51). In our study, LPS-stimulated hepatoma-cell increased phosphorylation of STAT1 and STAT3, which was AG490-sensitive. Indeed, by inhibiting JAK2 and consequently STAT1, addition of AG490 to pancreatic β-cell cultures has been shown to ameliorate cytokine-mediated cell death, thereby delaying the development of autoimmune diabetes (52). Interestingly, AG-490-facilitated inhibition of STAT-3 may increase ERK activation and survival of mouse proximal tubular epithelial cells during oxidative stress (53), consistent with the finding that STAT1 and STAT3 activation parallels the severity of IR liver injury (22). We favor an idea that STAT1 rather than STAT3 is exerting the dominant function in inducing liver IRI, in agreement with proposed by us novel mechanism by which HO-1 exerts adaptive cytoprotective/anti-inflammatory functions by blunting activation of STAT1 via type-1 IFN pathway in the context of innate TLR4 activation (15).
In summary, our present results document the role of JAK/STAT signaling pathway in cell death and tissue inflammation leading to liver damage due to IR. This study is also the first to directly demonstrate that specific targeting of JAK2 ameliorated tissue damage due to warm liver IR insult. Thus, this work supports the rationale for identifying inhibitors that specifically target JAK2 pathway as a potential therapeutic approach in combating liver IRI.
Acknowledgments
Grant support: NIH RO1 DK062357 and The Dumont Research Foundation. M.C.S.F. was supported by CAPES (Coordencao de Aperfeicoamento de Pessoal de Nivel Superior) and a Fellowship Grant from ISN (International Society of Nephrology).
Abbreviations
- IRI
ischemia/reperfusion injury
- ROS
reactive species oxygen
- JAK/STAT
Janus kinase/signal transducers and activators of transcription
- IFNs
interferons
- JAK2
Janus kinase-2
- TLR
toll-like receptor
- CXCL-10
chemokine (C-X-C motif) ligand 10
- WT
wild-type
- DMSO
dimethyl sulfoxide
- PBS
phosphate buffered saline
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- OCT
optimal cutting temperature
- HPF
high-power field
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- BMM
bone marrow-derived macrophages
- LPS
lipopolysaccharide
- CXCL-2
chemokine (C-X-C motif) ligand 2
- iNOS
inducible nitric oxide synthase
- HPRT
hypoxanthine phosphoribosyltransferase
References
- 1.Fondevila C, Busuttil RW, Kupiec-Weglinski JW. Hepatic ischemia/reperfusion injury - a fresh look. Exp Mol Pathol. 2003;74:86–93. doi: 10.1016/s0014-4800(03)00008-x. [DOI] [PubMed] [Google Scholar]
- 2.Jaeschke H, et al. Mechanisms of reperfusion injury after warm ischemia of the liver. J Hepatobiliary Pancreat Surg. 1998;5:402–408. doi: 10.1007/s005340050064. [DOI] [PubMed] [Google Scholar]
- 3.Rao PN, Liu T, Synder JT, Platt JL, Starzl TE. Reperfusion injury following cold ischemia activates rat liver Kupffer cells. Transplant Proc. 1991;23:666–669. [PMC free article] [PubMed] [Google Scholar]
- 4.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Pina E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR. Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell. 1984;38:745–755. doi: 10.1016/0092-8674(84)90270-8. [DOI] [PubMed] [Google Scholar]
- 6.Schindler C, Fu XY, Improta T, Aebersold R, Darnell JE., Jr Proteins of transcription factor ISGF-3: one gene encodes the 91-and 84-kDa ISGF-3 proteins that are activated by interferon alpha. Proc Natl Acad Sci U S A. 1992;89:7836–7839. doi: 10.1073/pnas.89.16.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luster AD, Unkeless JC, Ravetch JV. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- 8.Dudley AC, Thomas D, Best J, Jenkins A. The STATs in cell stress-type responses. Cell Commun Signal. 2004;2:8. doi: 10.1186/1478-811X-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maziere C, Conte MA, Maziere JC. Activation of JAK2 by the oxidative stress generated with oxidized low-density lipoprotein. Free Radic Biol Med. 2001;31:1334–1340. doi: 10.1016/s0891-5849(01)00649-9. [DOI] [PubMed] [Google Scholar]
- 10.Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–217. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- 11.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/s0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 12.O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109(suppl):S121–131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 13.Pardanani A. JAK2 inhibitor therapy in myeloproliferative disorders: rationale, preclinical studies and ongoing clinical trials. Leukemia. 2008;22:23–30. doi: 10.1038/sj.leu.2404948. [DOI] [PubMed] [Google Scholar]
- 14.Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 15.Tsuchihashi S, Zhai Y, Bo Q, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type-1 interferon signaling. Transplantation. 2007;83:1628–1634. doi: 10.1097/01.tp.0000266917.39958.47. [DOI] [PubMed] [Google Scholar]
- 16.Zwacka RM, Zhang Y, Halldorson J, Schlossberg H, Dudus L, Engelhardt JF. CD4+ T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J Clin Invest. 1997;100:279–289. doi: 10.1172/JCI119533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen XD, Ke B, Zhai Y, Amersi F, Gao F, Anselmo DM, Busuttil RW, Kupiec-Weglinski JW. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation. 2002;74:315–319. doi: 10.1097/00007890-200208150-00005. [DOI] [PubMed] [Google Scholar]
- 18.Banes AK, Shaw S, Jenkins J, Redd H, Amiri F, Pollock DM, Marrero MB. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am J Physiol Renal Physiol. 2004;286:F653–F659. doi: 10.1152/ajprenal.00163.2003. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury: modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- 21.Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, et al. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996;379:645–648. doi: 10.1038/379645a0. [DOI] [PubMed] [Google Scholar]
- 22.Langdale LA, Hoagland V, Benz W, Riehle KJ, Campbell JS, Liggitt DH, Fausto N. Suppressor of cytokine signaling expression with increasing severity of murine hepatic ischemia-reperfusion injury. J Hepatol. 2008;49:198–206. doi: 10.1016/j.jhep.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhai Y, Qiao B, Gao F, Shen X, Vardanian A, Busuttil RW, Kupiec-Weglinski JW. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. 2008;47:199–206. doi: 10.1002/hep.21970. [DOI] [PubMed] [Google Scholar]
- 24.Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, Luster AD, Busuttil RW, Kupiec-Weglinski JW. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47:207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- 25.Yang N, Luo M, Li R, Huang Y, Zhang R, Wu Q, Wang F, Li Y, Yu X. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol Dial Transplant. 2008;23:91–100. doi: 10.1093/ndt/gfm509. [DOI] [PubMed] [Google Scholar]
- 26.Leonard MO, Hannan K, Burne MJ, Lappin DW, Doran P, Coleman P, Stenson C, et al. 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure. J Am Soc Nephrol. 2002;13:1657–1662. doi: 10.1097/01.asn.0000015795.74094.91. [DOI] [PubMed] [Google Scholar]
- 27.Mascareno E, El-Shafei M, Maulik N, Sato M, Guo Y, Das DK, Siddiqui MA. JAK/STAT signaling is associated with cardiac dysfunction during ischemia and reperfusion. Circulation. 2001;104:325–329. doi: 10.1161/01.cir.104.3.325. [DOI] [PubMed] [Google Scholar]
- 28.Mascareno E, Beckles DL, Siddiqui MA. Janus kinase-2 signaling mediates apoptosis in rat cardiomyocytes. Vascul Pharmacol. 2005;43:327–335. doi: 10.1016/j.vph.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 29.Sandberg EM, Sayeski PP. Jak2 tyrosine kinase mediates oxidative stress-induced apoptosis in vascular smooth muscle cells. J Biol Chem. 2004;279:34547–34552. doi: 10.1074/jbc.M405045200. [DOI] [PubMed] [Google Scholar]
- 30.Wanner GA, Ertel W, Muller P, Hofer Y, Leiderer R, Menger MD, Messmer K. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock. 1996;5:34–40. doi: 10.1097/00024382-199601000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 32.Day YJ, Marshall MA, Huang L, McDuffie MJ, Okusa MD, Linden J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: Inhibition of chemokine induction. Am J Physiol Gastrointest Liver Physiol. 2004;286:G285–293. doi: 10.1152/ajpgi.00348.2003. [DOI] [PubMed] [Google Scholar]
- 33.Colletti LM, Green ME, Burdick MD, Strieter RM. The ratio of ELR+ to ELR− CXC chemokines affects the lung and liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 2000;31:435–445. doi: 10.1002/hep.510310225. [DOI] [PubMed] [Google Scholar]
- 34.Qi XF, Kim DH, Yoon YS, Jin D, Huang XZ, Li JH, Deung YK, Lee KJ. Essential involvement of cross-talk between IFN-gamma and TNF-alpha in CXCL10 production in human THP-1 monocytes. J Cell Physiol. 2009;220:690–697. doi: 10.1002/jcp.21815. [DOI] [PubMed] [Google Scholar]
- 35.Ganster RW, Taylor BS, Shao L, Geller DA. Complex regulation of human inducible nitric oxide synthase gene transcription by Stat 1 and NF-kappa B. Proc Natl Acad Sci U S A. 2001;98:8638–8643. doi: 10.1073/pnas.151239498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol. 1991;260:G355–362. doi: 10.1152/ajpgi.1991.260.3.G355. [DOI] [PubMed] [Google Scholar]
- 37.Hasegawa T, Malle E, Farhood A, Jaeschke H. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: attenuation by ischemic preconditioning. Am J Physiol Gastrointest Liver Physiol. 2005;289:G760–767. doi: 10.1152/ajpgi.00141.2005. [DOI] [PubMed] [Google Scholar]
- 38.Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392:565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- 39.Sakamoto E, Hato F, Kato T, Sakamoto C, Akahori M, Hino M, Kitagawa S. Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis 2. J Leukoc Biol. 2005;78:301–309. doi: 10.1189/jlb.1104690. [DOI] [PubMed] [Google Scholar]
- 40.Kohli V, Selzner M, Madden JF, Bentley RC, Clavien PA. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia-reperfusion injury in the rat liver. Transplantation. 1999;67:1099–1105. doi: 10.1097/00007890-199904270-00003. [DOI] [PubMed] [Google Scholar]
- 41.Gao W, Bentley RC, Madden JF, Clavien PA. Apoptosis of sinusoidal endothelial cells is a critical mechanism of preservation injury in rat liver transplantation. Hepatology. 1998;27:1652–1660. doi: 10.1002/hep.510270626. [DOI] [PubMed] [Google Scholar]
- 42.Hattori R, Maulik N, Otani H, Zhu L, Cordis G, Engelman RM, Siddiqui MA, Das DK. Role of STAT3 in ischemic preconditioning. J Mol Cell Cardiol. 2001;33:1929–1936. doi: 10.1006/jmcc.2001.1456. [DOI] [PubMed] [Google Scholar]
- 43.Neria F, Castilla MA, Sanchez RF, Gonzalez Pacheco FR, Deudero JJ, Calabia O, Tejedor A, Manzarbeitia F, Ortiz A, Caramelo C. Inhibition of JAK2 protects renal endothelial and epithelial cells from oxidative stress and cyclosporin A toxicity. Kidney Int. 2009;75:227–234. doi: 10.1038/ki.2008.487. [DOI] [PubMed] [Google Scholar]
- 44.Stephanou A, Latchman DS. Opposing actions of STAT-1 and STAT-3. Growth Factors. 2005;23:177–182. doi: 10.1080/08977190500178745. [DOI] [PubMed] [Google Scholar]
- 45.Stephanou A, Brar BK, Scarabelli TM, Jonassen AK, Yellon DM, Marber MS, Knight RA, Latchman DS. Ischaemia induced STAT-1 expression and activation plays a critical role in cardiac myocyte apoptosis. J Biol Chem. 2000;275:10002–10008. doi: 10.1074/jbc.275.14.10002. [DOI] [PubMed] [Google Scholar]
- 46.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Shen B, Li Y, Sun Y. STAT3 exerts two-way regulation in the biological effects of IL-6 in M1 leukemia cells. Leukemia Research. 2001;25:463–472. doi: 10.1016/s0145-2126(00)00157-0. [DOI] [PubMed] [Google Scholar]
- 48.Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, Akira S, Clarke AR, Watson CJ. Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999;13:2604–2616. doi: 10.1101/gad.13.19.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed-Choudhury J, Williams KT, Young LS, Adams DH, Afford SC. CD40 mediated human cholangiocyte apoptosis requires JAK2 dependent activation of STAT3 in addition to activation of JNK1/2 and ERK1/2. Cell Signal. 2006;18:456–68. doi: 10.1016/j.cellsig.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 50.Hong F, Jaruga B, Kim WH, Radaeva S, El-Assal ON, Tian Z, Nguyen VA, Gao B. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J Clin Invest. 2002;110:1503–1513. doi: 10.1172/JCI15841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaruga B, Hong F, Kim WH, Gao B. IFN-γ/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1044–1052. doi: 10.1152/ajpgi.00184.2004. [DOI] [PubMed] [Google Scholar]
- 52.Kim S, Kim HS, Chung KW, Oh SH, Yun JW, Im SH, Lee MK, Kim KW, Lee MS. Essential role for signal transducer and activator of transcription-1 in pancreatic beta-cell death and autoimmune type 1 diabetes of nonobese diabetic mice. Diabetes. 2007;56:2561–2568. doi: 10.2337/db06-1372. [DOI] [PubMed] [Google Scholar]
- 53.Arany I, Megyesi JK, Nelkin BD, Safirstein RL. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int. 2006;70:669–674. doi: 10.1038/sj.ki.5001604. [DOI] [PubMed] [Google Scholar]