

Abstract
Microvessels and neurons respond rapidly and simultaneously in focal regions of ischaemic injury in such a way as to suggest that the responses could be coordinated. The ability of neurons to modulate cerebral blood flow in regions of activation results from neurovascular coupling. But little is known about the microvessel-to-neuron direction of the relationship. The presence and participation of intervening glial cells implies the association of microvessels, glia, and neurons in a ‘neurovascular unit’. The interdependent functions of the cellular and matrix components of this theoretical unit have not been rigorously explored, except under conditions of injury where, for the most part, only single components or tissue samples have been studied. Whereas maintenance or timely re-establishment of flow reduces tissue and neuron injury in both humans and animal models, protection of neuron function in humans has not prevented the evolution of injury despite the inherent mechanisms of neurovascular coupling. However, occlusion of flow to the brain rapidly identifies regions of neuron-vascular vulnerability within the vascular territory-at-risk. These coalesce to become the mature ischaemic lesion. The failure, so far, of clinical trials of neuron protectant agents to achieve detectable tissue salvage could be explained by the vulnerability (and lack of protection) of essential components of the ‘unit’. This presentation summarizes evidence and thoughts on this topic. These support the need to understand component interactions within the neurovascular unit.
Keywords: clinical trials, haemorrhagic transformation, ischaemic stroke, neurons, neurovascular unit, proteases
Despite a growing comprehension of the molecular processes that cause ischaemic damage to neurons and glia, the simultaneous mechanisms that underlie the maturation of the ischaemic lesion to infarction, and the acute responses of microvessels in the ischaemic territory, there is the need to understand these processes together. Here, we discuss the rationale for both a conceptual and practical framework that links microvessel with neuron (neuron-vascular) interactions.
The neurovascular unit
The control and modulation of regional and local cerebral blood flow, in the absence of ischaemic injury, depends upon neurovascular coupling [1–4]. Microvessel responses reflect the presence of neuronal activation, requiring the function of intact neurons. Recently, significant new findings of alterations in microvessel structure (basal lamina matrix), protease generation, endothelial cell activation and astrocyte-endothelial cell adhesion suggest that communication might also be directed from microvessels to the neurons they supply, in view of the proximity of microvascular endothelial cells to the circumferential astrocyte end-feet [5–7]. The general observation that neurovascular communication is disrupted under conditions of ischaemia has led to the supposition that neuron-microvascular interactions can be described by a theoretical ‘neurovascular unit’. The conceptual ‘unit’ consists of microvessels [endothelial cells-basal lamina matrix-astrocyte end-feet (and pericytes)], astrocytes, neurons and their axons, and other supporting cells (e.g., microglia and oligodendroglia) that are likely to modulate the function of the ‘unit’. This provides a framework for considering bi-directional communication between neurons and their supply microvessels with the participation of the intervening astrocytes (Fig. 1). It may also offer a platform for understanding the evolution of CNS injury processes, and the success and appropriateness of clinical/experimental interventions. The resilience of the ‘unit’ to any reduction in blood flow or to flow cessation is unclear, but the processes involved in communications and injury responses are likely to be more complex than presently understood as adjacent units would be connected through their common microvessels (including astrocytes in a synctium) and through dendritic connections. This affords both cellular intercommunication and protected perfusion at the same time. The conceptual framework links microvessel and neuron function, and their responses, to injury; the structural arrangement links microvessel components with neurons via the common astrocytes.
Fig. 1.
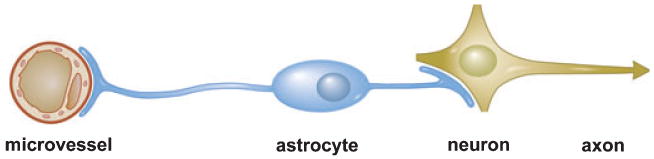
A schematic of the ‘neurovascular unit’. Denoting the inter-relationship of microvessels to their dependent neurons and axons, via astrocytes. Injury to any component is likely to affect the function of the entire unit.
Bases for the unitary view are that (i) ischaemic stroke is a vascular disorder with neurological consequences, (ii) neuron-targeted approaches have been uniformly unhelpful in clinical trials, (iii) neuron-vascular communication is proven, (iv) focal ischaemic injury occurs in the territory-at-risk (yet to be demonstrated in advance of the event in the human CNS), (v) neuron and microvascular responses to focal ischaemia are acute and simultaneous, and (vi) cell–cell and ultrastructural relationships during development and adulthood are generally predictable.
In this presentation we will: (i) consider the relevance of this conceptual framework to the ultrastructural and functional relationships of the microvessel to its neighbouring neuron, (ii) the simultaneous co-expression of matrix proteases by neurovascular cells and neurons, and (iii) the responses of treatment targets to the specific agents in preclinical models and clinical trials.
Microvessel–neuron relationships
During cerebral development, microvessels and neurons are arranged relative to one another by growth along extracellular matrix paths [8–12]. Astrocytes and endothelial cells interact to form the intervening basal lamina barrier and the inter-endothelial tight junctions as part of the permeability barrier of capillaries [13–19]. This has also been demonstrated by elegant xenograft experiments, where endothelial cells and astrocyte end-feet are required for the appearance of the permeability barrier phenotype [20]. In the cerebral grey matter, the arterial supply is arranged in a series of stacked hexagonal arrays descending from the pial supply to the dependent capillary beds [21–23]. This hierarchical organization of the arterial supply is something like a honeycomb that also extends to capillary branching that reflects the hexagonal arrangement of the arteries. Within the corpus striatum, the neuron-microvessel distribution is ordered in a more homogeneous fashion [24]. The capillary arrangement has branch points at approximately 30 μm intervals [24]. Within the white matter, capillaries are arranged in line with axons and comprise ∼10% of the capillary density found within the grey matter [7]. Hence, there are region-specific arrangements of the microvasculature. This accords with differences in regional cerebral blood flow (rCBF) whereby flow is lowest in the striatum and highest in the cortical grey matter. The relative arrangements and densities of the neurons (and neuron sub-types) to the ‘supply’ microvessels in these regions are not known.
Coordinate microvessel–neuron responses to focal ischaemia
Studies with the nonhuman primate model of middle cerebral artery occlusion (MCA:O) have provided two forms of evidence for acute coincident microvessel (m)–neuron (n) responses to focal ischaemia: the responses to focal ischaemia of the (m–n) distance distribution, and of matrix protease elaboration [24]. Both settings are promoted by cessation of vascular flow within the territory-at-risk, in this case within the lenticulostriate arteriolar bed when the proximal MCA is occluded. These two examples highlight the responses of microvessels and neurons in close proximity [as (m–n) pairs] that initially, at least, are heterogeneously distributed within the striatal territory-at-risk.
(m–n) Distance distributions
The responses of microvessel–neuron pairs to focal ischaemia are highly ordered and consistent (Fig. 2). In a study of the course of neuron injury at 2 h following ischaemia onset in the nonhuman primate, neurons furthest from their most proximate microvessel (within a ∼30 μm radius) were most likely to display injury [24]. In this period, no distortion of the tissue occurred due to the ischaemic injury (e.g., by oedema) and there was no evidence of haemorrhage. Injured neurons (n*) were scattered amongst neurons not displaying evidence of injury, indicating that the foci of injury were heterogeneously distributed within the ischaemic territory. The observation of neurons most distant from their nearest microvessel, that preferentially incorporated the deoxyribonucleotide dUTP into the segments of cellular DNA damage or scission (demonstrating cells with DNA injury), compared with neurons not displaying dUTP incorporation and therefore less injured, is not easily explained by the expected distribution of O2 diffusion, however [24]. The fall in tissue O2 with distance from the capillary would be insufficient to cause ischaemic injury throughout the 30 μm radius. Regarding selective vulnerability, the neuron subgroup that did not express glutamic acid decarboxylase (GAD−) displayed the same injury distance (m–n*) distribution as the overall group, but this was not true of the response of neurons expressing other neurotransmitters. The (m–n*) distribution could not be explained as either a consequence of leucocyte invasion or cytokine effects, because the former would have been expected to injure neurons closest to their nearest microvessels, and cytokine generation would have been expected to affect all neurons in a region no matter the distance from their supply microvessel(s) if they induced neuron degeneration equally. This suggests injury to other structural elements in the microvascular unit.
Fig. 2.
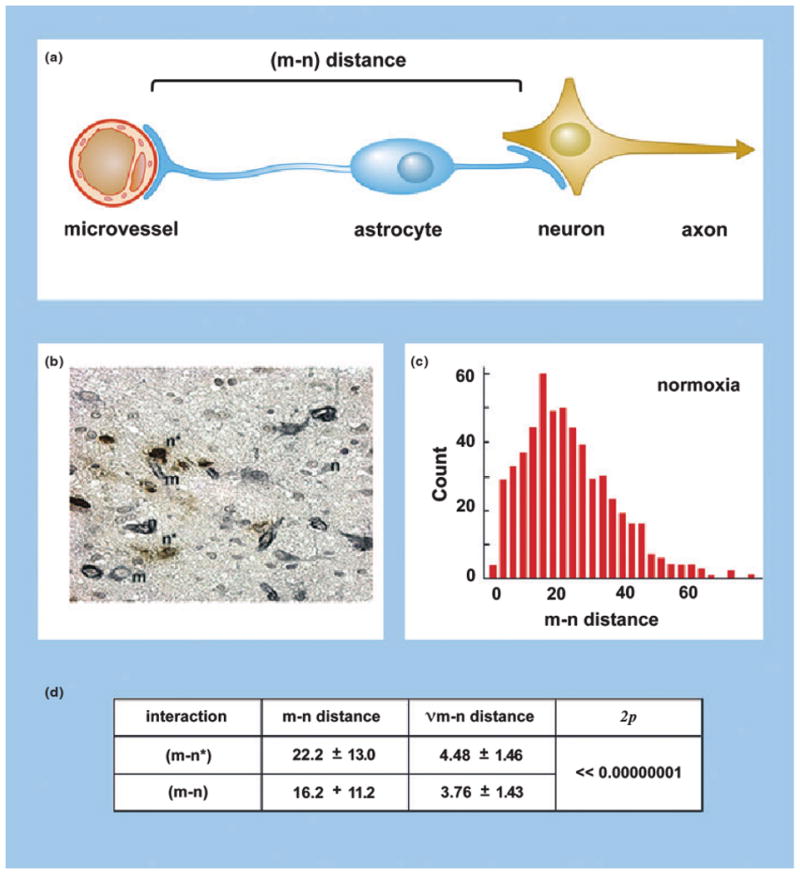
The effects of focal ischaemia on the neurovascular unit. (a) The distance between microvessels and neurons [(m–n) distance] within the striatum of the nonhuman primate (Papio anubis/cynocephalus) as a measure of neurovascular integrity. (b) Injured neurons (n*), uninjured neurons (n), and microvessels (m) scattered within the ischaemic core at 2 h following middle cerebral artery (MCA) occlusion in the nonhuman primate. (c) (m–n) Distance distribution based upon measurements of normoxic striatal neurons and their proximate microvessels. (d) Demonstration that neurons more distant from their nearest microvessel [(m–n*)] are significantly more likely to display injury than those at lesser distance [(m–n)] in the ischaemic striatum at 2 h post-MCA occlusion [24].
Microvascular matrix and matrix proteases
The basal lamina of cerebral microvessels provides a scaffold on which the endothelial and the glial compartments interact. The integrity of the microvasculature depends upon the proximity of astrocyte end-feet to the endothelium: both are required for the formation of the basal lamina matrix and permeability barriers [13, 20, 25]. The matrix limits the transmigration or leakage of blood cells: erythrocytes during haemorrhage and leucocytes in response to inflammatory stimuli (e.g., the inflammatory phase of ischaemia). The integrity of the microvasculature is also affected by the presence of pericytes located within the matrix, or vessel wall histiocytes within larger vessels [26].
Focal ischaemia initiates: (i) rapid loss of integrity of the matrix within the microvasculature, and (ii) rapid loss of matrix-adhesion receptors (or changes in their conformation). Loss of the basal lamina matrix also corresponds to the appearance of haemorrhagic transformation within the ischaemic territory [27]. A significant rapid loss of the β1-integrin subunits α1, α3 and α6 from endothelial cells, the integrin α6β4 from astrocytes, and αβ-dystroglycan from astrocyte end-feet occurs within the ischaemic core following MCA:O [28–30].
Loss of the matrix proteins laminin, collagen IV, cellular fibronectin and perlecan has been attributed to the rapid generation of matrix proteases in response to ischaemia. del Zoppo et al. have demonstrated the rapid appearance of members of four families of proteases in the ischaemic territory following MCA:O (nonhuman primate) on microvessels and nearby neurons: (i) the latent matrix metalloproteinases (pro-MMP)-2 and -9 [31, 32]: pro-MMP-9 is associated with the appearance of haemorrhagic transformation [31], whilst pro-MMP-2 is directly related to the appearance of neuron injury within the ischaemic core regions [31]; (ii) cathepsin L, a member of the cysteine protease family, is rapidly generated in select microvessels and neurons, accompanying degradation of perlecan and laminin within the microvascular basal lamina [33]; (iii) heparanase is also generated in the same temporal and topographic context[33]; and (iv) the serine protease urokinase (u-PA) is associated with its receptor u-PAR [32]. It seems likely that these matrixdegrading proteases can participate in matrix-adhesion receptor changes within the microvasculature. As one recent example, incubation with the general MMP activity inhibitors GM6001 or 1,10-phenanthroline prevented the loss of β-dystroglycan from murine primary cerebral astrocytes under conditions of experimental ischaemia, suggesting that MMPs participate in this adhesion receptor change [7].
The common mechanism(s) responsible for the appearance of the matrix proteases (urokinase (u-PA), cathepsin L, heparanase, and pro-MMP-2) on both microvessels and neurons within the microvascular unit are not yet known [31–33]. Their rapid and simultaneous association with microvessels and proximate neurons reinforce the impression; however, that events within different elements of the microvascular unit within the target territory are somehow coordinated during initiated focal ischaemia.
In summary: (i) microvessel responses and neuron injury occur in the same time frame and the same sub-regions of ischaemic injury, (ii) an ordered relationship exists between microvessels and neighbouring neurons within the susceptible territory, (iii) rapid significant alterations in the matrix of the vascular and in the nonvascular compartments of the ischaemic territory occur, and (iv) the loss of one type of matrix-adhesion receptor that accompanies microvascular matrix degradation can be prevented, in part, by inhibition using known Zn2+ chelating agents.
These observations suggest the hypothesis that focal ischaemia can initiate coordinated responses within the microvasculature and nearby neurons in which both elements appear to behave as a unit.
One of the implications of this hypothesis is that neurons and the microvascular endothelium can communicate in both directions. Rather than a unilateral unidirectional dependence of regional microvascular flow and dynamics upon neuron signalling, the proximity of the endothelium to astrocyte end-feet and the bi-functional nature of their matrix-adhesion receptors imply that communication is possible across the matrix. Another implication is that events that stimulate cell activation (e.g., inflammation) may have differential impact on the components of the neurovascular unit. There are further implications for the testing of proposed interventions in clinical trials and their consequences depending upon whether the target is flow through the microvessel bed or neuron integrity. The ‘unitary’ conceptual framework predicts that there would be a limit to recovery, if flow were not re-established, because of the vulnerability of sensitive components of the ‘unit’. Strategies that attempt to preserve one component of the ‘unit’, such as glia or neurons might be unhelpful (or perhaps harmful), if flow is not also involved. This requires understanding specific cellular vulnerabilities and how the cell components interact with each other under conditions of normoxia and of ischaemia.
Inflammatory stimuli
One example of differential responses within the neurovascular structure is the effect of inflammatory mediators on tissue integrity. All components of the neurovascular unit appear to respond to inflammation [34]. These include the endothelium, astrocytes, the vascular extracellular matrix, pericytes, and neurons and their axons. Microglia, oligodendroglia and mast cell function are modulated by inflammatory stimuli. Many of the processes known to be initiated by ischaemia, that affect the neurovascular unit, including inflammatory stimuli are listed in Table 1. As in focal cerebral ischaemia, inflammatory responses are mediated/generated in part through the cytokines IL-1β and TNF-α that appear initially within 2–6 h of ischaemia [35–37]. The effects in individual cell populations (e.g., endothelium, astrocytes, pericytes, microglia, mast cells and neurons) have been examined. Endothelial cells facilitate cellular inflammation (e.g., by participation in firm adhesion of polymorphonuclear (PMN) leucocytes, and facilitation of transmigration) and generate and respond to proinflammatory stimuli. But, how individual cells within the unit communicate with each other during post-ischaemic inflammatory responses is not clear: a unifying synthesis of inflammatory responses has not yet been developed.
Table 1.
Processes involved in ischaemic injury of the CNS
| Metabolic events |
| Generation of free radicals |
| NO generation |
| Protein oxidation |
| Lipid peroxidation |
| Depolarization by neurons in evolving infarction and peri-infarct area |
| Acidosis (lactoacidosis) |
| Ca2+-mediated cell demise |
| Failure of Ca2+ exclusion mechanisms |
| Increase in cytosolic Ca2+ |
| Activation of lipolytic pathways |
| Alterations in protein phosphorylation |
| Depletion of ATP stores with depression of glucose metabolism |
| Impaired mitochondrial function |
| Proteolysis |
| Protein synthesis |
| Cellular events (acute) |
| Activation of cell signalling pathways |
| Decreased endothelial cell matrix-adhesion receptor expression |
| Decreased astrocyte matrix-adhesion receptor expression |
| Detachment of astrocyte end-feet from vascular basal lamina matrix |
| Degradation of vascular basal lamina matrix components |
| Expression of matrix proteases |
| Matrix metalloproteinases |
| Cathepsins |
| Heparanase |
| Serine proteases (e.g., urokinase) |
| Increased microvascular permeability |
| Astrocyte swelling |
| Neuron swelling |
| Release of glutamate from neurons |
| Depolarization by neurons in evolving infarction and peri-infarct area |
| Expression of cytokines |
| Generation of platelet activating factor (PAF) |
| Activation and appearance of inflammation |
| Activation of endothelial cell leucocyte adhesion receptors |
| Activation of PMN leucocytes and monocytes |
| Adhesion and transmigration of PMN leucocytes |
| Platelet activation |
| Microglial cell activation, shape alterations, and migration |
| Tissue injury |
| Oedema formation |
| Cellular swelling |
| Focal ‘no-reflow’ within ischaemic regions |
| Increased microvascular permeability with leakage |
| Angiogenesis with capillary bud formation |
| Cell demise |
| Haemorrhagic transformation |
| Liquefaction and cavitation |
The neurovascular unit and acute interventions in ischaemic stroke
Considerable effort has been expended to treat patients acutely after the onset of ischaemic stroke, despite a lack of characterization of the unitary responses of cells or cell groups within the unit to ischaemia, when it was shown that neurological benefit could be gained with arterial recanalization. Prospective controlled randomized trials and fundamental experimental work have examined two classes of treatments in the early moments following focal ischaemia: (i) ‘neuroprotectant’ agents, and (ii) anti-thrombotic agents. Amongst the neuron protectant group tested have been agents with NMDA receptor antagonism (e.g., cerestat, citicholine), free radical scavenger (e.g., tirilazad mesylate, NXY-059), or anti-inflammatory immune inhibitor (e.g., enlimomab) properties. Amongst anti-thrombotic agents used for acute intervention are select anti-platelet agents (e.g., abciximab, eptifibatide), and plasminogen activators (e.g., rt-PA, single chain urokinase (scu-PA).
Neuron protectants
Agents that appear to prevent the demise of cultured neurons or to limit ischaemic injury in small animal models of focal cerebral ischaemia have been termed ‘neuroprotectants’. Amongst the post-ischaemic injury mechanisms specifically targeted are Ca2+ regulation and transit, neurotransmitter release, and cell death pathways. Whilst fundamental studies have identified compounds that salvage neurons in preclinical model systems, the undertaking of clinical trials of these compounds have, for various reasons, proved unsuccessful. Much has been written about the reasons for these apparent failures, to which we should like to add a further comment.
Although several examples can be chosen, the recently terminated programme to develop NXY-059 for acute intervention in ischaemic stroke provides some insight into the relevance of the neurovascular unit [38–42]. NXY-059, a bis-sulfamyl analogue of the nitrone spin trap compound phenylbutylnitrone (PBN), was shown to reduce injury in small animal focal ischaemia models, including both transient and focal models in rodents [43]. Synthesized NXY-059, in a rabbit multi-hit model, suggested decreased injury in the presence of rt-PA [44]. Marmosets treated with NXY-059 displayed improved behavioural outcome compared to controls [43]. Advanced to stroke patients, a dose range study demonstrated that NXY-059 was safe in patient volunteers [38]. In short, based on the assumptions of the first STAIR conference, that an ascending phylogenic array of stroke models could adequately define success in clinical outcome, large-scale placebo-controlled trials were planned [43]. With this background, two phase III prospective blinded control trials were undertaken. The SAINT-1 trial, a prospective placebo-controlled randomized multi-centre study of NXY-059 on stroke outcome hinted at benefit at 90 days post-treatment [39]. The follow-on study, SAINT-2, unequivocally demonstrated no benefit compared to placebo [41]. Despite a long and vigorous development period, this compound, whilst apparently safe, showed no efficacy in two clinical populations. Green summarized his interpretation of this string of preclinical studies supported by the sponsor: ‘it is the first neuroprotectant to have entered clinical trials having fulfilled the STAIR criteria. The preclinical profile has enabled a direct translation from the findings in animal models to the clinical trial design in terms of time window and exposure, for the first time in the history of neuroprotective drug development [43]’.
But, limitations in the applicability of the preclinical data to the pathophysiology of cerebral ischaemia were not considered: (i) the generation of free radicals at the microvessel-neuropil interface [45, 46], (ii) unanswered questions regarding the volume of distribution of the agent and its ability to cross the blood–brain barrier, and (iii) concerns regarding the translation of results in small animal models of focal ischaemia to ischaemic stroke in humans [47]. NXY-059 did not cross the permeability barrier into the CNS to the abluminal face of the ischaemic microvasculature where free radical generation is known to occur [45]. Nonetheless, a reasonable sequential clinical test development of NXY-059 was undertaken.
Acute intervention with other agents targeting neurons has produced neutral or negative results in placebo-controlled trials, in contrast to demonstrable benefit in cell culture and small animal models of experimental ischaemia. Explanations for these discrepancies suggest a number of questions: why have agents shown to protect neurons in model systems been uniformly unsuccessful in clinical trials? Are there characteristics of human cerebral responses to ischaemia that current brain injury models cannot reproduce? Two broad categories of concerns about the failure to successfully translate benefits from animal model systems into the clinical arena in ischaemic stroke are (i) limitations in clinical trial design, and (ii) limitations associated with the model systems. In the clinical design, for NMDA receptor inhibitors, failure of translation was in part because of the failure to use doses beneficial in models, but associated with expected somnolence in patients. The failure of tirilazad mesylate in clinical trials may have in part been due to the failure to include reperfusion in the trial design. But other explanations are possible and likely. Amongst the limitations with regard to in vitro cell culture and suspension models as well as anaesthetized small animal preparations, the failure to identify specific target cells and the interactions of those cells under normal function may have contributed to these failures. In the absence of a clear understanding of the interactions of the cells within the neurovascular unit, and their behaviour as targets to potential interventions during ischaemic injury that would affect all components, it is likely that affecting the upstream blood flow characteristics of the unit could be most beneficial. Can restitution of flow through an acutely thrombosed cerebral artery limit injury progression in all patients?
Anti-thrombotic agents
Ischaemic stroke is a vascular disorder. Whilst neuron injury from other causes can mimic the neurological abnormalities of focal ischaemia, neuron injury alone without impairment of the arterial supply does not generate the infarcted lesion typical of focal ischaemia. Hence, strategies that could preserve or re-establish flow can provide benefit. To this end, anti-platelet interventions are employed for secondary prevention of additional ischaemic events after a signal transient ischaemic attack (TIA) or ischaemic stroke, whilst anticoagulants are employed for primary prevention of thromboembolic events from nonvalvular atrial fibrillation and select prosthetic valve devices. The potential role of anti-thrombotic agents in microvascular disease, typified by lacunar lesions or neuropsychiatric lupus, has not been defined. When no clear aetiology is available for an ischaemic stroke, well-controlled oral anticoagulation appears to have no advantage over aspirin [48].
Acute infusion of the plasminogen activator rt-PA in carefully selected patients within 3 h of symptom onset is associated with a significant 10–13% increase in the proportion of patients with little or no residual deficit in patients treated [49]. Recently, it has been reported that well-selected patients can also be treated 3.0–4.5 h after symptom onset [50]. Those studies assume that the improvement in outcome was directly due to recanalization through the microvascular bed of the ischaemic zone, but a direct relationship was not looked for. In early studies employing arterial imaging, recanalization of carotid artery territory occlusions was achieved in 34–59% of patients treated by intravenous infusion techniques within 8 h of symptom onset [51–54], whereas early studies using catheter-directed SK or u-PA delivery achieved recanalization of symptomatic carotid artery territory occlusions in 46–90% of patients treated within the same time frame [55, 56]. Haemorrhagic transformation occurred in 29–53% of treated patients in those studies [51–54]. A prospective placebo-controlled trial by Mori et al. was the first to demonstrate significant improvement in neurological outcome in relation to reperfusion [54].
Those studies demonstrated both the feasibility and the relative safety of the plasminogen activator rt-PA (duteplase) to re-establish flow in the CNS and improve neurological outcome. They suggested, in more contemporary terms, that preservation of flow to/through the microvessel components of the neurovascular unit could preserve neuron function. However, there is heterogeneity in recanalization within series of patients based on occlusion locations of the brain-supplying artery: acute recanalization occurs more frequently with MCA division and branch occlusions, than the more proximal internal carotid artery occlusions [51]. But, the responses of neurons to vascular reperfusion are very relevant. Whilst some differences in outcomes amongst separate prospective controlled clinical trials can be explained by differences in trial conduct, other features that reflect vascular and tissue responses to arterial reflow probably also operate. These could, indeed, reflect differences in the responses of neurovascular units within the injured territory.
For instance, whilst prospective blinded-placebo-controlled trials have demonstrated significant improvement with acute plasminogen activator use, there were significant differences in outcome amongst the trials that suggest differences in tissue response. In the two-part, four-armed, placebo-controlled outcome study of rt-PA in patients entered within 3 h from ischaemic stroke symptom onset demonstrated a significant 11–13% absolute increase over placebo in each of four measures of outcome [49].
Two phase III prospective, randomized placebo-controlled safety and efficacy studies of intravenous rt-PA (alteplase) given within 6 h of symptom onset, the European Cooperative Acute Stroke Study (ECASS) and ECASS-II, indicated no significant difference in disability outcome between the two groups [57, 58]. With an adjustment to ensure a consistent application of the entry criteria (to limit apparent injury by CT evaluation to <0.33 of the MCA territory) an absolute improvement of 3.7% in mRS = 0–1 was observed in ECASS-II [58]. The median entry NIHSS scores of patients in ECASS and ECASS-II were very similar, suggesting that for the most part the study populations presented with comparable stroke severity. Cerebral haemorrhage causing death or deterioration was also more frequent in the rt-PA group than the placebo group (11.7% vs. 3.1%).
European Cooperative Acute Stroke Study-III, a prospective blinded-placebo-controlled study of rt-PA given to patients treated between 3.0 and 4.5 h after symptom onset demonstrated a significant 7.2% improvement in favourable outcome (mRS = 0–1) amongst patients treated with rt-PA [50]. That trial confirms and supports the observation that exposure of the ischaemic territory with this plasminogen activator within the first hours of ischaemia onset can produce detectable consistent improvement in clinical outcome in a subset of patients. Given the variance around the point estimates of the odds ratios between the NINDS-sponsored trial and ECASS-III, the per-patient odds of beneficial neurological outcome were not different between the two studies.
This variance in the experience amongst the prospective clinical trials hints at a relevant feature of the brain response to injury that encompasses both brain tissue and neurovascular susceptibility. The contributors to outcome appear to include microvascular patency, restitution of flow, direct effects of interventional agents, and intrinsic features of the vulnerable brain. Studies of MCA occlusion and restitution of flow in the nonhuman primate were the first to demonstrate that large artery perfusion is associated with decreased injury in a humanoid species; however, injury volume depended in part on the exposure to anaesthesia [59, 60]. Microvessel patency is altered by processes that initiate ischaemia. In as much as the neuron viability depends exquisitely on flow in nearby and adjacent supply capillaries, preservation of capillary flow is fundamental to the preservation and rapid recovery of neurological function. During experimental focal ischaemia occlusion of supply microvessels occurs in a heterogeneous pattern with ‘open’ microvessels adjacent to obstructed microvessels in regions of neuron injury [5]. Inhibition of fibrin formation, platelet activation, or endothelial cell adhesion of PMN leucocytes results in the preservation of microvessel patency and (in settings where it has been tested) neurological recovery [61–64].
Another clinical example of variance is demonstrated in the prospective double-blind placebo-controlled level I dose-finding study (prourokinase in acute cerebral thromboembolism, PROACT) that compared recombinant scu-PA (6 mg) with placebo in patients treated within 6 h of symptom onset who had demonstrated M1 and M2 segment MCA occlusions [65]. The significant increase in MCA recanalization observed in the scu-PA group was heparin-dependent. A follow-on study, PROACT-2, prospectively tested the effect of unblinded direct delivery of recombinant scu-PA (9 mg) against no instrumentation on recanalization and disability outcome [66], but demonstrated no benefit when the outcome mRS = 0–1 was reported, compared to the control group. Technical issues in both studies with individual patients could also have contributed to the variance in outcome, but were not sufficiently commented upon.
The consistency of patient selection is an important contribution to the heterogeneity in patient outcome. At least two of the trials mentioned were underpowered to achieve the projected treatment benefit at the outset, and a further study did not attempt to achieve a homogeneous population. Deviations from patient selection criteria in several studies give some insight into tissue susceptibility. During ECASS, ∼60 patients with evidence of prior focal cerebral injury were entered in violation of the prescribed entry criteria. This subgroup, as evidenced by the outcomes in the subgroup that received placebo, demonstrated greater residual disability than the overall study population, and significantly greater early mortality when treated with rt-PA. The frequency of haemorrhagic transformation, a measure of microvessel or arteriolar integrity, was significantly increased in the rt-PA group. Those observations imply that the degree of injury within the neurovascular unit is fundamental to the responses of the injured tissue to rt-PA or placebo.
A current problem is how to clinically determine the extent of injury of various regions within the ischaemic territory. Experimental model studies, and positron emission tomography (PET) and magnetic resonance imaging studies of patients, suggest that early post-ischaemia the injury is homogeneous, whilst cellmicrovessel studies in experimental ischaemia demonstrate that neurons and endothelial cell β1-integrin responses are heterogeneous [30, 67].
Haemorrhagic transformation
The prospective clinical studies of acute plasminogen activator delivery have suggested tissue signatures of cerebral haemorrhage risk. Haemorrhagic transformation is defined as either haemorrhagic infarction (HI, petechial to confluent petechial haemorrhages), parenchymal haemorrhage (PH, haematomas causing mass effect and most often clinical worsening), or both [51, 68, 69].
Haemorrhagic infarction appears to follow from degradation of the microvessel basal lamina matrix within the ischaemic core, which permits local leakage of blood elements in the tissue [27]. The loss of microvascular matrix corresponds to the appearance of the four families of matrix proteases by proximity, timing, sources, and the up-regulation of specific protease activation systems [27, 31–33, 70].
Parenchymal haemorrhage has been attributed to structural degradation of arterioles damaged by ischaemia when they are exposed to arterial pressure [68]. The risk of detectable haemorrhage increases in the presence of anti-thrombotics, with the highest risk of PH associated with the use of plasminogen activators, and the lowest with anti-platelet agents. Haemorrhage could be accentuated when un-inhibited plasmin degrades extravascular fibrin, thereby preventing the sustained formation of a fibrin-based and haemostatic thrombus [51]. Clinically, PH frequency depends upon a number of known risk factors (Table 2). Contributors to the risk of haemorrhage in the setting of plasminogen activator treatment of ischaemic stroke include excessive time from the onset of symptoms to treatment, low body mass index, diastolic hypertension, older age and the use of rt-PA [49, 51, 65, 71–73]. Data from individual studies have shown that when patients received rt-PA (duteplase) as late as 8 h after symptom onset there was a monotonic increase in the frequencies of HI and PH [51]. That observation explains both earlier experimental and clinical observations with streptokinase and urokinase. The appearance of ‘early signs of ischaemia’ on the initial CT scan is also associated with increased risk of haemorrhage and demise [57]. Because the substrates of ischaemic injury include microvessel and vascular matrix integrity within the neurovascular unit, it is predicted that haemorrhage risk reflects the fundamental risk in the placebo-treated population.
Table 2.
Haemorrhagic transformation in the setting of plasminogen activator treatment
| Factor | Agent |
|---|---|
| Time from symptom onset | rt-PA (d) |
| Diastolic hypertension | rt-PA |
| Low body mass | rt-PA |
| Age | rt-PA |
| Atrial fibrillation | rt-PA |
| ‘Early signs’ of ischaemia | rt-PA |
| rscu-PA | |
| Haemorrhage | rt-PA |
Parenchymal haemorrhage can significantly impact survival, but can also affect neurological (neuron) function. This appears to be at least population-dependent.
In the NINDS-sponsored study, the frequency of PH was significantly greater amongst those patients receiving rt-PA (6.4%) than amongst those who received placebo (0.6%) at 3 months. Whilst overall mortality was unchanged, intracerebral haemorrhage contributed to demise in the rt-PA group [49]. The PH frequency associated with rt-PA exposure in ECASS was higher, as was that of the placebo group [57]. Similarly, in ECASS-II, PH in both the active agent and the placebo groups exceeded that of the NINDS study, but were intermediate with respect to the ECASS study [58]. A survey of the CNS haemorrhagic events in recent trials of acute thrombolysis indicates that the increased incidence of PH amongst patients treated acutely with rt-PA is directly related to the incidence of PH in the placebo population, and that this varies from study to study. Therefore, one might postulate that the incidence of PH, and associated neurovascular unit injury, reflect attributes of brain tissue susceptibility to injury of individual patients or patient groups, and therefore the heterogeneity of this susceptibility. The nature of these attributes could be taken as the ‘environment’ of the neurovascular unit.
Environments of the neurovascular unit
Both the known contributors to the increased risk of haemorrhagic transformation (section Haemorrhagic transformation) and the evidence of microvessel-neuron distribution features during focal ischaemia (section (m–n) Distance distributions) suggest that the integrity of the neurovascular unit depends upon this environment. One set of events that likely contributes to the change in neurovascular environment is that which contributes to the loss in the microvessel permeability barrier. Heo et al. first demonstrated that haemorrhagic transformation is associated with pro-MMP-9 expression in a human-relevant system [31]. The mechanisms for this are not apparent, although several clinical [74–77] and small animal experimental studies [31, 78, 79] have supported those observations.
A fundamental aspect of the microvessel ‘environment’, independent of neuron involvement (or of changes in neuron integrity which could be a direct consequence), is the impact of focal ischaemia on microvessel reactivity: (i) the alterations in the permeability barrier, and (ii) the focal ‘no-reflow’ phenomenon.
Adhesion receptor changes
Focal cerebral ischaemia, as early as 2 h after onset, induces a rapid loss of β1-integrin expression by endothelial cells and astrocytes (Fig. 3) [29, 67, 80]. Because these events coincide with increased cerebrovascular permeability [81], loss of endothelial and astrocyte adhesion to the microvascular basal lamina may be early pivotal events that contribute to loss of the microvascular permeability barrier. In the neurovascular unit, the proximity of astrocyte end-feet to the abluminal surface of the endothelium suggests roles in integrin signalling by the matrix in both cell compartments that support their maintenance of the permeability barrier.
Fig. 3.
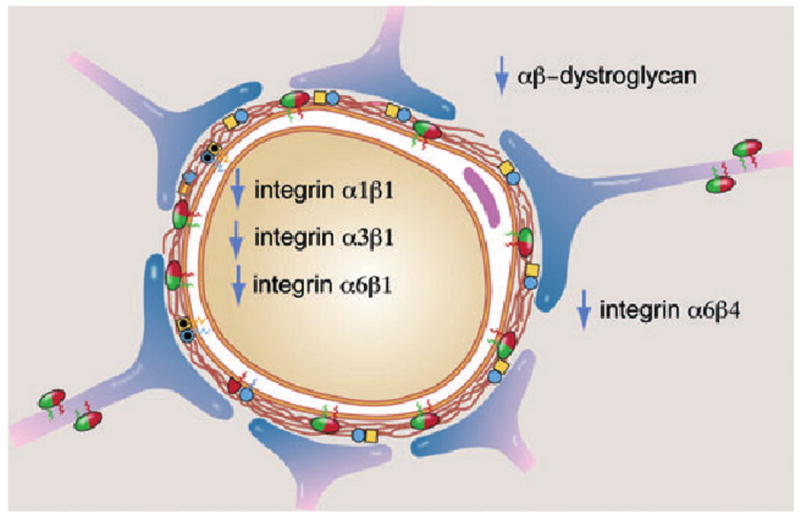
Impact of ischaemia on the expression of matrix integrin receptors by microvessel endothelium, and matrix integrins and dystroglycan receptors on astrocyte end-feet in a model cerebral capillary. All changes occur within 2 h of MCA occlusion in the nonhuman primate striatum.
To define these events at baseline, we have examined how integrin expression by endothelial cells and astrocytes is regulated by matrix proteins present in the basal lamina, and then by ischaemia mimicked in vitro by oxygen-glucose deposition (OGD). OGD is a form of experimental ischaemia applied to cell cultures that incorporates restrictions in O2 supply and nutrient availability, but not the cessation of flow or pH changes observed in focal cerebral ischaemia.
Previous studies with other cell types have shown that cellular expression of integrins can be strongly influenced by the matrix ligands on which they are cultured [82, 83]. The major findings are that (i) whereas astrocyte β1-integrin expression is relatively unaffected by exposure to the matrix substrate, the expressions of subunits β1 and α5 by endothelium are promoted by all matrix ligands, particularly perlecan, (ii) OGD induces significant changes in integrin expression of both astrocyte and endothelial cells, and (iii) during OGD the murine cell responses largely recapitulate the basic changes in microvascular integrin expression observed during focal cerebral ischaemia in the nonhuman primate.
During OGD, β1-integrin expression increases, which is supported by the up-regulation of β1-transcription by astrocytes, and is recapitulated by the increase in β1-integrin protein in microvessels in the ischaemic sub-region [67]. On isolated primary endothelial cells, OGD induced an unexpected twofold increased expression of the β1-integrin subunit in one study. Indeed, with the exception of the integrin αvβ3, focal cerebral ischaemia in vivo induces loss of all endothelial integrins including β1 [30, 67, 80]. But, the β1-integrin responses by endothelial cells in vitro are consistent with the previous finding of increased microvessel-associated β1-integrin mRNA expression within sub-regions peripheral to ischaemic cores, but not within the core regions themselves [67]. This suggests that brain endothelial cells (and astrocytes) can respond to ischaemia by attempting to increase β1-integrin expression levels. One implication of this finding is that the conditions initiated by focal ischaemia effect changes in microvascular β1-integrin expression by processes that are likely post-transcriptional, as suggested by the in vitro astrocyte responses. Most importantly these early events are distributed heterogeneously, involving discrete sub-regions of core injury surrounded by boundaries of microvessel activity.
Focal ischaemia in experimental systems alters endothelial cell-astrocyte relationships via its matrix receptors [7, 28, 30, 67]. The implications for ischaemic stroke are that (i) efforts to preserve or rapidly reestablish flow through the threatened microvascular bed are likely to reduce neuron injury within a regional network, (ii) ‘neuron protection’ per se may not be sufficient if the astrocyte-endothelial cell relationships are disrupted by ischaemia, (iii) preservation of the matrix-matrix receptor interactions could contribute to preservation of neuron/neurovascular function, and (iv) preservation of astrocyte function would seem essential for maintaining the normal function of the neurovascular unit. Given our natural limitations in detecting small improvements in function clinically efforts that successfully maintain neurovascular function in the face of focal ischaemia may not be detectable in the clinical setting. However, experience with acute interventions so far should give us a view of whether this direction of enquiry has merit.
Focal microvessel obstruction
Within hours following proximal MCA occlusion in the nonhuman primate, obstruction of microvessels in the striatum is observed [61, 62]. This focal ‘no-reflow’ results when the endothelium is activated, expresses the leucocyte adhesion receptors P-selectin and intercellular adhesion molecule-1 (ICAM-1), such that PMN leucocytes are activated and lodge within microvessels in the territory-at-risk. These events are distributed heterogeneously, with occluded microvessels observed amongst those that are patent. Microvessel obstruction can be prevented by inhibition of PMN leucocyte β2-integrin–ICAM-1 interactions [62]. The obstructions also contain activated platelets and fibrin, variously [63, 64]. The deposition of fibrin within the microvasculature originates from exposure of perivascular tissue factor (TF) to the plasma, and can be significantly decreased by blocking TF-factor VIIa interactions that initiate thrombin generation via the extrinsic system [63]. Platelet activation entails interaction of the platelet surface integrin receptor αIIBβ3 with fibrin(ogen), an interaction that is inhibited in a dose-dependent manner by organic inhibitors or arginine-glycine-aspartic acid (RGD)-containing peptides [64, 84]. With increased concentrations of the inhibitor, significant symptomatic haemorrhage results. Whether inhibiting PMN leucocyte adherence within the microvasculature, preventing platelet and fibrin deposition, or both leads to a reduction in neuron injury has not yet been studied. But, separate experiments demonstrate that the acute use of low molecular weight heparins (LMWHs) (e.g., enoxaparin) in rodent models of MCA occlusion, significantly decreases residual injury volume and improves behavioural outcome [85].
It is certain that focal ischaemia initiates major alterations in cerebral microvessel integrity that coincide with increased permeability, exposure of the perivascular tissue to plasma (that initiates fibrin formation), and the effects of haemorrhagic transformation. Furthermore, within the same territory-at-risk, the endothelium is activated, leading to leucocyte adhesion and activated platelets that contribute to microvessel obstruction at the same time that neurons display injury. Early on those events are heterogeneously distributed.
Summary
Cell and tissue model studies indicate the importance of microvessel–neuron relationships in the acute setting of focal ischaemia. The coordinate and potentially unitary nature of these relationships is suggested by known matrix responses to focal ischaemia. As a framework for understanding the responses to acute intervention during ischaemic stroke, further basic information is required that adequately relate microvessel reactivity and changes in neuron integrity in mammalian systems. The observations of matrix-matrix receptor interactions within cerebral microvessels and the rapid expression of matrix-sensitive proteases under normoxic and ischaemic conditions provide starting points for dissecting the direction and consistency of microvessel–neuron communication. Those ongoing studies do not yet examine how these processes affect neuron excitability and membrane function. But, they do indicate at how many levels functions within the neurovascular framework are still not understood. One implication of this paucity of information and the clinical trial experience so far is that interventions that solely target ‘neuron protection’ may be wholly inadequate to achieve clinical benefit. Strategies that extend to structural integrity and astrocyte function within the neurovascular unit may be revealing. But, this will require a more fundamental understanding of the cell–cell, tissue functional, and neurovascular inter-relationships.
Footnotes
Conflict of interest statement: No conflict of interest was declared.
References
- 1.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–60. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 2.Zonta M, Angulo M, Gobbo S, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]
- 3.Nedergaard M, Ransom BR, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–30. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Koehler RC, Roman RJ, Harder DR. Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 2009;32:160–9. doi: 10.1016/j.tins.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 5.del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23:879–94. doi: 10.1097/01.WCB.0000078322.96027.78. [DOI] [PubMed] [Google Scholar]
- 6.del Zoppo GJ, Milner R. Integrin-matrix interactions in the cerebral microvasculature. Arterioscler Thromb Vasc Biol. 2006;26:1966–75. doi: 10.1161/01.ATV.0000232525.65682.a2. [DOI] [PubMed] [Google Scholar]
- 7.Milner R, Hung S, Wang X, Spatz M, del Zoppo GJ. The rapid decrease in astrocyte-associated dystroglycan expression by focal cerebral ischemia is protease-dependent. J Cereb Blood Flow Metab. 2008;28:812–23. doi: 10.1038/sj.jcbfm.9600585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liesi P. Do neurons in the vertebrate CNS migrate on laminin? EMBO J. 1985;4:1163–70. doi: 10.1002/j.1460-2075.1985.tb03755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engvall E, Davis GE, Dickerson K, Ruoslahti E, Varon S, Manthorpe M. Mapping of domains in human laminin using monoclonal antibodies: localization of the neurite-promoting site. J Cell Biol. 1986;103:2457–65. doi: 10.1083/jcb.103.6.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herken R, Gotz W, Thies M. Appearance of laminin, heparan sulphate proteoglycan and collagen type IV during initial stages of vascularisation of the neuroepithelium of the mouse embryo. J Anat. 1990;169:189–95. [PMC free article] [PubMed] [Google Scholar]
- 11.David S, Braun PE, Jackson DL, Kottis V, McKerracher L. Laminin overrides the inhibitory effects of peripheral nervous system and central nervous system myelin-derived inhibitors of neurite growth. J Neurosci Res. 1995;42:594–602. doi: 10.1002/jnr.490420417. [DOI] [PubMed] [Google Scholar]
- 12.Grant DS, Kleinman HK. Regulation of capillary formation by laminin and other components of the extracellular matrix. EXS. 1997;79:317–33. doi: 10.1007/978-3-0348-9006-9_13. [DOI] [PubMed] [Google Scholar]
- 13.Bernstein JJ, Getz R, Jefferson M, Kelemen M. Astrocytes secrete basal lamina after hemisection of rat spinal cord. Brain Res. 1985;327:135–41. doi: 10.1016/0006-8993(85)91507-0. [DOI] [PubMed] [Google Scholar]
- 14.The Persantine-Aspirin Reinfarction Study Research Group. Persantine and aspirin in coronary heart disease. Circulation. 1980;62:449–61. doi: 10.1161/01.cir.62.3.449. [DOI] [PubMed] [Google Scholar]
- 15.Nagano N, Aoyagi M, Hirakawa K. Extracellular matrix modulates the proliferation of rat astrocytes in serum-free culture. GLIA. 1993;8:71–6. doi: 10.1002/glia.440080202. [DOI] [PubMed] [Google Scholar]
- 16.Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol. 1993;123:1777–88. doi: 10.1083/jcb.123.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh M, Nagafuchi A, Yonemura S, Kitani-Yasuda T, Tsukita S, Tsukita S. The 220-kD protein colocalizing with cadherins in non-epithelial cells is identical to ZO-1, a tight junction-associated protein in epithelial cells: cDNA cloning and immunoelectron microscopy. J Cell Biol. 1993;121:491–502. doi: 10.1083/jcb.121.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuse M, Itoh M, Hirase T, et al. Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol. 1994;127:1617–26. doi: 10.1083/jcb.127.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147:891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hurwitz AA, Berman JW, Rashbaum WK, Lyman WD. Human fetal astrocytes induce the expression of blood-brain barrier specific proteins by autologous endothelial cells. Brain Res. 1993;625:238–43. doi: 10.1016/0006-8993(93)91064-y. [DOI] [PubMed] [Google Scholar]
- 21.Bär T. Morphometric evaluation of capillaries in different laminae of rat cerebral cortex by automatic image analysis: changes during development and aging. In: Cervos-Navarro J, editor. Advances in Neurology. New York: Raven Press; 1978. pp. 1–9. [PubMed] [Google Scholar]
- 22.Bär T. The vascular system of the cerebral cortex. Adv Anat Embryol Cell Biol. 1980;59:1–62. doi: 10.1007/978-3-642-67432-7. [DOI] [PubMed] [Google Scholar]
- 23.Bär T. Patterns of vascularization in the developing cerebral cortex. Ciba Found Symp. 1983;100:20–36. doi: 10.1002/9780470720813.ch3. [DOI] [PubMed] [Google Scholar]
- 24.Mabuchi T, Lucero J, Feng A, Koziol JA, del Zoppo GJ. Focal cerebral ischemia preferentially affects neurons distant from their neighboring microvessels. J Cereb Blood Flow Metab. 2005;25:257–66. doi: 10.1038/sj.jcbfm.9600027. [DOI] [PubMed] [Google Scholar]
- 25.Webersinke G, Bauer H, Amberger A, Zach O, Bauer HC. Comparison of gene expression of extracellular matrix molecules in brain microvascular endothelial cells and astrocytes. Biochem Biophys Res Commun. 1992;189:877–84. doi: 10.1016/0006-291x(92)92285-6. [DOI] [PubMed] [Google Scholar]
- 26.Dore-Duffy P. Pericytes: pluripotent cells of the blood brain barrier. Curr Pharm Des. 2008;14:1581–93. doi: 10.2174/138161208784705469. [DOI] [PubMed] [Google Scholar]
- 27.Hamann GF, Okada Y, del Zoppo GJ. Hemorrhagic transformation and microvascular integrity during focal cerebral ischemia/reperfusion. J Cereb Blood Flow Metab. 1996;16:1373–8. doi: 10.1097/00004647-199611000-00036. [DOI] [PubMed] [Google Scholar]
- 28.Haring HP, Akamine P, Habermann R, Koziol JA, del Zoppo GJ. Distribution of integrin-like immunoreactivity on primate brain microvasculature. J Neuropathol Exp Neurol. 1996;55:236–45. doi: 10.1097/00005072-199602000-00012. [DOI] [PubMed] [Google Scholar]
- 29.Wagner S, Tagaya M, Koziol JA, Quaranta V, del Zoppo GJ. Rapid disruption of an astrocyte interaction with the extracellular matrix mediated by integrin alpha 6 beta 4 during focal cerebral ischemia/reperfusion. Stroke. 1997;28:858–65. doi: 10.1161/01.str.28.4.858. [DOI] [PubMed] [Google Scholar]
- 30.Milner R, Hung S, Wang X, Berg GI, Spatz M, del Zoppo GJ. Responses of endothelial cell and astrocyte matrix-integrin receptors to ischemia mimic those observed in the neurovascular unit. Stroke. 2008;39:191–7. doi: 10.1161/STROKEAHA.107.486134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–33. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Chang DI, Hosomi N, Lucero J, et al. Activation systems for matrix metalloproteinase-2 are upregulated immediately following experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:1408–19. doi: 10.1097/01.WCB.0000091765.61714.30. [DOI] [PubMed] [Google Scholar]
- 33.Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL, del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke. 2004;35:998–1004. doi: 10.1161/01.STR.0000119383.76447.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takano T, Han X, Deane R, Zlokovic B, Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer's disease. Ann N Y Acad Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concommitant cortical expression of TNF-a and IL-1b mRNA following transient focal ischemia. Mol Chem Neuropathol. 1994;23:103–14. doi: 10.1007/BF02815404. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Barone FC, Aiyar NV, Feuerstein GZ. Increased interleukin-1 receptor and receptor antagonist gene expression after focal stroke. Stroke. 1997;28:155–61. doi: 10.1161/01.str.28.1.155. [DOI] [PubMed] [Google Scholar]
- 37.Wang X, Yue TL, Barone FC, Feuerstein GZ. Monocyte chemoattractant protein-1 messenger RNA expression in rat ischemic cortex. Stroke. 1995;26:661–5. doi: 10.1161/01.str.26.4.661. [DOI] [PubMed] [Google Scholar]
- 38.Lees KR, Barer D, Ford GA, et al. Tolerability of NXY-059 at higher target concentrations in patients with acute stroke. Stroke. 2003;34:482–7. doi: 10.1161/01.str.0000053032.14223.81. [DOI] [PubMed] [Google Scholar]
- 39.Lees KR, Zivin JA, Ashwood T, et al. NXY-059 for acute ischemic stroke. N Engl J Med. 2006;354:588–600. doi: 10.1056/NEJMoa052980. [DOI] [PubMed] [Google Scholar]
- 40.AstraZeneca On-line Press Release. AstraZeneca announces SAINT II Trial results showed no efficacy in acute ischaemic stroke. [26 October 2006]; Available at: http://www.astrazeneca.com/media/latest-press-releases/2006/5279?itemId=3891714.
- 41.Shuaib A, Lees KR, Lyden P, et al. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357:562–71. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- 42.Koziol JA, Feng AC. On the analysis and interpretation of outcome measures in stroke clinical trials: lessons from the SAINT I study of NXY-059 for acute ischemic stroke. Stroke. 2006;37:2644–7. doi: 10.1161/01.STR.0000241106.81293.2b. [DOI] [PubMed] [Google Scholar]
- 43.Green AR, Ashwood T. Free radical trapping as a therapeutic approach to neuroprotection in stroke: experimental and clinical studies with NXY-059 and free radical scavengers. Curr Drug Targets CNS Neurol Disord. 2005;4:109–18. doi: 10.2174/1568007053544156. [DOI] [PubMed] [Google Scholar]
- 44.Lapchak PA, Araujo DM, Song D, Wei J, Purdy R, Zivin JA. Effects of the spin trap agent disodium-[tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (generic NXY-059) on intracerebral hemorrhage in a rabbit Large clot embolic stroke model: combination studies with tissue plasminogen activator. Stroke. 2002;33:1665–70. doi: 10.1161/01.str.0000017145.22806.aa. [DOI] [PubMed] [Google Scholar]
- 45.Kontos CD, Wei EP, Williams JI, Kontos HA, Povlishock JT. Cytochemical detection of superoxide in cerebral inflammation and ischemia in vivo. Am J Physiol. 1992;263:H1234–42. doi: 10.1152/ajpheart.1992.263.4.H1234. [DOI] [PubMed] [Google Scholar]
- 46.Heo JH, Han SW, Lee SK. Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med. 2005;39:51–70. doi: 10.1016/j.freeradbiomed.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 47.del Zoppo GJ. Why do all drugs work in animals but none in stroke patients? A Drugs promoting cerebral blood flow. J Intern Med. 1995;237:79–88. doi: 10.1111/j.1365-2796.1995.tb01144.x. [DOI] [PubMed] [Google Scholar]
- 48.Mohr JP, Thompson JLP, Lazar RM, et al. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke Warfarin-Aspirin Recurrent Stroke Study Group. N Engl J Med. 2001;345:1444–51. doi: 10.1056/NEJMoa011258. [DOI] [PubMed] [Google Scholar]
- 49.The NINDS and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–7. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 50.Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359:1317–29. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 51.del Zoppo GJ, Poeck K, Pessin MS, et al. Recombinant tissue plasminogen activator in acute thrombotic and embolic stroke. Ann Neurol. 1992;32:78–86. doi: 10.1002/ana.410320113. [DOI] [PubMed] [Google Scholar]
- 52.Sato PH, Hall ED. Tirilazad mesylate protects vitamins C and E in brain ischemia-reperfusion injury. J Neurochem. 1992;58:2263–8. doi: 10.1111/j.1471-4159.1992.tb10972.x. [DOI] [PubMed] [Google Scholar]
- 53.Yamaguchi T. Intravenous rt-PA in acute embolic stroke. In: Hacke W, del Zoppo GJ, Hirschberg M, editors. Thrombolytic Therapy in Acute Ischemic Stroke. Heidelberg: Springer-Verlag; 1991. pp. 168–74. [Google Scholar]
- 54.Mori E, Yoneda Y, Ohkawa S, et al. Double-blind placebo-controlled trial of intravenous recombinant tissue plasminogen activator (rt-PA) in acute carotid stroke. Neurology. 1991;41:347. doi: 10.1212/wnl.42.5.976. [DOI] [PubMed] [Google Scholar]
- 55.del Zoppo GJ, Ferbert A, Otis S, et al. Local intra-arterial fibrinolytic therapy in acute carotid territory stroke: a pilot study. Stroke. 1988;19:307–13. doi: 10.1161/01.str.19.3.307. [DOI] [PubMed] [Google Scholar]
- 56.Mori E, Tabuchi M, Yoshida T, Yamadori A. Intracarotid urokinase with thromboembolic occlusion of the middle cerebral artery. Stroke. 1988;19:802–12. doi: 10.1161/01.str.19.7.802. [DOI] [PubMed] [Google Scholar]
- 57.Hacke W, Kaste M, Fieschi C, et al. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke The European Cooperative Acute Stroke Study (ECASS) JAMA. 1995;274:1017–25. [PubMed] [Google Scholar]
- 58.Hacke W, Kaste M, Fieschi C, et al. Randomised double-blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II) lancet. 1998;352:1245–51. doi: 10.1016/s0140-6736(98)08020-9. [DOI] [PubMed] [Google Scholar]
- 59.del Zoppo GJ, Copeland BR, Harker LA, et al. Experimental acute thrombotic stroke in baboons. Stroke. 1986;17:1254–65. doi: 10.1161/01.str.17.6.1254. [DOI] [PubMed] [Google Scholar]
- 60.Young AR, Touzani O, Derlon JM, Sette G, MacKenzie ET, Baron JC. Early reperfusion in the anesthetized baboon reduces brain damage following middle cerebral artery occlusion: a quantitative analysis of infarction volume. Stroke. 1997;28:632–7. doi: 10.1161/01.str.28.3.632. [DOI] [PubMed] [Google Scholar]
- 61.del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–84. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- 62.Mori E, del Zoppo GJ, Chambers JD, Copeland BR, Arfors KE. Inhibition of polymorphonuclear leukocyte adherence suppresses no-reflow after focal cerebral ischemia in baboons. Stroke. 1992;23:712–8. doi: 10.1161/01.str.23.5.712. [DOI] [PubMed] [Google Scholar]
- 63.Okada Y, Copeland BR, Fitridge R, Koziol JA, del Zoppo GJ. Fibrin contributes to microvascular obstructions and parenchymal changes during early focal cerebral ischemia and reperfusion. Stroke. 1994;25:1847–53. doi: 10.1161/01.str.25.9.1847. [DOI] [PubMed] [Google Scholar]
- 64.Abumiya T, Fitridge R, Mazur C, et al. Integrin alpha(IIb)-beta(3) inhibitor preserves microvascular patency in experimental acute focal cerebral ischemia. Stroke. 2000;31:1402–10. doi: 10.1161/01.str.31.6.1402. [DOI] [PubMed] [Google Scholar]
- 65.del Zoppo GJ, Higashida RT, Furlan AJ, et al. PROACT: a phase II randomized trial of recombinant pro-urokinase by direct arterial delivery in acute middle cerebral artery stroke. Stroke. 1998;29:4–11. doi: 10.1161/01.str.29.1.4. [DOI] [PubMed] [Google Scholar]
- 66.Furlan AJ, Higashida R, Wechsler L, Schulz G, PROACT II Investigators PROACT II: recombinant prourokinase (r-ProUK) in acute cerebral thromboembolism: initial Trial Results. Stroke. 1999;30:202. [Google Scholar]
- 67.Tagaya M, Haring HP, Stuiver I, et al. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. 2001;21:835–46. doi: 10.1097/00004647-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 68.Fisher CM, Adams RD. Observations on brain embolism with special reference to the mechanism of hemorrhagic infarction. J Neuropathol Exp Neurol. 1951;10:92–4. [PubMed] [Google Scholar]
- 69.Fisher CM, Adams RD. Observations on brain embolism with special reference to hemorrhage infarction. In: Furlan AJ, editor. The Heart and Stroke Exploring Mutual Cerebrovascular and Cardiovascular Issues. New York: Springer-Verlag; 1987. pp. 17–36. [Google Scholar]
- 70.Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995;26:2120–6. doi: 10.1161/01.str.26.11.2120. [DOI] [PubMed] [Google Scholar]
- 71.Ueda T, Hatakeyama T, Kumon Y, Sakaki S, Uraoka T. Evaluation of risk of hemorrhagic transformation in local intra-arterial thrombolysis in acute ischemic stroke by initial SPECT. Stroke. 1994;25:298–303. doi: 10.1161/01.str.25.2.298. [DOI] [PubMed] [Google Scholar]
- 72.Larrue V, von Kummer R, del Zoppo GJ, Bluhmki E. Hemorrhagic transformation in acute ischemic stroke: potential contributing factors in the European Cooperative Acute Stroke Study. Stroke. 1997;28:957–60. doi: 10.1161/01.str.28.5.957. [DOI] [PubMed] [Google Scholar]
- 73.Larrue V, von Kummer RR, Muller A, Bluhmki E. Risk factors for severe hemorrhagic transformation in ischemic stroke patients treated with recombinant tissue plasminogen activator: a secondary analysis of the European-Australasian Acute Stroke Study (ECASS II) Stroke. 2001;32:438–41. doi: 10.1161/01.str.32.2.438. [DOI] [PubMed] [Google Scholar]
- 74.Abilleira S, Montaner J, Molina CA, Monasterio J, Castillo J, Alvarez-Sabin J. Matrix metalloproteinase-9 concentration after spontaneous intracerebral hemorrhage. J Neurosurg. 2003;99:65–70. doi: 10.3171/jns.2003.99.1.0065. [DOI] [PubMed] [Google Scholar]
- 75.Montaner J, Alvarez-Sabin J, Molina CA, et al. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke. 2001;32:2762–667. doi: 10.1161/hs1201.99512. [DOI] [PubMed] [Google Scholar]
- 76.Montaner J, Molina CA, Monasterio J, et al. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- 77.Rosell A, Ortega-Aznar A, Alvarez-Sabin J, et al. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- 78.Tejima E, Zhao BQ, Tsuji K, et al. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab. 2007;27:460–8. doi: 10.1038/sj.jcbfm.9600354. [DOI] [PubMed] [Google Scholar]
- 79.Tsuji K, Aoki T, Tejima E, et al. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36:1954–9. doi: 10.1161/01.STR.0000177517.01203.eb. [DOI] [PubMed] [Google Scholar]
- 80.Tagaya M, Liu KF, Copeland B, et al. DNA scission after focal brain ischemia Temporal differences in two species. Stroke. 1997;28:1245–54. doi: 10.1161/01.str.28.6.1245. [DOI] [PubMed] [Google Scholar]
- 81.Abumiya T, Lucero J, Heo JH, et al. Activated microvessels express vascular endothelial growth factor and integrin alpha(v)-beta3 during focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:1038–50. doi: 10.1097/00004647-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 82.Streuli CH, Bissell MJ. Expression of extracellular matrix components is regulated by substratum. J Cell Biol. 1990;110:1405–15. doi: 10.1083/jcb.110.4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Milner R, Campbell IL. The extracellular matrix and cytokines regulate microglial integrin expression and activation. J Immunol. 2003;170:3850–8. doi: 10.4049/jimmunol.170.7.3850. [DOI] [PubMed] [Google Scholar]
- 84.Choudhri TF, Hoh BL, Zerwes HG, et al. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor-mediated platelet aggregation. J Clin Invest. 1998;102:1301–10. doi: 10.1172/JCI3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stutzmann JM, Mary V, Wahl F, Grosjean-Plot O, Uzan A, Pratt J. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev. 2002;8:1–30. doi: 10.1111/j.1527-3458.2002.tb00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]