

Abstract
Background
The lymphatic endothelium is an important semi-permeable barrier separating lymph from the interstitial space. However, there is currently a limited understanding of the lymphatic endothelial barrier and the mechanisms of lymph formation. The objectives of this study were to investigate the potential active role of lymphatic endothelial cells in barrier regulation, and to test whether the endothelial cell agonists VEGF-A and VEGF-C can alter lymphatic endothelial barrier function.
Methods and Results
Cultured adult human dermal microlymphatic endothelial cells (HMLEC-d) and human umbilical vein endothelial cells (HUVEC) were respectively used as models of lymphatic and vascular endothelium. Transendothelial electrical resistance (TER) of endothelial monolayers served as an index of barrier function. Cells were treated with VEGF-A, VEGF-C, or the VEGFR-3 selective mutant VEGF-C156S. MAZ51 was used to inhibit VEGFR-3 signaling. The results show that while VEGF-A causes a time-dependent decrease in TER in HUVEC, there is no response in HMLEC-d. In contrast, VEGF-C and VEGF-C156S cause a similar decrease in TER in HMLEC-d that is not observed in HUVEC. These results corresponded to the protein expression of VEGFR-2 and VEGFR-3 in these cell types, determined by Western blotting. In addition, the VEGF-C- and VEGF-C156S-induced TER changes were inhibited by MAZ51.
Conclusions
The results indicate differential responses of the lymphatic and vascular endothelial barriers to VEGF-A and VEGF-C. Furthermore, our data suggest that VEGF-C alters lymphatic endothelial function through a mechanism involving VEGFR-3.
Keywords: Lymph Formation, Vascular Endothelial Growth Factors, Endothelium
INTRODUCTION
The endothelial cells that form initial lymphatic vessels and the inner lining of collecting lymphatics act as a barrier between the lymphatic vessel lumen and the surrounding tissues. The structural role of lymphatic endothelial cells in maintaining normal lymphatic function and fluid homeostasis is well recognized. However, relatively little is understood about the specific cellular and molecular mechanisms that regulate lymph formation and propulsion, and what active role lymphatic endothelial cells may have in such processes.
Our current understanding of normal lymph formation involves a positive hydrostatic pressure gradient favoring diffusion of fluids and solutes from the interstitial space to the lumen of the initial lymphatic1. Fluid and solutes enter the initial lymphatics through sub-microscopic primary one-way valves in the endothelium that prevent backflow of fluid to the interstitial space as the luminal volume increases2–4. Subsequent opening of a larger, secondary valve that connects the initial lymphatic to its downstream collecting lymphatic, allows emptying of the newly formed lymph and restarts the cycle of fluid entry1. In this paradigm, the endothelial cells have been assumed to have a predominantly passive, structural role. However, we know from several studies of blood vessels that endothelial cells typically play an active role in regulation of the transvascular exchange of fluids and solutes5–7. Thus, we hypothesized that lymphatic endothelial cells may also possess the ability to alter the lymphatic barrier and thus potentially influence the rate of lymph formation.
Studying lymph formation in vivo is challenging because so many endocrine, neurogenic, myogenic, and hemodynamic factors in the surrounding tissue can potentially alter the gradient between interstitial and intralymphatic fluid pressure. Moreover, there is also the difficulty of selectively targeting lymphatic endothelial cells with pharmacological agents or genetic treatments in vivo. One approach to overcome these challenges is to utilize isolated, cultured lymphatic endothelial cells as a model of the lymphatic endothelial barrier in a similar fashion as cultured vascular endothelial cell models of microvascular permeability8–10. Thus, the overall objective of this study was to evaluate the barrier characteristics of cultured human microlymphatic endothelial cells under basal conditions and after stimuli that could potentially alter the lymphatic barrier.
Vascular endothelial growth factors (VEGFs) are endothelial agonists that possess vasoactive as well as angiogenic properties11–14. The first member of the VEGF family to be discovered, VEGF-A (also VEGF-165), binds to the receptors VEGFR-1 and VEGFR-2, and through the latter of these receptors causes vasodilation and elevates microvascular permeability, in addition to promoting angiogenesis14–16. VEGF-C binds to VEGFR-2 with less affinity than VEGF-A, and has also been shown to cause mild vasodilation and increase permeability in vivo17. VEGF-C also binds to VEGFR-3, located primarily on lymphatic endothelium, through which it can promote lymphangiogenesis18. The importance of VEGFR-3 in normal lymphatic development and function is highlighted by reports showing that certain mutations of this receptor cause primary lymphedema19–21.
VEGF-A and VEGF-C are normally present in the circulation, with serum levels respectively estimated to be as high as 380 pg/ml and 10 ng/ml (0.6 nM) in healthy patients, and even higher in patients who are obese or have lung disease or cancer 22–26. However, the physiological and pathological roles of these VEGFs have not been completely characterized, particularly how they could affect lymphatic barrier function and lymph formation. The purpose of this study was to study the barrier characteristics of cultured adult human dermal microlymphatic endothelial cells (HMLEC-d), under basal conditions, and after stimulus with VEGF-A and VEGF-C. We utilized measurement of transendothelial electrical resistance (TER) as an index of endothelial barrier function. HUVEC monolayers were used a model of vascular endothelium for comparison studies. Here, we describe the differential responses to VEGF-A and VEGF-C we observed in HMLEC-d and HUVEC, as well as our investigation of the role of VEGFR-3 in lymphatic endothelial barrier function.
MATERIALS AND METHODS
Materials
Cryopreserved adult human dermal microlymphatic endothelial cells (HMLEC-d), human umbilical vein endothelial cells (HUVEC), Clonetics microvascular endothelial growth medium (EGM-2MV) and endothelial basal medium (EBM) were obtained from Cambrex (East Rutherford, NJ). Recombinant human VEGF-A, VEGF-C, and VEGF-C-Cys-156-Ser (VEGF-C156S) were purchased from R&D Systems (Minneapolis, MN). MAZ51 was purchased from EMD-Calbiochem (San Diego, CA). Rabbit anti-prox-1 antibody (ab11941) was purchased from Novus Biologicals (Littleton, CO). Rabbit anti-LYVE-1 (H-156) and goat-anti-VE-cadherin (C-19) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-VEGFR-3 (clone 9D9F9) and mouse anti-VEGFR-2 (clone CH-11) were purchased from Chemicon/Upstate (Charlottesville, VA). Rabbit anti-podoplanin (PA1143A) was from Cell Sciences, Inc. (Canton, MA) and mouse anti-PECAM-1 (clone WM59) was from GeneTex (San Antonio, TX). Cy2 and Cy3-labeled secondary antibodies were from Jackson Immunoresearch (West Grove, PA). HRP-conjugated anti-mouse IgG was purchased from Promega (Madison, WI). Vectashield mounting medium was from Vector Laboratories (Burlingame, CA).
Cell Culture and TER Measurement
HMLEC-d and HUVEC were routinely maintained in EGM-2MV. TER measurement was performed using an Electrical Cell Motility System (CET, Inc., Coralville, IA) as previously reported9. In brief, cells were seeded at 105 cells/cm2 onto gelatin-coated gold electrode arrays (Applied Biophysics, Troy, NY). Medium served as the electrolyte, and barrier function was dynamically measured by determining the impedance of the cell-covered electrodes. A 1-V, 4000-Hz AC signal was supplied through a 1-MΩ resistor to approximate a constant-current source. The in-phase voltage (proportional to resistance) and the out-of-phase voltage (proportional to capacitive resistance) were measured. Cells were grown overnight on the electrode arrays, and any endothelial monolayers with baseline resistance values below 5000 Ω were excluded from the study. The next morning, medium was replaced with serum-free EBM, and cells were incubated a minimum of 2 hours prior to experimental treatments. Cells were treated with the indicated concentrations of VEGF-A, VEGF-C, VEGF-C156S, or MAZ51. The optimal concentrations used were based upon our dose-response study and work reported by others8, 27–31. Endothelial barrier function is presented as a fractional change in TER.
Immunofluorescence Labeling and Microscopy
Cells were grown on gelatin-coated round #1 coverslips (VWR) and allowed to reach confluence. The cells were then fixed at 25 °C in 4% paraformaldehyde in PBS for 5 min., followed by permeablization with 0.5% Triton X-100 in PBS for 10 min. The cells were then incubated for 1 h at 25 °C in a 1% BSA blocking solution followed by overnight incubation at 4 °C with primary antibody. After washing 3X with PBS, cells were incubated for 1 h at 25 °C with Cy2 or Cy3-labeled secondary antibody, and washed again 3X with PBS. Nuclei were labeled with Hoechst 33342. The coverslips were mounted on glass slides with Vectashield, and images were obtained with a Zeiss Axiovert 200M fluorescence microscope and AxioCam MRm black and white digital camera (Carl Zeiss, Thornwood, NY). Digital images were collected using Zeiss Axiovision 4.5 software.
Western Blotting
Protein was obtained by lysis of cells in ice-cold RIPA lysis buffer (Upstate) containing 1X HALT protease inhibitor cocktail (Pierce, Rockford, IL) for 15 min. Protein concentrations were determined with the Bradford assay, and equal amounts of protein were loaded for SDS-PAGE in 6% acrylamide gels. Mobility was assessed using NexusPointer protein marker #BNPM50 (BioNexus, Oakland, CA). Proteins were transferred to nitrocellulose for immunoblotting with anti-VEGFR-2 or anti-VEGFR-3 antibodies followed by appropriate HRP-coated secondary antibodies. Bands were visualized by chemiluminescence.
Data Analysis
TER data obtained from multiple wells of different electrode arrays were normalized to baseline, averaged, and reported as normalized resistance or the fractional change in resistance. The concentration-response data are presented as a percentage of the maximum response to VEGF-C. Concentration-response relationships were calculated and plotted using Deltagraph 5.5.2 software. Data are presented as the mean ± s.e.m. The significance between groups was determined by t-tests, or one-way ANOVA followed by Bonferroni t-tests for multiple comparisons where appropriate. Significance was accepted at P<0.05.
RESULTS
As a first step in establishing a cultured cell-based model of the lymphatic endothelial barrier, we sought to characterize the lymphatic endothelial phenotype of HMLEC-d. To achieve this, we tested for the presence of four markers used to identify lymphatic endothelial cells: Prox-1, LYVE-1, VEGFR-3, and podoplanin. These markers were all present in HMLEC-d as shown in Fig. 1. These cells also prominently displayed at their cell-cell junctions the endothelial-specific adhesion proteins VE-cadherin and PECAM-1, which play important roles in the maintenance of endothelial barrier function (Fig. 1).
Fig. 1.
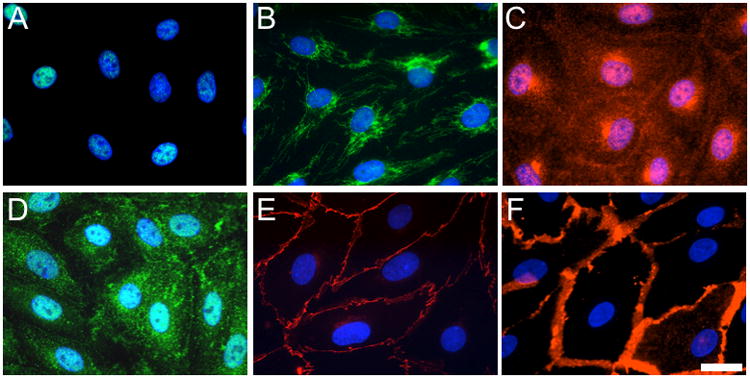
Expression of lymphatic endothelial markers in cultured HMLEC-d. The green labeling in panels A and B respectively show localization of prox-1 and LYVE-1. Panel C shows VEGFR-3 labeling in red. Podoplanin is labeled green in panel D. Panels E and F respectively show VE-cadherin and PECAM-1 labeled in red. Blue reflects nuclear labeling by Hoechst 33342. All images are representative of at least three experiments. Bar = 20 μm.
We next examined whether the endothelial barrier formed by HMLEC-d in culture is functionally comparable to that seen with cultured vascular endothelial cells, using TER as an index of barrier function. We first tested the ability of the endothelial monolayers to respond to VEGF-A (Fig. 2), which increases endothelial permeability both in vivo and in cultured endothelial cell models8, 14–16, 31. In HUVEC monolayers, VEGF-A caused a time-dependent decrease in TER, indicating an opening of the endothelial barrier, followed by a recovery characterized by elevated TER. HMLEC-d, however, did not respond to VEGF-A treatment (Fig. 2).
Fig. 2.
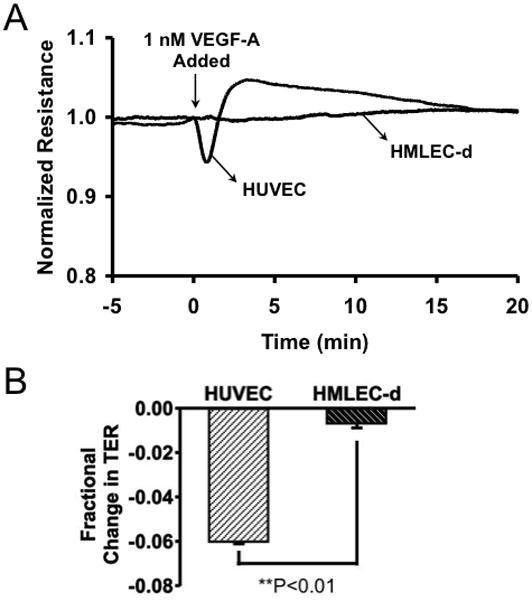
VEGF-A alters barrier function in HUVEC, but not in HMLEC-d. A. Tracings of the time-courses of HUVEC and HMLEC-d TER before and during exposure to 1 nM VEGF-A are shown. The tracings represent the averages of N=4 wells each. Panel B shows a comparison of the mean fractional change in TER 1 min. after VEGF-A was added. **P<0.01 HUVEC vs. HMLEC-d.
We also tested the ability of VEGF-C to alter TER in both HMLEC-d and HUVEC (Fig. 3). In this case, both HMLEC-d and HUVEC responded, although differently. In HUVEC, the response was somewhat similar to VEGF-A, however the initial decrease in TER was of a much lower magnitude, with the elevated TER during recovery being roughly the same as with VEGF-A treatment. In HMLEC-d, VEGF-C caused a steady decline in TER with a maximal change after about 40 minutes, followed by a gradual recovery to the initial TER level (Fig. 3).
Fig. 3.
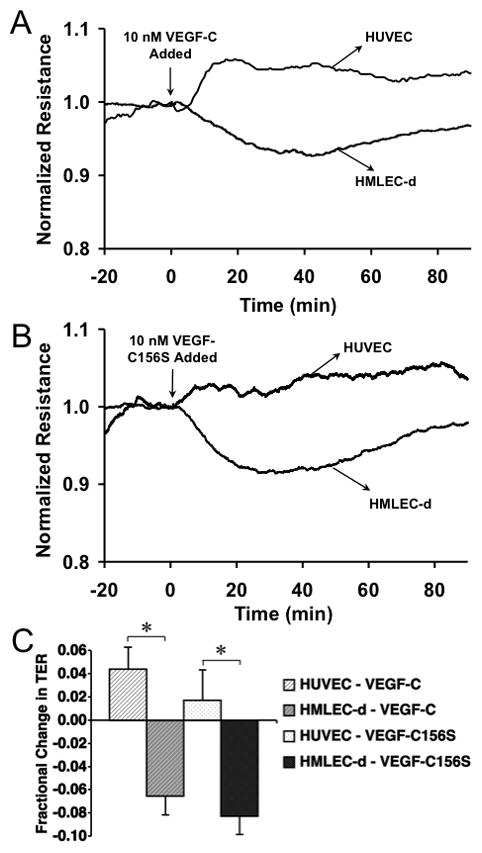
VEGF-C causes differential changes in TER in HMLEC-d and HUVEC. A. The tracings show average TER of HMLEC-d and HUVEC before and during treatment with 10 nM VEGF-C (N=4 each). Panel B shows the HMLEC-d and HUVEC TER before and during treatment with 10 nM VEGF-C156S (N=4 each). C. The mean fractional changes in TER 30 minutes after the addition of VEGF-C or VEGF-C156S are shown. *P<0.05 between the indicated groups.
To better understand these differential responses by HMLEC-d and HUVEC, we determined the expression of VEGFR-2 and VEGFR-3 in these two cell types by Western blotting (Fig. 4). The blot for VEGFR-2 shows a much stronger band for VEGFR-2 in HUVEC than in HMLEC-d. On the other hand, the blot for VEGFR-3 shows that expression of this receptor was much greater in HMLEC-d than in HUVEC. Only a very faint middle band (175 kDa) corresponding to a precursor form of VEGFR-332, 33 was apparent in the HUVEC lysate (Fig. 4).
Fig. 4.
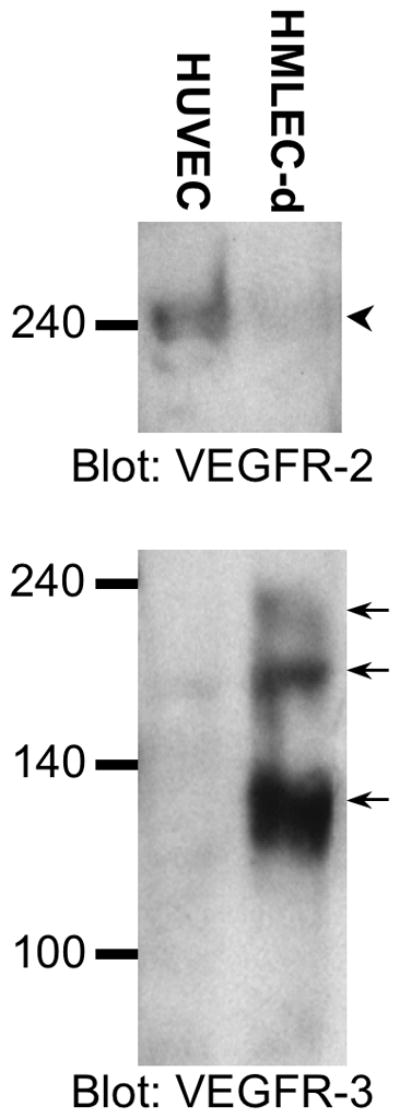
HMLEC-d and HUVEC differentially express VEGFR-2 and VEGFR-3. The upper blot shows a strong band for VEGFR-2 in HUVEC and a very weak band in HMLEC-d (arrowhead). The lower blot shows strong bands for VEGFR-3 (arrows) in the HMLEC-d lane and one weak VEGFR-3 band in the HUVEC lane (middle arrow). The bands at ~195, ~175, and ~125 kDa reflect the uncleaved, mature form, a unglycosylated precuror, and cleaved form of VEGFR-3, respectively32, 33. The blots are representative of three separate experiments.
Next, we further determined whether the VEGF-C-induced changes in HMLEC-d TER are due to a VEGFR-3-dependent mechanism (Fig. 5). There were no significant differences between the time-courses of the responses of HMLEC-d to VEGF-C and the VEGFR-3 selective agonist, VEGF-C156S (Fig. 5A). In addition, the concentration-response relationships of these two agonists were also quite similar (Fig. 5B). Furthermore, when we pretreated HMLEC-d with the VEGFR-3 inhibitor MAZ51, the response to VEGF-C156S was abolished (Fig. 6).
Fig. 5.
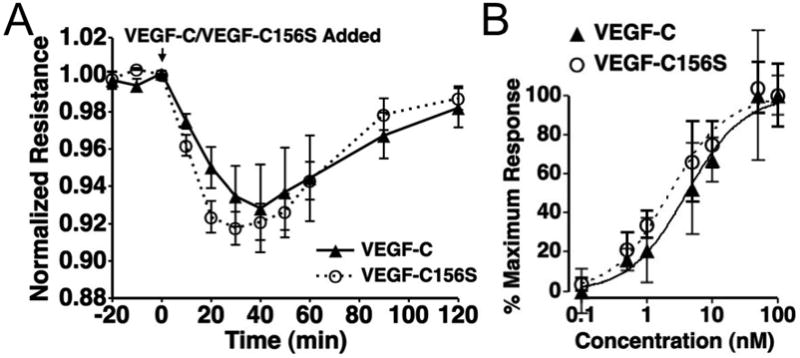
The time-course and dose-response characteristics of VEGF-C- and VEGF-C156S-induced TER changes in HMLEC-d are similar A. No significant differences in the time courses of VEGF-C and VEGF-C156S were apparent. N=4 for both treatments. B. VEGF-C and VEGF-C156s also showed very similar dose response characteristics, with both curves in the same order of magnitude and with VEGF-C156S having a similar Emax value and VEGF-C. The estimated EC50s for VEGF-C and VEGF-C156S are 4.13 and 2.25 nM, respectively. There were no significant differences between the responses to VEGF-C and VEGF-C156S at each concentration tested. N=4 for each concentration tested.
Fig. 6.
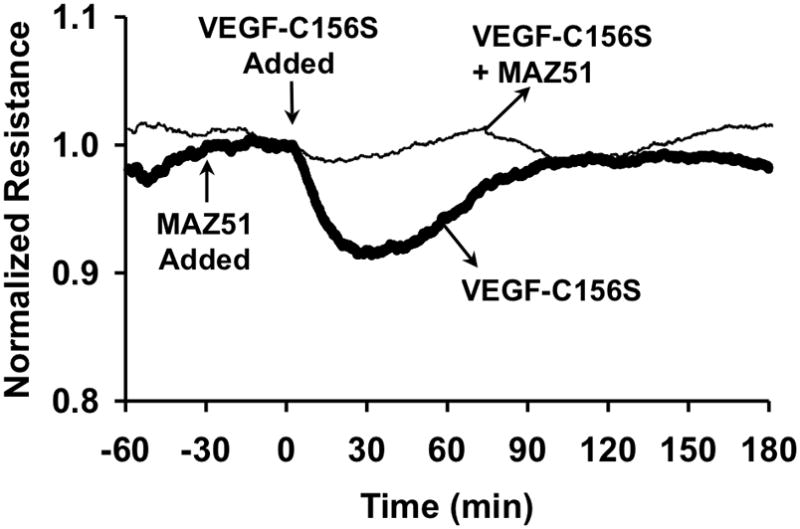
Inhibition of VEGFR-3 blocks VEGF-C156S-induced changes in TER in HMLEC-d. MAZ51 (5 μM) was added 30 minutes prior to VEGF-C156S (10 nM), and itself did not alter TER. However, MAZ51-treated HMLEC-d did not display decreased TER after treatment with VEGF-C156S, unlike cells that were not treated with MAZ51. The tracings show the average TER from both groups. N=4 for both groups.
DISCUSSION
In the present study, we demonstrate the novel finding that VEGF-C alters the barrier function of cultured lymphatic endothelial cells in a concentration- and time-dependent manner. We also show that this response is not present in vascular endothelial cells having little or no detectable expression of functional VEGFR-3. In addition, we show that the VEGF-C induced changes in TER can be mimicked by selective activation of VEGFR-3 with VEGF-C156S, and that inhibition of VEGFR-3 with MAZ51 abolishes this response. Taken together, the data suggest that VEGF-C-induced changes in lymphatic endothelial barrier function are dependent upon a mechanism that involves VEGFR-3.
Previously, in the mechanism of lymph formation, lymphatic endothelial cells have predominantly been considered as passive structures with one-way primary valves for the convective diffusion of fluid and solutes from the interstitial space into the initial lymphatic lumen1–4. Our data, however, implicate a possible active role of lymphatic endothelial cells in this process. By actively altering barrier function in response to various stimuli, endothelial cells may regulate the hydraulic conductivity of water and diffusive permeability coefficients of various solutes into initial lymphatics. Considering our current finding that VEGF-C decreases lymphatic TER, suggesting elevated permeability, we speculate that in vivo VEGF-C may possibly promote more rapid lymph formation when the concentration gradients that drive lymph formation are in a steady state.
While our finding that VEGF-C can alter the barrier function of HMLEC-d may be novel, the concept of lymphatic endothelial cells modulating normal, physiologic function of lymphatic vessels is already established. Several studies have demonstrated the importance of signaling from lymphatic endothelial cells to lymphatic smooth muscle in the regulation of phasic and tonic contractility34–36. In addition, recent reports suggest that lymphatic endothelial cells serve as sensors of local lymph flow and transduce signals to lymphatic smooth muscle37, 38.
Relatively little is known about the barrier function of the lymphatic endothelium. One study reports that in isolated iliac lymphatics, solutes smaller than 4.4 kDa can pass through the lymphatic barrier, yet larger molecules do not, suggesting a permselective endothelial barrier39. However, it is not yet clear what stimuli may modulate the permselectivity of the lymphatic endothelium. Lymphatic endothelial cells may respond in similar ways to some agents that alter vascular endothelial barrier function. Nevertheless, our results showing differential expression of VEGFR-2 and VEGFR-3 between vascular and lymphatic endothelial cells, similar to a previous report32, as well as different functional responses to VEGF-A and VEGF-C strongly indicate that this cannot be assumed. It is also worth noting that the abundant presence of VEGFR-3 on lymphatic endothelial cells, and their responsiveness to VEGF-C may also have a profound impact on the network of signaling pathways and cellular structures that define the responsiveness of the lymphatic endothelium40.
We chose TER measurement to assess barrier function because it has been previously established as a reliable index of permeability of vascular endothelial monolayers9, which is verified by the fact that the response we observed to VEGF-A reflects previously published findings8, 14–16, 31. TER also has the advantage insofar that rapid, dynamic changes can be measured, whereas solute flux studies are generally less sensitive because they are limited to determining average permeability coefficients over longer time periods required to accumulate enough tracer to accurately measure flux. Using a cell culture model also has the advantages of the ability to exclusively study lymphatic endothelial cells, and having tight control over hydrostatic and chemical factors that cannot be easily controlled in vivo.
In summary, we present novel data showing that VEGF-C can alter the barrier function of lymphatic endothelial cells through a VEGFR-3 dependent mechanism. Much work remains to be done to elucidate the cellular and molecular mechanisms that determine lymph formation. Our findings suggest that the endothelial cells lining the initial lymphatic vessels are not static structures, but rather possess the ability to actively regulate the translymphatic flux of fluids and solutes.
Acknowledgments
The authors thank Mr. Chase Clanton for his technical assistance during this study. This work was supported by NIH grants R01 HL73324, R01 HL61507, and R01 HL70752, and NIH Fellowship F32 HL76079.
References
- 1.Adair TH, Guyton AC. Lymph formation and its modification in the lymphatic system. In: Johnston MG, editor. Experimental Biology of the Lymphatic Circulation. New York: Elsevier; 1985. pp. 13–44. [Google Scholar]
- 2.Schmid-Schonbein GW. The second valve system in lymphatics. Lymphat Res Biol. 2003;1:25–9. doi: 10.1089/15396850360495664. discussion 9–31. [DOI] [PubMed] [Google Scholar]
- 3.Mendoza E, Schmid-Schonbein GW. A model for mechanics of primary lymphatic valves. J Biomech Eng. 2003;125:407–14. doi: 10.1115/1.1568128. [DOI] [PubMed] [Google Scholar]
- 4.Trzewik J, Mallipattu SK, Artmann GM, Delano FA, Schmid-Schonbein GW. Evidence for a second valve system in lymphatics: endothelial microvalves. Faseb J. 2001;15:1711–7. doi: 10.1096/fj.01-0067com. [DOI] [PubMed] [Google Scholar]
- 5.Yuan SY. Signal transduction pathways in enhanced microvascular permeability. Microcirculation. 2000;7:395–403. [PubMed] [Google Scholar]
- 6.Huxley VH, Rumbaut RE. The microvasculature as a dynamic regulator of volume and solute exchange. Clin Exp Pharmacol Physiol. 2000;27:847–54. doi: 10.1046/j.1440-1681.2000.03344.x. [DOI] [PubMed] [Google Scholar]
- 7.Michel CC, Curry FE. Microvascular permeability. Physiol Rev. 1999;79:703–61. doi: 10.1152/physrev.1999.79.3.703. [DOI] [PubMed] [Google Scholar]
- 8.Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, 2nd, Duran WN. VEGF Increases Endothelial Permeability by Separate Signaling Pathways Involving ERK-1/2 and Nitric Oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–100. doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- 9.Breslin JW, Sun H, Xu W, Rodarte C, Moy AB, Wu MH, Yuan SY. Involvement of ROCK-mediated endothelial tension development in neutrophil-stimulated microvascular leakage. Am J Physiol Heart Circ Physiol. 2006;290:H741–50. doi: 10.1152/ajpheart.00238.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breslin JW, Yuan SY. Involvement of RhoA and Rho kinase in neutrophil-stimulated endothelial hyperpermeability. Am J Physiol Heart Circ Physiol. 2004;286:H1057–62. doi: 10.1152/ajpheart.00841.2003. [DOI] [PubMed] [Google Scholar]
- 11.Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132. doi: 10.1007/978-3-642-59953-8_6. [DOI] [PubMed] [Google Scholar]
- 12.Tammela T, Enholm B, Alitalo K, Paavonen K. The biology of vascular endothelial growth factors. Cardiovasc Res. 2005;65:550–63. doi: 10.1016/j.cardiores.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol. 2001;280:C1358–66. doi: 10.1152/ajpcell.2001.280.6.C1358. [DOI] [PubMed] [Google Scholar]
- 14.Bates DO, Harper SJ. Regulation of vascular permeability by vascular endothelial growth factors. Vascul Pharmacol. 2003;39:225–37. doi: 10.1016/s1537-1891(03)00011-9. [DOI] [PubMed] [Google Scholar]
- 15.Wu HM, Yuan Y, Zawieja DC, Tinsley J, Granger HJ. Role of phospholipase C, protein kinase C, and calcium in VEGF-induced venular hyperpermeability. Am J Physiol. 1999;276:H535–42. doi: 10.1152/ajpheart.1999.276.2.H535. [DOI] [PubMed] [Google Scholar]
- 16.Aramoto H, Breslin JW, Pappas PJ, Hobson RW, 2nd, Duran WN. Vascular endothelial growth factor stimulates differential signaling pathways in in vivo microcirculation. Am J Physiol Heart Circ Physiol. 2004;287:H1590–8. doi: 10.1152/ajpheart.00767.2003. [DOI] [PubMed] [Google Scholar]
- 17.Hillman NJ, Whittles CE, Pocock TM, Williams B, Bates DO. Differential effects of vascular endothelial growth factor-C and placental growth factor-1 on the hydraulic conductivity of frog mesenteric capillaries. J Vasc Res. 2001;38:176–86. doi: 10.1159/000051044. [DOI] [PubMed] [Google Scholar]
- 18.Taipale J, Makinen T, Arighi E, Kukk E, Karkkainen M, Alitalo K. Vascular endothelial growth factor receptor-3. Curr Top Microbiol Immunol. 1999;237:85–96. doi: 10.1007/978-3-642-59953-8_5. [DOI] [PubMed] [Google Scholar]
- 19.Karkkainen MJ, Ferrell RE, Lawrence EC, Kimak MA, Levinson KL, McTigue MA, Alitalo K, Finegold DN. Missense mutations interfere with VEGFR-3 signalling in primary lymphoedema. Nat Genet. 2000;25:153–9. doi: 10.1038/75997. [DOI] [PubMed] [Google Scholar]
- 20.Brice G, Child AH, Evans A, Bell R, Mansour S, Burnand K, Sarfarazi M, Jeffery S, Mortimer P. Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet. 2005;42:98–102. doi: 10.1136/jmg.2004.024802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans AL, Bell R, Brice G, Comeglio P, Lipede C, Jeffery S, Mortimer P, Sarfarazi M, Child AH. Identification of eight novel VEGFR-3 mutations in families with primary congenital lymphoedema. J Med Genet. 2003;40:697–703. doi: 10.1136/jmg.40.9.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silha JV, Krsek M, Sucharda P, Murphy LJ. Angiogenic factors are elevated in overweight and obese individuals. Int J Obes (Lond) 2005;29:1308–14. doi: 10.1038/sj.ijo.0802987. [DOI] [PubMed] [Google Scholar]
- 23.Seyama K, Kumasaka T, Souma S, Sato T, Kurihara M, Mitani K, Tominaga S, Fukuchi Y. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphat Res Biol. 2006;4:143–52. doi: 10.1089/lrb.2006.4.143. [DOI] [PubMed] [Google Scholar]
- 24.Mitsuhashi A, Suzuka K, Yamazawa K, Matsui H, Seki K, Sekiya S. Serum vascular endothelial growth factor (VEGF) and VEGF-C levels as tumor markers in patients with cervical carcinoma. Cancer. 2005;103:724–30. doi: 10.1002/cncr.20819. [DOI] [PubMed] [Google Scholar]
- 25.Krzystek-Korpacka M, Matusiewicz M, Diakowska D, Grabowski K, Blachut K, Banas T. Up-regulation of VEGF-C secreted by cancer cells and not VEGF-A correlates with clinical evaluation of lymph node metastasis in esophageal squamous cell carcinoma (ESCC) Cancer Lett. 2006 doi: 10.1016/j.canlet.2006.08.011. In Press. [DOI] [PubMed] [Google Scholar]
- 26.Krenn K, Klepetko W, Taghavi S, Lang G, Schneider B, Aharinejad S. Recipient Vascular Endothelial Growth Factor Serum Levels Predict Primary Lung Graft Dysfunction. Am J Transplant. 2007 doi: 10.1111/j.1600-6143.2006.01673.x. [DOI] [PubMed] [Google Scholar]
- 27.Marchio S, Primo L, Pagano M, Palestro G, Albini A, Veikkola T, Cascone I, Alitalo K, Bussolino F. Vascular endothelial growth factor-C stimulates the migration and proliferation of Kaposi’s sarcoma cells. J Biol Chem. 1999;274:27617–22. doi: 10.1074/jbc.274.39.27617. [DOI] [PubMed] [Google Scholar]
- 28.Kirkin V, Mazitschek R, Krishnan J, Steffen A, Waltenberger J, Pepper MS, Giannis A, Sleeman JP. Characterization of indolinones which preferentially inhibit VEGF-C- and VEGF-D-induced activation of VEGFR-3 rather than VEGFR-2. Eur J Biochem. 2001;268:5530–40. doi: 10.1046/j.1432-1033.2001.02476.x. [DOI] [PubMed] [Google Scholar]
- 29.Kirkin V, Thiele W, Baumann P, Mazitschek R, Rohde K, Fellbrich G, Weich H, Waltenberger J, Giannis A, Sleeman JP. MAZ51, an indolinone that inhibits endothelial cell and tumor cell growth in vitro, suppresses tumor growth in vivo. Int J Cancer. 2004;112:986–93. doi: 10.1002/ijc.20509. [DOI] [PubMed] [Google Scholar]
- 30.Joukov V, Kumar V, Sorsa T, Arighi E, Weich H, Saksela O, Alitalo K. A recombinant mutant vascular endothelial growth factor-C that has lost vascular endothelial growth factor receptor-2 binding, activation, and vascular permeability activities. J Biol Chem. 1998;273:6599–602. doi: 10.1074/jbc.273.12.6599. [DOI] [PubMed] [Google Scholar]
- 31.Moy AB, Blackwell K, Wu MH, Granger HJ. Growth factor- and heparin-dependent regulation of constitutive and agonist-mediated human endothelial barrier function. Am J Physiol Heart Circ Physiol. 2006;291:H2126–35. doi: 10.1152/ajpheart.00185.2006. [DOI] [PubMed] [Google Scholar]
- 32.Bando H, Brokelmann M, Toi M, Alitalo K, Sleeman JP, Sipos B, Grone HJ, Weich HA. Immunodetection and quantification of vascular endothelial growth factor receptor-3 in human malignant tumor tissues. Int J Cancer. 2004;111:184–91. doi: 10.1002/ijc.20211. [DOI] [PubMed] [Google Scholar]
- 33.Pajusola K, Aprelikova O, Pelicci G, Weich H, Claesson-Welsh L, Alitalo K. Signalling properties of FLT4, a proteolytically processed receptor tyrosine kinase related to two VEGF receptors. Oncogene. 1994;9:3545–55. [PubMed] [Google Scholar]
- 34.Mizuno R, Dornyei G, Koller A, Kaley G. Myogenic responses of isolated lymphatics: modulation by endothelium. Microcirculation. 1997;4:413–20. doi: 10.3109/10739689709146805. [DOI] [PubMed] [Google Scholar]
- 35.Mizuno R, Koller A, Kaley G. Regulation of the vasomotor activity of lymph microvessels by nitric oxide and prostaglandins. Am J Physiol. 1998;274:R790–6. doi: 10.1152/ajpregu.1998.274.3.R790. [DOI] [PubMed] [Google Scholar]
- 36.von der Weid PY, Crowe MJ, Van Helden DF. Endothelium-dependent modulation of pacemaking in lymphatic vessels of the guinea-pig mesentery. J Physiol. 1996;493 (Pt 2):563–75. doi: 10.1113/jphysiol.1996.sp021404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gashev AA, Davis MJ, Zawieja DC. Inhibition of the active lymph pump by flow in rat mesenteric lymphatics and thoracic duct. J Physiol. 2002;540:1023–37. doi: 10.1113/jphysiol.2001.016642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koller A, Mizuno R, Kaley G. Flow reduces the amplitude and increases the frequency of lymphatic vasomotion: role of endothelial prostanoids. Am J Physiol. 1999;277:R1683–9. doi: 10.1152/ajpregu.1999.277.6.R1683. [DOI] [PubMed] [Google Scholar]
- 39.Ono N, Mizuno R, Ohhashi T. Effective permeability of hydrophilic substances through walls of lymph vessels: roles of endothelial barrier. Am J Physiol Heart Circ Physiol. 2005;289:H1676–82. doi: 10.1152/ajpheart.01084.2004. [DOI] [PubMed] [Google Scholar]
- 40.Yong C, Bridenbaugh EA, Zawieja DC, Swartz MA. Microarray analysis of VEGF-C responsive genes in human lymphatic endothelial cells. Lymphat Res Biol. 2005;3:183–207. doi: 10.1089/lrb.2005.3.183. [DOI] [PubMed] [Google Scholar]