

Abstract
Cardiovascular disease, such as atherosclerosis, has been associated with reduced bone mineral density and fracture risk. A major etiologic factor in atherogenesis is believed to be oxidized phospholipids. We previously found that these phospholipids inhibit spontaneous osteogenic differentiation of marrow stromal cells, suggesting that they may account for the clinical link between atherosclerosis and osteoporosis. Currently, anabolic agents that promote bone formation are increasingly used as a new treatment for osteoporosis. It is not known, however, whether atherogenic phospholipids alter the effects of bone anabolic agents, such as bone morphogenetic protein (BMP)-2 and parathyroid hormone (PTH). Therefore we investigated the effects of oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (ox-PAPC) on osteogenic signaling induced by BMP-2 and PTH in MC3T3-E1 cells. Results showed that ox-PAPC attenuated BMP-2 induction of osteogenic markers alkaline phosphatase and osteocalcin. Ox-PAPC also inhibited both spontaneous and BMP-induced expression of PTH receptor. Consistently, pretreatment of cells with ox-PAPC inhibited PTH-induced cAMP production and expression of immediate early genes Nurr1 and IL-6. Results from immunofluorescence and Western blot analyses showed that inhibitory effects of ox-PAPC on BMP-2 signaling were associated with inhibition of SMAD 1/5/8 but not p38-MAPK activation. These effects appear to be due to ox-PAPC activation of the ERK pathway, as the ERK inhibitor PD98059 reversed ox-PAPC inhibitory effects on BMP-2-induced alkaline phosphatase activity, osteocalcin expression, and SMAD activation. These results suggest that atherogenic lipids inhibit osteogenic signaling induced by BMP-2 and PTH, raising the possibility that hyperlipidemia and atherogenic phospholipids may interfere with anabolic therapy.
Cardiovascular disease has been associated with lower bone mineral density, especially in the presence of atherosclerosis. Aortic atherosclerosis has been found to predict, age independently, bone loss and fracture risk (1-3). Plasma low density lipoprotein (LDL)2 cholesterol levels have been inversely correlated with bone mineral density (4). Recent evidence suggests that postmenopausal women with atherogenic lipid profiles have a greater risk of osteopenia than those with normal lipid profiles, paralleling the risk of cardiovascular disease (5). In animal models, atherosclerosis-susceptible mice fed a high fat diet have reduced bone density and bone marrow osteocalcin expression compared with those on a chow diet (6).
A major etiologic factor in atherogenesis is believed to be oxidatively modified lipoproteins and phospholipids. A mildly oxidized form of low density lipoprotein (MM-LDL) and one of its biologically active components, 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (ox-PAPC), have been shown to trigger atherogenic responses in endothelial cells (7, 8). In vitro, these atherogenic lipids inhibit spontaneous osteoblastic differentiation and mineralization of calvarial preosteoblasts and bone marrow stromal cells (9, 10).
Atherogenic phospholipids and lipoproteins have been shown to activate p42/44 mitogen-activated MAPK, i.e. the ERK pathway in endothelial cells and vascular smooth muscle cells (11, 12). These lipids also activate the ERK pathway in bone marrow stromal cells. Activation of the ERK pathway has been shown to negatively regulate osteoblastic differentiation (9). Suppression of ERK activation by dominant negative Ras expression in the calvaria of 1-day-old mice results in increased mineralization in the calvaria (13). In addition, bone morphogenetic protein (BMP)-induced osteogenesis requires down-regulation of the ERK pathway in bone marrow stromal cells (14).
Currently, anabolic agents that promote osteoblastic differentiation leading to increased bone formation have opened a new therapeutic approach for treating osteoporosis. BMPs are potent anabolic agents and play a role in postnatal bone formation. The osteogenic potential of BMP-2 has been shown in both animal models and in clinical studies. It has been used in clinical treatment of non-union fractures as well as osteoporosis (15-17). Upon ligand binding, BMP-2 signals are mediated by activation of SMAD 1, 5, and 8. In addition to the SMAD proteins, activation of the p38-MAPK pathway is important for BMP-2-induced signaling, and it influences osteoblastic differentiation (18). Another important anabolic factor is parathyroid hormone (PTH). When given intermittently, PTH has been shown to increase bone mineral density (19-21). In contrast, continuous administration of PTH has the opposite effect (22). Thus, the anabolic response of PTH has been difficult to study in vitro. It has been shown that anabolic effects of PTH in vivo are mediated by the cAMP-dependent protein kinase A pathway (23).
In this report, we extend our previous findings to address the effects of oxidized phospholipids on osteogenic activity of BMP-2. Results showed that ox-PAPC attenuates BMP-induced osteoblastic differentiation and PTH receptor expression in calvarial preosteoblasts, MC3T3-E1 cells. In addition, results showed that ox-PAPC inhibits PTH signaling including cAMP production and expression of immediate early genes, suggesting that atherogenic lipids interfere with effects of anabolic agents. These findings raise the possibility that hyperlipidemia may adversely affect anabolic therapies for osteoporosis.
EXPERIMENTAL PROCEDURES
Materials
MC3T3-E1, a murine preosteoblastic calvarial cell line, was obtained from Riken Cell Bank (Japan). BMP-2 was obtained from R&D Systems and Peprotech. PAPC was from Avanti-Polar Lipids. PAPC was oxidized and its oxidation state verified as described previously (24). Purified human high density lipoproteins (HDL) SB203580 and PD98059 were from Calbiochem. Antibodies to phosphorylated ERK, p38-MAPK, and total p38-MAPK and ERK were from Cell Signaling. Antibodies for total SMAD 1/5/8 were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture
MC3T3-E1 cells were plated at 40,000/cm2. β-glycerophosphate (5 mm) and ascorbic acid (50 μg/ml) were added as part of the BMP-2 treatment. These osteogenic supplements were not used in experiments without BMP-2. In the experiments using ox-PAPC pretreatment, cells were incubated with ox-PAPC for a total of 6 – 8 days unless otherwise indicated. Assays were performed at different time points as appropriate for the earliest expected appearance of each osteoblastic differentiation marker (25, 26). Alkaline phosphatase activity was assayed at 2 days and osteocalcin expression at 3–4 days after BMP-2 treatment.
Alkaline Phosphatase Activity
Cells were plated at 40,000/cm2. One day after plating, cells were treated with the indicated reagents in α-minimal essential medium (Mediatech) supplemented with 10% fetal bovine serum, 5 mm β-glycerophosphate (Sigma), and 50 μg/ml ascorbic acid (Sigma). Alkaline phosphatase activity in a whole cell lysate was assayed in quadruplicate and normalized to total protein, determined by the Bradford method (27).
Real-time RT-PCR
Total RNA was isolated using TRIzol reagent (Invitrogen), DNase-treated RNA was reverse transcribed, and real-time PCR was performed using the Mx3005P (Stratagene). Sequences for the primers are as follows: Nurr-1 (sense) 5′-ACTCCAATAACTCTGCTGAAG-3′, (antisense) 5′-TGCTAAACTTGACAAACTCTG-3′; β-actin (sense) 5′-AGAGGGAAATCGT-GCGTGAC-3′, (antisense) 5′-CAATAGTGAT-GACCTGGCCGT-3′; osteocalcin (sense) 5′-CCGGGAGCAGTGTGAGCTTA-3′, (antisense) 5′-TAGATGCGTTTGTAGGCGGTC-3′; PTH receptor (sense) 5′-TCAGGGACACTGTGGCAGATC-3′, (antisense) 5′-GCACCTCACCATTGCAGAAAC-3′; IL-6 (sense) 5′-TGTATGAAC-AACGATGATGCACTT-3′, (antisense) 5′-GGTACTCCAGAAGACCAGAGGAAAT-3′.
cAMP Assay
Cells were pretreated with ox-PAPC 1 day after seeding. After 6–7 days of treatment, cells were treated with 3-isobutyl-1-methylxanthine (0.5 mm) for 15 min, followed by treatment with PTH (10−8 m) for 30 min. Cyclic AMP accumulation in the whole cell extracts was measured by a cAMP EIA kit (Cayman Chemical) according to the manufacturer's protocol. The data are represented as an average of 3 wells ± S.D.
Western Blot Analysis
Whole cell lysates were prepared using lysis buffer supplemented with phosphatase and protease inhibitors, and Western analysis was performed using standard protocols.
Immunocytochemistry
Cells were washed, fixed in 10% formalin, and blocked in 5% normal serum. Cells were probed with primary antibody pSMAD1/5/8 (1:100; Cell Signaling) overnight at 4 °C and visualized by the secondary antibody conjugated to Alexa Fluor 488 (1:200; Invitrogen). ProLong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (Invitrogen P36935) was used for mounting.
Data Analysis
Each experiment was performed in at least triplicate wells for each condition, and each experiment was repeated at least three times. Data are expressed as mean ± S.E. from all experiments where noted, otherwise as mean ± S.D. from a representative experiment. Means were compared using one-way analysis of variance, with comparison of different groups by Fisher's protected least significant difference test. A value of p < 0.05 was considered significant.
RESULTS
Effect of Ox-PAPC on BMP-induced Osteoblastic Differentiation
To determine the effects of ox-PAPC on BMP-induced osteoblastic differentiation, MC3T3-E1 cells were cotreated with vehicle, ox-PAPC, and/or BMP-2 for 2 days, after which alkaline phosphatase activity was assessed. Results showed that ox-PAPC decreased the alkaline phosphatase activity induced by BMP-2 (Fig. 1A). Quantitative real-time RT-PCR analysis showed that pretreatment of cells with ox-PAPC for 4 days, followed by cotreatment with BMP-2 for 4 additional days, also inhibited BMP-2 induction of osteocalcin (Fig. 1A). In additional studies, we found that cotreatment (without ox-PAPC pretreatment) was sufficient for the inhibitory effects (Fig. 1B). Treatment of cells with native PAPC (negative control) did not (Fig. 1B), suggesting that oxidative modification is required for the inhibitory effects.
FIGURE 1. Effect of ox-PAPC on BMP-induced osteoblastic differentiation.

A, alkaline phosphatase activity and osteocalcin expression in response to BMP-2 and ox-PAPC. For the alkaline phosphatase activity assay, cells were cotreated for 2 days with vehicle control, BMP-2 (50 ng/ml), and/or ox-PAPC (10 μg/ml). For osteocalcin expression, RNA was isolated from cells pretreated with ox-PAPC (10 μg/ml) for 4 days, followed by treatment with ox-PAPC and/or BMP-2 (50 ng/ml) for an additional 4 days. Expression was assessed by real-time RT-PCR and normalized to β-actin. B, osteocalcin expression in response to BMP-2, ox-PAPC, and native PAPC. RNA was isolated from cells treated for 3 days with BMP-2 (50 ng/ml) along with either ox-PAPC (10 μg/ml) or PAPC (10 μg/ml). Expression was assessed by real-time RT-PCR and normalized to β-actin. C, PTH receptor (PTHR) expression in response to BMP-2 and ox-PAPC. RNA was isolated from cells as in panel A. Expression was assessed by real-time RT-PCR and normalized to β-actin. Results are shown as mean ± S.E. from all experiments. *, p < 0.0001; #, p < 0.005.
Because BMP-2 induces PTH receptor expression (28), we next assessed the ox-PAPC effect on PTH receptor expression. Results showed that PTHR expression induced by BMP-2 was also attenuated by ox-PAPC (Fig. 1C).
Effect of Ox-PAPC on PTH Signaling
PTH and its related peptides induce anabolic bone formation in vivo when administered intermittently (19, 20). Because our results showed that ox-PAPC attenuated BMP-2-induced PTHR expression, we further assessed the effect of ox-PAPC on PTH signaling. Treatment of MC3T3-E1 cells with ox-PAPC alone for 6 days inhibited PTHR expression (Fig. 2A). In addition, pretreatment of cells with ox-PAPC for 7 days inhibited PTH induction of cAMP (Fig. 2B). We next assessed the effects of ox-PAPC on PTH induction of immediate early genes mediated both in vitro and in vivo via the cAMP-dependent protein kinase A pathway (29, 30). Results showed that pretreatment of cells with ox-PAPC inhibited expression of immediate early genes Nurr1 and IL-6 induced by PTH at 2 h (Fig. 2C). Ox-PAPC treatment alone showed some induction of IL-6 expression (Fig. 2C).
FIGURE 2. Effect of ox-PAPC on PTH signaling.

A, PTH receptor expression in response to ox-PAPC. RNA was isolated from cells treated for 6 days with ox-PAPC (10 μg/ml). Expression was assessed by real-time RT-PCR and normalized to β-actin. B, cAMP production in response to PTH and ox-PAPC. cAMP accumulation in the medium of cells pretreated for 7 days with ox-PAPC, followed by treatment for an additional 30 min with vehicle, PTH (10−8 m), and/or ox-PAPC (10 μg/ml). C, Nurr-1 and IL-6 expression in response to PTH and ox-PAPC. RNA isolated from cells pretreated for 6 days with ox-PAPC, followed by treatment for an additional 2 h with vehicle, PTH (10−8 m), and/or ox-PAPC (10 μg/ml). Expression was assessed by real-time RT-PCR and normalized to β-actin. Results are shown as mean ± S.E. from all experiments. *, p < 0.0001; #, p < 0.005; **, p < 0.05.
Effect of Ox-PAPC on BMP-2 Signaling Pathway
Osteogenic effects of BMP-2 have been shown to be mediated by the SMAD and p38-MAPK pathways (18). Consistently, we found that treatment of MC3T3-E1 cells with SB203580, an inhibitor of p38-MAPK, attenuated BMP-induced alkaline phosphatase activity (data not shown). To assess whether the inhibitory effect of ox-PAPC is due to inhibition of BMP-2-induced p38-MAPK, cells were treated with BMP-2 and/or ox-PAPC. Results showed that BMP-2 treatment alone induced p38-MAPK phosphorylation, which was not attenuated by ox-PAPC (Fig. 3A). Cotreatment with ox-PAPC and SB203580, a specific p38-MAPK inhibitor, caused further inhibition of alkaline phosphatase activity (Fig. 3B, compare bars 2, 3, and 4). Altogether, these results suggest that the inhibitory effect of ox-PAPC on BMP-2 signaling is either downstream of p38-MAPK or not via inhibition of the p38-MAPK pathway. We next assessed the effect of ox-PAPC on BMP-induced SMAD activation by Western analysis and immunofluorescence. Cells were treated with BMP-2 and ox-PAPC for 1.5 h, the time point previously shown for SMAD activation (14). Results showed that ox-PAPC attenuated BMP-2 activation of SMAD 1/5/8 phosphorylation (Fig. 3, C and D).
FIGURE 3. Effect of ox-PAPC on p38-MAPK and ERK.

A, activation of p38-MAPK in response to BMP-2 and ox-PAPC. Western analysis of phosphorylated p38-MAPK from cultures treated with vehicle, BMP-2 (100 ng/ml), and/or ox-PAPC (10μg/ml) for 30 min (left panel)or3 h(right panel). Total p38-MAPK was used as a loading control. B, alkaline phosphatase activity in response to BMP-2, ox-PAPC, and SB203580. Cells were pretreated for 30 min with SB203580 (10 μm), followed by treatment for 2 days with vehicle, BMP-2 (100 ng/ml), ox-PAPC (10 μg/ml), and/or SB203580 (10 μm) as indicated. *, p < 0.0001. C, activation of SMADs in response to BMP-2 and ox-PAPC. Western analysis of phosphorylated SMAD 1/5/8 in cells treated for 1.5 h with vehicle, BMP-2 (100 ng/ml), ox-PAPC (30 μg/ml), or BMP-2/ox-PAPC. Total SMAD 1/5/8 was used as a loading control. D, immunofluorescent (fluorescein isothiocyanate) staining of phosphorylated SMAD 1/5/8 (green; left panels) in cells treated for 1.5 h with vehicle (a), ox-PAPC (10 μg/ml) (b), BMP-2 (50 ng/ml) (c), or BMP-2 and ox-PAPC (d). 4′,6-Diamidino-2-phenylindole staining (blue; right panels) was used to visualize cell nuclei (magnification ×400).
In marrow stromal cells, MM-LDL activated the ERK pathway (9), which negatively regulates matrix mineralization both in vitro and in vivo (13). Therefore, we assessed the effect of ox-PAPC on ERK activation. Results showed that ox-PAPC induced ERK phosphorylation, slightly at 30 min and more significantly at 3 h, but BMP-2 did not (Fig. 4A). Consistently, both Western analysis and immunofluorescence showed that treatment of cells with an ERK inhibitor, PD98059, partially reversed the inhibitory effects of ox-PAPC on BMP-2 activation of SMAD 1/5/8 (Fig. 4, B and C). In addition, treatment of cells with PD98059 reversed the BMP-induced alkaline phosphatase activity and osteocalcin expression (Fig. 4, D and E).
FIGURE 4. Effect of ERK inhibition on ox-PAPC inhibitory effects.
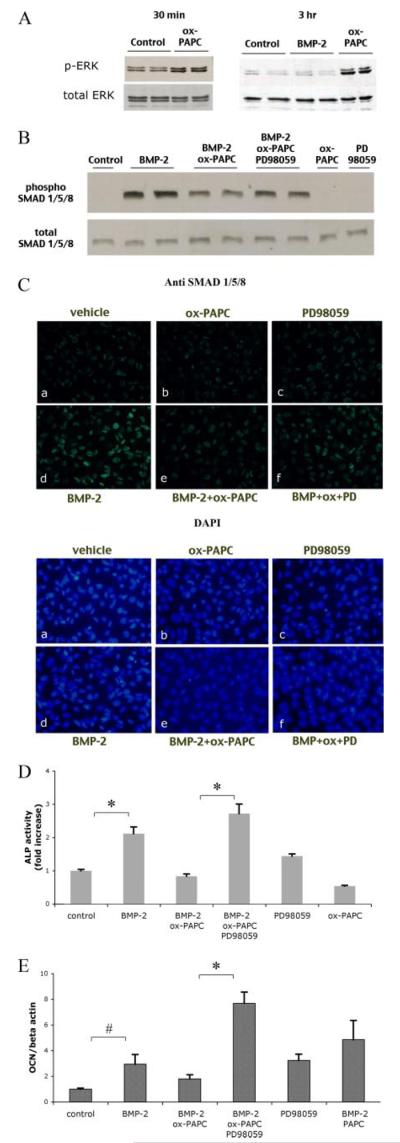
A, activation of ERK in response to BMP-2 and ox-PAPC. Western analysis of phosphorylated ERK (p-ERK) from cell cultures treated for 30 min (left panel) or 3 h(right panel) with BMP-2 (50 ng/ml) or ox-PAPC (10 μg/ml). Total ERK was used as a loading control. B, activation of SMADs in response to BMP-2, ox-PAPC, and PD98059. Western analysis of phosphorylated SMAD 1/5/8 in cells treated for 1.5 h with vehicle, BMP-2 (100 ng/ml), ox-PAPC (30 μg/ml), or PD98059 (5 μm), BMP-2/ox-PAPC, or BMP-2/ox-PAPC/PD98059. Total SMAD 1/5/8 was used as a loading control. C, immunofluorescent (fluorescein isothiocyanate) staining of phosphorylated SMAD 1/5/8 (green; upper panels) in cells pretreated for 30 min with PD98059, followed by treatment for an additional 1.5 h with vehicle (a), ox-PAPC (b), PD98059 (c), BMP-2 (d), BMP-2 and ox-PAPC (e), BMP-2, ox-PAPC, and PD98059 (f) in medium supplemented with 0.5% fetal bovine serum. 4 ,6-Diamidino-2-phenylindole staining (blue; lower panels) was used to visualize cell nuclei (magnification ×400). D, alkaline phosphatase activity in response to BMP-2, ox-PAPC, and PD98059. Cells were treated for 2 days with BMP-2 (50 ng/ml), ox-PAPC (10 μg/ml), and/or PD98059 (5 μm) as indicated. E, osteocalcin expression in response to BMP-2, ox-PAPC, and PD98059. Cells were cotreated for 3 days with vehicle, BMP-2 (50 ng/ml), ox-PAPC (10 μg/ml), PAPC (10 μg/ml), or PD98059 (5 μm) as indicated. *, p < 0.0001; #, p < 0.05.
Oxidant Stress Does Not Mediate Ox-PAPC Inhibitory Effects
Because oxidant stress has been shown to inhibit osteoblastic differentiation via the ERK pathway (31), and oxidant stress mediates MM-LDL inhibitory effects on differentiation of osteoblasts (32), we next examined whether antioxidants were able to reverse ox-PAPC inhibitory effects. Cells were pretreated with the antioxidant particle HDL (100 μg/ml) for 24 h prior to cotreatment with BMP-2 and ox-PAPC. Results showed that HDL treatment did not reverse the inhibitory effect of ox-PAPC on BMP-2-induced alkaline phosphatase activity (Fig. 5). Treatment with other antioxidants, Trolox (100 μm) or pyrrolidinedithiocarbamate (10 μm), also had no significant effect on ox-PAPC inhibitory effects (data not shown).
FIGURE 5. Effect of antioxidant particle HDL on ox-PAPC inhibitory effects.

Alkaline phosphatase activity in response to BMP-2, ox-PAPC, and HDL. Cells were pretreated for 24 h with HDL (100 μg/ml), followed by treatment for an additional 2 days with vehicle, BMP-2 (50 ng/ml), ox-PAPC (10 μg/ml), and/or HDL as indicated. *, p < 0.0001.
DISCUSSION
The present study extends our previous findings concerning spontaneous osteogenesis to the clinically important area of anabolic stimulation. In previous studies, we found that atherogenic lipoproteins and phospholipids, including mildly oxidized LDL and ox-PAPC, inhibit spontaneous osteogenesis in marrow stromal cells via the ERK pathway (9), thus providing a possible link for the epidemiological association of osteoporosis with atherosclerosis. The present results indicate that ox-PAPC also inhibits osteoblastic differentiation induced by BMP-2 in mouse calvarial preosteoblasts. The inhibitory effects of ox-PAPC on BMP-2 signaling appear to be through inhibition of SMADs via ERK, and not p38-MAPK (Fig. 6).
FIGURE 6. Proposed inhibitory mechanisms of ox-PAPC on BMP-2 and PTH signaling pathways.

Ox-PAPC appears to inhibit BMP-2 signaling by attenuating SMAD 1/5/8 activation via ERK and inhibits PTH signaling by attenuating induction of cAMP.
The ERK signaling pathway has been shown to play a role in osteoblastic differentiation. Some have reported that ERK activation is required for induction of alkaline phosphatase activity (33-36), whereas others have shown that ERK activation has inhibitory effects on osteoblastic differentiation (9, 13, 37). Furthermore, the ERK pathway has been shown to mediate the inhibitory effects of oxidative stress (31). The inhibitory mechanism of ERK has been shown to involve attenuation of BMP-induced nuclear translocation of SMAD 1, 5, and 8 (14). In the present study, we also found that ox-PAPC reduces BMP-2 activation of SMADs (Fig. 6). Consistent with this, inhibitors of ERK reverse the inhibitory effects of ox-PAPC on BMP-2 effects, including activation of SMAD 1, 5, and 8, thus supporting the inhibitory role of ERK in osteoblastic differentiation.
We have previously found that MM-LDL induces reactive oxygen species, which inhibit osteoblastic differentiation of MC3T3-E1 cells (32). In addition, oxidant stress has been shown to inhibit osteoblastic differentiation via the ERK pathway (31). Therefore, we tested whether antioxidants reverse the inhibitory effects of ox-PAPC. Although ox-PAPC is an active component of MM-LDL, our results show that inhibitors of oxidant stress did not attenuate ox-PAPC effects on BMP-2 induction of alkaline phosphatase activity, suggesting that components other than ox-PAPC in MM-LDL mediate oxidant stress or, alternatively, that BMP-2-induced osteoblastic differentiation is independent of oxidant stress.
Inhibitory effects of ox-PAPC on BMP-2 may stem from inhibition of PTH receptor expression or vice versa since studies from other investigators demonstrate an interaction between BMP-2 and PTH receptor signaling. For example, BMP-2 induces PTH receptor expression (28), whereas PTH-related protein enhances BMP-2 induction of osteogenesis (38). Our results indicate that ox-PAPC also inhibits PTH receptor induction by BMP-2 and consequently that it inhibits PTH-induced cAMP production and expression of immediate early genes Nurr1 and IL-6 (Fig. 6). Interestingly, the data also suggest that preosteoblasts may be more sensitive to the inhibitory effects of ox-PAPC, since ox-PAPC inhibited PTH receptor expression to a greater extent in cells that did not receive the osteogenic supplements (Fig. 2A versus Fig. 1C).
Prolonged expression of inflammatory cytokines enhances bone resorption and thus contributes to osteoporosis (39). Previously, ox-PAPC has been shown to induce inflammatory genes, including IL-6, in endothelial cells and monocytes both in vitro and in vivo (8, 40). Our results show that chronic ox-PAPC treatment increased IL-6 expression in preosteoblastic calvarial cells. These findings suggest the potential role of atherogenic lipids in osteoclastic resorption by induction of inflammatory cytokines by osteoblasts. This is in agreement with our previous findings where oxidized lipids enhance osteoclastic differentiation of marrow preosteoclasts both directly and in coculture with stromal/osteoblasts (41, 42). In summary, these findings raise the possibility that hyperlipidemia may interfere with the therapeutic efficacy of anabolic agents.
Acknowledgments
We thank Dr. A. Watson and E. Robison and A. Wagner for expert assistance with liquid chromatography/mass spectrometry, Dr. F. Parhami for suggestions, and Dr. C. Magyar for expert assistance with fluorescence microscopy.
Footnotes
This work was supported by National Institutes of Health Grants DK076009-01 (to Y. T.) and HL081202 (to L. L. D.) and by Molecular Cellular and Integrative Physiology Training Grant NIGMS T32 GM065823 (to S. M.).
The abbreviations used are: LDL, low density lipoprotein; ox-PAPC, oxidized-1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; BMP, bone morphogenetic protein; PTH, parathyroid hormone; HDL, high density lipoprotein; RT-PCR, reverse transcription PCR.
REFERENCES
- 1.Bagger YZ, Tanko LB, Alexandersen P, Qin G, Christiansen C. J. Intern. Med. 2006;259:598–605. doi: 10.1111/j.1365-2796.2006.01640.x. [DOI] [PubMed] [Google Scholar]
- 2.Jie KG, Bots ML, Vermeer C, Witteman JC, Grobbee DE. Calcif. Tissue Int. 1996;59:352–356. doi: 10.1007/s002239900139. [DOI] [PubMed] [Google Scholar]
- 3.Barengolts EI, Berman M, Kukreja SC, Kouznetsova T, Lin C, Chomka EV. Calcif. Tissue Int. 1998;62:209–213. doi: 10.1007/s002239900419. [DOI] [PubMed] [Google Scholar]
- 4.Yamaguchi T, Sugimoto T, Yano S, Yamauchi M, Sowa H, Chen Q, Chihara K. Endocr. J. 2002;49:211–217. doi: 10.1507/endocrj.49.211. [DOI] [PubMed] [Google Scholar]
- 5.Orozco P. Eur. J. Epidemiol. 2004;19:1105–1112. doi: 10.1007/s10654-004-1706-8. [DOI] [PubMed] [Google Scholar]
- 6.Parhami F, Tintut Y, Beamer WG, Gharavi N, Goodman W, Demer LL. J. Bone Miner. Res. 2001;16:182–188. doi: 10.1359/jbmr.2001.16.1.182. [DOI] [PubMed] [Google Scholar]
- 7.Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ, Fogelman AM. Proc. Natl. Acad. Sci. U. S. A. 1990;87:5134–5138. doi: 10.1073/pnas.87.13.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furnkranz A, Schober A, Bochkov VN, Bashtrykov P, Kronke G, Kadl A, Binder BR, Weber C, Leitinger N. Arterioscler. Thromb. Vasc. Biol. 2005;25:633–638. doi: 10.1161/01.ATV.0000153106.03644.a0. [DOI] [PubMed] [Google Scholar]
- 9.Parhami F, Jackson SM, Tintut Y, Le V, Balucan JP, Territo M, Demer LL. J. Bone Miner. Res. 1999;14:2067–2078. doi: 10.1359/jbmr.1999.14.12.2067. [DOI] [PubMed] [Google Scholar]
- 10.Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, Berliner JA, Demer LL. Arterioscler. Thromb. Vasc. Biol. 1997;17:680–687. doi: 10.1161/01.atv.17.4.680. [DOI] [PubMed] [Google Scholar]
- 11.Kronke G, Bochkov VN, Huber J, Gruber F, Bluml S, Furnkranz A, Kadl A, Binder BR, Leitinger N. J. Biol. Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- 12.Velardez MO, Ogando D, Franchi AM, Duvilanski BH. Mol. Cell. Endocrinol. 2001;172:7–12. doi: 10.1016/s0303-7207(00)00399-3. [DOI] [PubMed] [Google Scholar]
- 13.Kono SJ, Oshima Y, Hoshi K, Bonewald LF, Oda H, Nakamura K, Kawaguchi H, Tanaka S. Bone. 2007;40:68–74. doi: 10.1016/j.bone.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 14.Osyczka AM, Leboy PS. Endocrinology. 2005;146:3428–3437. doi: 10.1210/en.2005-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen D, Zhao M, Mundy GR. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- 16.Johnson EE, Urist MR, Finerman GA. Clin. Orthop. Relat. Res. 1988;230:257–265. [PubMed] [Google Scholar]
- 17.Urist MR, Behnam K, Kerendi F, Raskin K, Nuygen TD, Shamie AN, Malinin TI. Connect. Tissue Res. 1997;36:9–20. doi: 10.3109/03008209709160210. [DOI] [PubMed] [Google Scholar]
- 18.Lee KS, Hong SH, Bae SC. Oncogene. 2002;21:7156–7163. doi: 10.1038/sj.onc.1205937. [DOI] [PubMed] [Google Scholar]
- 19.Dempster DW, Cosman F, Parisien M, Shen V, Lindsay R. Endocr. Rev. 1993;14:690–709. doi: 10.1210/edrv-14-6-690. [DOI] [PubMed] [Google Scholar]
- 20.Ma YF, Jee WS, Ke HZ, Lin BY, Liang XG, Li M, Yamamoto N. J. Bone Miner. Res. 1995;10:496–505. doi: 10.1002/jbmr.5650100322. [DOI] [PubMed] [Google Scholar]
- 21.Shao JS, Cheng SL, Charlton-Kachigian N, Loewy AP, Towler DA. J. Biol. Chem. 2003;278:50195–50202. doi: 10.1074/jbc.M308825200. [DOI] [PubMed] [Google Scholar]
- 22.Iida-Klein A, Lu SS, Kapadia R, Burkhart M, Moreno A, Dempster DW, Lindsay R. J. Endocrinol. 2005;186:549–557. doi: 10.1677/joe.1.06270. [DOI] [PubMed] [Google Scholar]
- 23.Keller H, Kneissel M. Bone. 2005;37:148–158. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. J. Biol. Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 25.Date T, Doiguchi Y, Nobuta M, Shindo H. J. Orthop. Sci. 2004;9:503–508. doi: 10.1007/s00776-004-0815-2. [DOI] [PubMed] [Google Scholar]
- 26.Stein GS, Lian JB, Stein JL, Van Wijnen AJ, Montecino M. Physiol. Rev. 1996;76:593–629. doi: 10.1152/physrev.1996.76.2.593. [DOI] [PubMed] [Google Scholar]
- 27.Abedin M, Lim J, Tang TB, Park D, Demer LL, Tintut Y. Circ. Res. 2006;98:727–729. doi: 10.1161/01.RES.0000216009.68958.e6. [DOI] [PubMed] [Google Scholar]
- 28.Ju W, Hoffmann A, Verschueren K, Tylzanowski P, Kaps C, Gross G, Huylebroeck D. J. Bone Miner. Res. 2000;15:1889–1899. doi: 10.1359/jbmr.2000.15.10.1889. [DOI] [PubMed] [Google Scholar]
- 29.Pirih FQ, Aghaloo TL, Bezouglaia O, Nervina JM, Tetradis S. Biochem. Biophys. Res. Commun. 2005;332:494–503. doi: 10.1016/j.bbrc.2005.04.132. [DOI] [PubMed] [Google Scholar]
- 30.Pirih FQ, Nervina JM, Pham L, Aghaloo T, Tetradis S. Biochem. Biophys. Res. Commun. 2003;306:144–150. doi: 10.1016/s0006-291x(03)00931-8. [DOI] [PubMed] [Google Scholar]
- 31.Bai XC, Lu D, Bai J, Zheng H, Ke ZY, Li XM, Luo SQ. Biochem. Biophys. Res. Commun. 2004;314:197–207. doi: 10.1016/j.bbrc.2003.12.073. [DOI] [PubMed] [Google Scholar]
- 32.Mody N, Parhami F, Sarafian TA, Demer LL. Free Radic. Biol. Med. 2001;31:509–519. doi: 10.1016/s0891-5849(01)00610-4. [DOI] [PubMed] [Google Scholar]
- 33.Chae HJ, Jeong BJ, Ha MS, Lee JK, Byun JO, Jung WY, Yun YG, Lee DG, Oh SH, Chae SW, Kwak YG, Kim HH, Lee ZH, Kim HR. Immunopharmacol. Immunotoxicol. 2002;24:31–41. doi: 10.1081/iph-120003401. [DOI] [PubMed] [Google Scholar]
- 34.Lai CF, Chaudhary L, Fausto A, Halstead LR, Ory DS, Avioli LV, Cheng SL. J. Biol. Chem. 2001;276:14443–14450. doi: 10.1074/jbc.M010021200. [DOI] [PubMed] [Google Scholar]
- 35.Lou J, Tu Y, Li S, Manske PR. Biochem. Biophys. Res. Commun. 2000;268:757–762. doi: 10.1006/bbrc.2000.2210. [DOI] [PubMed] [Google Scholar]
- 36.Xiao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, Franceschi RT. J. Biol. Chem. 2000;275:4453–4459. doi: 10.1074/jbc.275.6.4453. [DOI] [PubMed] [Google Scholar]
- 37.Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H, Itoh K. J. Bone Miner. Res. 2002;17:1785–1794. doi: 10.1359/jbmr.2002.17.10.1785. [DOI] [PubMed] [Google Scholar]
- 38.Chan GK, Miao D, Deckelbaum R, Bolivar I, Karaplis A, Goltzman D. Endocrinology. 2003;144:5511–5520. doi: 10.1210/en.2003-0273. [DOI] [PubMed] [Google Scholar]
- 39.Rodan GA. Bone. 1992;13(Suppl. 1):S3–S6. doi: 10.1016/s8756-3282(09)80003-3. [DOI] [PubMed] [Google Scholar]
- 40.Kadl A, Huber J, Gruber F, Bochkov VN, Binder BR, Leitinger N. Vascul. Pharmacol. 2002;38:219–227. doi: 10.1016/s1537-1891(02)00172-6. [DOI] [PubMed] [Google Scholar]
- 41.Tintut Y, Morony S, Demer LL. Arterioscler. Thromb. Vasc. Biol. 2004;24:e6–e10. doi: 10.1161/01.ATV.0000112023.62695.7f. [DOI] [PubMed] [Google Scholar]
- 42.Tintut Y, Parhami F, Tsingotjidou A, Tetradis S, Territo M, Demer LL. J. Biol. Chem. 2002;277:14221–14226. doi: 10.1074/jbc.M111551200. [DOI] [PubMed] [Google Scholar]