

Abstract
A hallmark of hemostasis is that cells and proteins involved in the formation of a blood clot remain in a quiescent state and are only activated following an appropriate stimulus. The homologous proteins factors V and VIII cannot participate to any significant degree in their macromolecular enzyme complexes and are thus considered procofactors. Activity is generated following limited proteolysis, indicating that the conversion of the procofactors to factor Va and factor VIIIa must result in structural changes that impart cofactor function. The proteolytic events that lead to the activation of these proteins have been extensively characterized over the past three decades. However, a fundamental understanding of the mechanism(s) by which these proteins are kept as inactive procofactors and how specific bond cleavage facilitates the conversion to the active cofactor state is only starting to become known. These molecular processes undoubtedly play critical regulatory roles, evolved to maintain normal hemostasis since factor Va and factor VIIIa have a tremendous influence on thrombin generation. This review will detail our current understanding of the molecular process of procofactor activation and highlight structural features that play a major role in factor V and factor VIII activation.
Keywords: Factor V, Factor VIII, prothrombin activation, FX activation, procofactor, proteolytic activation, hemostasis
Introduction
The majority of blood coagulation factors are synthesized as inactive precursors that only express activity following discrete and limited proteolysis. A defined conformational change ensues which allows these proteins to assemble on cellular surfaces localized at the site of injury where they optimally function. This molecular strategy allows for a high level of temporal and spatial regulation as well as protection against naturally circulating inhibitors which typically target the active protein conformation. It is well established that the serine protease zymogens of coagulation (e.g. FVII, FIX, FX, prothrombin, etc.) follow a general activation strategy which is shared by all serine proteases and is typified by trypsinogen and chymotrypsinogen [1]. The process requires cleavage following Arg15 (the bond between Arg15 and Ile16) which generally removes an activation peptide and exposes a new N-terminus in the catalytic domain beginning with Ile16. The new N-terminal sequence then folds back into the catalytic domain and inserts into the N-terminal binding cleft in a sequence-specific manner forming a salt bridge between the α-NH2 group of Ile16 and Asp194 in the interior of the catalytic domain. This transition is associated with numerous changes in the structure and ultimately leads to the maturation of the active serine protease. In contrast to this molecular strategy, the mechanism underlying the activation of the two major procofactors in blood coagulation, factor V (FV) and factor VIII (FVIII) are substantially different.
Factor V and FVIII are structurally and functionally homologous proteins and play a central role in the hemostatic process. Once activated these proteins serve analogous functions as cofactors in the blood coagulation system [2]. Activated FV (FVa) assembles with the serine protease factor Xa (FXa) while activated FVIII (FVIIIa) is a cofactor for factor IXa (FIXa); these protein complexes assemble in the presence of Ca2+ ions on a negatively charged membrane surface to form the prothrombinase and intrinsic tenase complexes, respectively [3,4]. The prothrombinase complex catalyzes the conversion of prothrombin to thrombin, while intrinsic Xase catalyzes the proteolytic conversion of FX to FXa; both pivotal steps in the coagulation cascade [5]. The individual contribution of the serine proteases FIXa and FXa to overall thrombin generation is relatively minor, as incorporation of FVa and FVIIIa into the macromolecular enzyme complexes enhances the reaction rate by several orders of magnitude [5]. The importance of these cofactors is further underscored by clinical findings, which indicate that FV and FVIII deficiency states lead to parahemophilia and hemophilia A, respectively [6,7].
Following the discovery of FV and FVIII in the 1930-40s, it was quickly recognized that they require proteolytic activation to fully participate in coagulation [8-11]. Over the past three decades considerable effort and progress has been made defining their mode of activation [12-14]; however, key mechanistic details remain to be uncovered. This review focuses on our current understanding of FV and FVIII activation and discusses the various structural elements that assist in keeping these proteins in an inactive procofactor state.
The Factor V procofactor to cofactor transition
Factor V is a large (Mr = 330,000), multidomain (A1-A2-B-A3-C1-C2), single chain glycoprotein that circulates in blood at a concentration of ∼20 nM (∼10 μg/mL) [12,14]. Of the total FV pool in whole blood, ∼20% is stored in the α-granules of platelets and secreted upon platelet activation [15]. At physiological plasma concentrations, the procofactor FV cannot assemble or function in the prothrombinase complex; proteolytic processing within the B domain is an absolute requirement for the expression of cofactor function [12,16-19]. Thrombin is considered the key physiological activator of FV and cleaves three peptide bonds (Arg709, Arg1018, and Arg1545) within the B domain, thereby facilitating B domain removal (Figure 1) [16,20-22]. While not completely defined, thrombin appears to interact with FV through the heavy and light chains [23,24]. The binding site within the light chain is thought to reside within the C2 domain and appears important for proteolysis at all three thrombin cleavage sites [24]. The resulting active cofactor species, FVa, is a heterodimer composed of a heavy chain (A1-A2; Mr = 105,000) and a light chain (A3-C1-C2; Mr = 71/74,000), which are associated through Ca2+ ions (Figure 1) [16,20-22,25]. The heavily glycosylated B domain, spanning amino acids 710-1545, is released as two large fragments (Mr = 71,000 and Mr = 150,000) [16,21,22,26].
Figure 1. Schematic representation of FV and FVa.
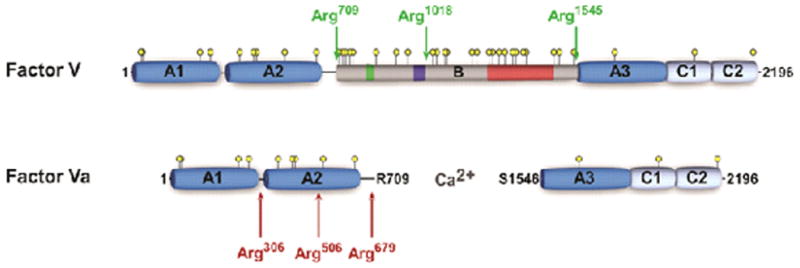
Schematic A1-A2-B-A3-C1-C2 domain representation of human FV and FVa. Thrombin cleavage sites are indicated by green arrows and APC cleavage sites by red arrows. Yellow circles represent potential N-linked glycosylation sites, the green box indicates a 2× 17 amino acid repeat region, the dark blue box corresponds to the basic sequence 963-1008 implicated in preserving the FV procofactor state, and the red box represents a 31× 9 amino acid tandem repeat region.
In addition to thrombin, various proteases have been identified that cleave FV to generate a cofactor species with variable amounts of activity. For example, several groups have established the FXa activates FV in a membrane- and Ca2+-dependent fashion following cleavage at or near Arg709 and Arg1018, and possibly at other sites in the light chain depending on reaction conditions [19,27,28]. Other proteases include, to name a few, activators from Daboia russelli (Russell's viper), Daboia lebetina, and Naja naja oxiana venoms [16,29-32], calpain [33]}, plasmin [34], platelet proteases [35,36], meizothrombin [37], as well as elastase and cathepsin G [38-41]. These later two proteases are of potential significance as they are released from polymorphonuclear leukocytes at extravascular tissue sites and could amplify thrombin generation through FV activation. This in combination with FV released from activated platelets may play a major role in the initiation phase of cell-based coagulation events.
While previously appreciated, the laboratories of Mann and Esmon firmly established that FV requires proteolytic processing at multiple sites to effect activation [22,42]. In the following decades, numerous studies have attempted to define the contribution of the individual cleavage sites to the development of FV cofactor activity. Although somewhat conflicting results have been obtained, it is generally acknowledged that proteolysis by thrombin follows a kinetically preferred order of bond cleavage: Arg709 is cleaved first, followed by cleavage at Arg1018, and Arg1545. Furthermore, most data support the finding that maximal activation of FV requires proteolysis at Arg1545. Cleavage at Arg709 and Arg1018 yields a FV derivative with significant, but partial cofactor activity [21,26,43,44], whereas individual cleavage at these sites does not lead to any substantial increase in cofactor activity [28,43,44]. Support for the contribution of cleavage at Arg1545 to maximal FV cofactor activity came from mutagenesis studies, which demonstrated that isolated cleavage at this site is sufficient for complete activation [28,43,44]. This is also consistent with the observation that proteolysis by Russell's viper venom (RVV-V) and Daboia lebetina venom (LVV-V), which cleave FV at Arg1545, results in full activation [16,32,45-47]. Thus release of the B domain from the FV light chain appears to be a necessary requirement for the expression of cofactor activity, which is facilitated by proteolysis of the two preceding activation sites in the B domain. Collectively, the results suggest that the FV B domain somehow keeps FV in an inactive state and its removal by proteolysis contributes to the activation mechanism.
The B domain preserves the procofactor state of factor V
The human FV B domain is 836 amino acids long, comprises ∼50% of the mass of the protein, and has no homology to any other known protein, including the FVIII B domain (Figure 1) [20,48]. The B domain is heavily glycosylated and has unusual regions of tandem repeats, of which the function remains to be elucidated. Electron microscopy and physical studies have suggested that the B domain appears as a bulky extension to a globular core, assumed to be the heavy/light chains [49,50]. Because of these unusual properties and lack of importance to FVa procoagulant activity, less attention has been paid to investigating its functional significance. Some studies have suggested that the B domain may play a role in the anticoagulant function of FV by stimulating the APC-mediated inactivation of FVIIIa (reviewed here [51]). As suggested above, one role must be to maintain FV as an inactive procofactor. Recent work from our laboratory as well as others has shed some light on how the FV B domain regulates the function of FV. An important first observation came from the Kane laboratory who showed that a B-domainless derivative of FV (FVdes811-1491 or FV-810) has constitutive, but partial activity compared to FVa [43,52]. These studies suggested that the B domain somehow prevents expression of procoagulant activity prior to proteolytic processing [43]. More recently, our laboratory has further investigated the molecular properties of this B domain-deleted FV variant. We found that purified FV-810 as well as a thrombin-resistant derivative interact with membrane-bound FXa with high affinity and are functionally equivalent to FVa in the absence of intentional proteolysis [53]. These findings indicate that proteolysis within the B domain, while necessary, is incidental to the mechanism by which cofactor function is actually realized. Instead, proteolytic activation of FV simply eliminates steric and/or conformational constraints imposed by the B domain that interfere with discrete binding interactions essential to the FVa cofactor activity, for example the FXa binding site. Removal of these inhibitory constraints through recombinant truncation bypasses the requirement for proteolysis to activate the molecule. Using a panel of progressively finer B domain-truncated variants, we were able to identify a discrete region of the B domain that appears to play a critical role in stabilizing the procofactor state [54]. Part of this B domain region (residues 963-1008) is unusually basic with 18 out of 46 residues being Arg or Lys and is well conserved across the vertebrate lineage [55]. As expected, disruption of this B domain region by mutagenesis or through deletion yielded derivatives with cofactor-like properties in the absence of intentional proteolysis, indicating that the length of the B domain per se is not a primary factor in preserving the procofactor state. Thrombin-mediated proteolysis of FV facilitates removal of these inhibitory B domain sequences; however, it is likely that other, as yet to be identified components of the B domain also play a role in preserving the procofactor state.
Insight into B domain function from non-mammalian forms of FV
Whereas most of the B domain sequence is highly variable throughout vertebrate evolution, several short motifs are strongly conserved, including the basic region 963-1008 detailed above [55-57]. An exception to these findings has been found in an unusual form of FV derived from the venom of some Australian Elapidae family members (O. microlepidotus, P. textilis, and O. scutellatus), which are among the most venomous snakes in the world [58]. A unique feature of these snakes is that approximately 5-40% of their venom consists of a large prothrombin activating complex comprising a cofactor FVa-like subunit and a serine protease FXa-like subunit, which share high sequence homology with mammalian FVa and FXa (55-60%) [59-63]. Remarkably, the FV homologues expressed in the elapid venom have extraordinarily short B domains: 46 versus ∼600-800 residues in mammals, and they lack the basic region. This intriguing observation prompted us to assess the functional properties of purified recombinant venom-derived P. textilis FV (pt-FV) [59]. Consistent with our previous observations, we were able to show that the absence of the basic region correlates with the expression of procoagulant FV activity, indicating that venom FV is expressed as a constitutionally active FV variant [59]. As such, this is the first FV species observed thus far that exists as an active cofactor. Notably, this protein can also function in the absence of anionic membranes and is completely resistant to inactivation by APC, despite APC-mediated proteolysis within the heavy chain at the equivalent Arg506 and Arg709 sites [59]. We speculate that a unique disulfide bond between the A2 and A3 domains of pt-FV enhances its structural stability and prevents dissociation of the A2 domain upon APC cleavage [59]. Thus, pt-FV represents an exceptional example of a protein that has adapted into a potent biological weapon for host defense and envenomation of prey.
Mutations in Factor V associated with disease states
Some mutations in FV have been reported to increase the risk of developing thrombosis (for a review see [64]), whereas others can lead to a bleeding disorder (reviewed in [65]). No mutations or polymorphisms associated with disease states have been observed at the FV thrombin cleavage sites [66].
APC-resistant factor V
One of the most commonly observed genetic risk factors for thrombosis is FVLeiden, which is partially resistant to inactivation by APC due to a Gln substitution at the APC cleavage site Arg506 [67-71]. Individuals who are heterozygous for this mutation have a 3-8-fold increased risk of developing thrombosis, whereas the risk for homozygous individuals is 50-80-fold higher as compared to individuals with normal FV [72,73]. Several other mutations and polymorphisms that have been shown to also result in some degree of APC resistance are reviewed elsewhere [74].
Parahemophilia
Factor V deficiency (parahemophilia) is an autosomal recessive bleeding disorder which was first described in the 1940s by Paul Owren in Norway [10]. It is a rare bleeding disorder that affects one in a million individuals and is characterized by low or undetectable FV activity and/or antigen levels (reviewed in [75]). Patients with undetectable levels of FV (<1%) due to homozygous nonsense, frameshift, or missense mutations exhibit the gamut of phenotypes from asymptomatic to severe bleeding. Two thirds of all mutations causing FV deficiency are nonsense mutations in the FV gene [65]. Even though mRNA containing a premature stop codon is normally degraded by nonsense mediated decay, it has been suggested that trace amounts of FV may be expressed following ribosomal slippage or somatic inversion events [76]. Of the reported missense mutations, most are clustered in the A and C domains, while none are found in the B domain [66]. Deficiency in FV presents a conundrum because the phenotype is variable and correlates unexpectedly poorly with FV levels in plasma [65,77-81]. These observations are generally not consistent with the fundamental role played by FV in coagulation and with findings in the FV knockout mice which have a lethal phenotype [82]. However, observations made in the past two years point to an important role for platelet FV that may help explain these observations.
Approximately one fifth of the total FV pool is stored in the α-granules of platelets from which it is secreted upon platelet activation [15]. While megakaryocytes can synthesize FV [83-85], the vast majority of platelet FV is endocytosed from the plasma pool by megakaryocytes [86-88]. Following endocytosis via a specific receptor-mediated process [89,90], FV is modified intracellularly such that it is functionally unique compared to its plasma-derived counterpart [88,91]. Recently, Duckers et al. were able to correlate the levels of platelet FV with thrombin generation in FV-deficient patients. They showed that patients deficient in FV resulting from missense mutations have sufficient functional FV in their platelets to guarantee thrombin generation and protect them against major bleeding [92]. Furthermore, the same group demonstrated that the FV requirement for thrombin generation is considerably lower in these patients due to markedly reduced levels of the anticoagulant protein tissue factor pathway inhibitor (TFPI) in FV-deficient plasma [93]. Whether the disease phenotype correlates with the platelet FV levels and whether a similar mechanism may explain the relatively mild phenotype in patients that have an introduced stop codon in the FV gene remains to be determined.
The procofactor to cofactor transition of factor VIII
Factor VIII circulates as a large (Mr ≈ 330,000), multidomain (A1-A2-B-A3-C1-C2) heterodimer resulting from limited proteolysis at the B-A3 junction and at additional sites in the B domain [13]. This heterodimer consists of a variably sized heavy chain (A1-A2-B; 200-90 kDa) and a light chain (A3-C1-C2; 80 kDa) that are noncovalently associated (Figure 2). The A domains are bordered by short segments (∼30-40 amino acids) of negatively charged residues, known as the acidic regions a1 (337-372), a2 (711-740), and a3 (1649-1689) (Figure 2). Whereas regions a2 and a3 are more or less well conserved in FV (Figure 3), the a1 region of FVIII is absent from both FV and ceruloplasmin [94-96], the latter being a ferroxidase with an A1-A2-A3 domain structure that originates from the same ancestral protein as FV and FVIII [97,98]. The FVIII acidic regions are thought to function, in part, as binding sites for thrombin [13,99,100]. In addition, the a3 region and regions within both C domains have been suggested to mediate the tight interaction of the FVIII heterodimer with its carrier protein von Willebrand factor (VWF) [101-104]. This interaction serves various important roles in FVIII physiology, as it has been reported to stabilize the heterodimeric structure of FVIII as well as to prevent proteolysis by FXa and APC [105].
Figure 2. Schematic representation of FVIII, FVIIIa, and FVIII-SQ.
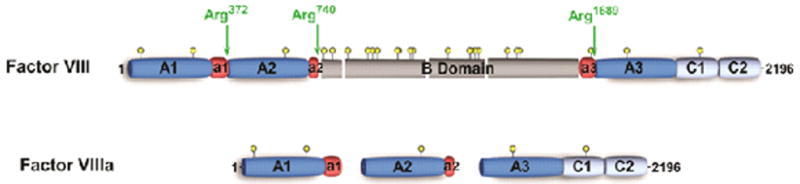
Schematic A1-A2-B-A3-C1-C2 domain representation of human FVIII and FVIIIa. The acidic regions denoted by a1, a2, and a3 are indicated in red, thrombin cleavage sites are indicated by green arrows, and yellow circles represent potential N-linked glycosylation sites.
Figure 3. Alignment of the acidic regions a1, a2, and a3 of FVIII.

The acidic regions a1, a2, and a3 from human FVIII were aligned with human FV and human ceruloplasmin (AlignX Module; Invitrogen Carlsbad, CA, USA). Residues fully conserved between all three molecules are shown in green, partially conserved amino acids are indicated in yellow, and the negatively charged residues characterizing the acidic regions are shown in red. The boundaries of the acidic regions are indicated by arrows and thrombin cleavage sites are indicated by arrows and depicted in blue.
The FVIII heterodimer is an inactive procofactor and must be subjected to limited proteolysis to effect full cofactor activity [11,106-112]. Factor VIII can be proteolytically activated by both thrombin and FXa [13]. Thrombin cleaves three peptide bonds at Arg372, Arg740, and Arg1689, thereby generating FVIIIa, which is a heterotrimer composed of the A1 (50 kDa; 1-372), A2 (43 kDa; 373-740), and the light chain (A3-C1-C2; 73 kDa; 1689-2332) (Figure 2) [112,113]. Activation of FVIII results in a transient ∼20-50-fold increase in biological activity which decays over a short period of time due to A2 domain dissociation from A1/A3-C1-C2, a mechanism which contributes to the regulation of FVIIIa cofactor activity [114-116].
Numerous studies have examined the role of the individual thrombin cleavage sites in the expression of FVIIIa cofactor activity. Similar to thrombin-mediated activation of FV, the evidence obtained supports an ordered cleavage pathway, with cleavage at Arg740 occurring first, followed by cleavage at Arg1689, and subsequently at Arg372. The specific role of the individual cleavage sites in the expression of FVIIIa cofactor activity will be discussed in the following sections.
Cleavage at Arg740 and Arg1689
Although cleavage at Arg740 appears to be of little consequence to the development of cofactor function [117], it is thought to facilitate subsequent proteolysis at Arg372 and Arg1689 [118]. Surprisingly however, its significance in FVIII activation is not reflected in the hemophilia A patient population, as there have been no reports of missense mutations at position 740 (http://hadb.org.uk). Cleavage of the light chain at Arg1689 results in dissociation of FVIII from VWF which allows for association of the cofactor with anionic phospholipids and FIXa [119,120]. Whether proteolysis at this site directly contributes to the potentiation of FVIIIa cofactor activity remains controversial. There is some evidence that this cleavage partially increases cofactor activity [121,122]; however, Pipe and Kaufman have shown using a single chain FVIII derivative (IR8) that cofactor activity can be obtained even in the absence of the Arg1689 cleavage site [123].
Cleavage at Arg372
The results obtained with various FVIII derivatives as well as other biochemical studies and naturally occurring mutations indicate that cleavage at Arg372 is essential to procofactor activation [117,124-127]. Biochemical data suggest that cleavage at this site exposes a functional FIXa binding site which promotes rapid FX activation by cofactor-bound FIXa [128]. Based on these and other observations, it was suggested that acidic region a1 and possibly a portion of the a3 region (1649-1689) could obscure functionally important surface areas such as a FIXa binding site; results that are in line with functional studies [128]. Interestingly, acidic region a1 of FVIII is noticeably absent from FV (missing from exon 7) possibly pointing to a unique function in FVIII (Figure 3) [94,95]. Furthermore, recent structural data on B domain-deleted FVIII indicate that this part of FVIII is highly flexible as no electron density was observed in this region [129,130]. Alternatively, cleavage at Arg372 could induce a change in conformation that is critical for the expression of FVIIIa cofactor function. Evidence for this comes from studies employing cross linking agents and apolar probes as well as circular dichroism experiments. These studies support the idea that there are subtle, yet measureable changes in conformation in the vicinity of the A2 domain when FVIII is activated to FVIIIa [131-133]. Future biochemical and structural studies are needed to resolve the precise mechanism by which cleavage at Arg372 facilitates the FVIII procofactor to cofactor transition.
The Factor VIII B domain
The FVIII B domain, like that of FV, is very large (908 residues), encoded by a single exon, heavily glycosylated, and is also removed following thrombin-mediated proteolysis; however it does not share sequence homology with the FV B domain. Yet, unlike FV, several groups have established that removal of most of the B domain yields a derivative that remains as an inactive procofactor [123,134-137]. Thus, the FVIII B domain does not appear to play a role in preserving FVIII as a procofactor. As such, the molecular mechanisms that regulate or prevent the potential cofactor activities of FV and FVIII are surprisingly different.
Mutations in Factor VIII associated with disease states
In contrast to the dual effects that mutations in FV can have on the hemostatic balance (e.g. procoagulant vs. anticoagulant), no FVIII mutations have been described thus far that are linked to a prothrombotic state. Even though APC resistance is strongly correlated with an enhanced risk of developing thrombosis, no mutations at the equivalent APC inactivation sites in FVIII have been observed in a cohort of patients with venous thrombosis [138]. In addition, recombinant FVIII variants carrying substitutions at one of the two APC cleavage sites did not show an APC resistant phenotype [139]. It was found, though, that high levels of FVIII are associated with an increased risk of venous thrombosis (reviewed here [140]). To date, no genetic variation in the FVIII gene has been identified that might account for this phenotype.
Hemophilia A
A deficiency or functional defect in FVIII is at the basis of the X-linked congenital bleeding disorder known as hemophilia A (for a review see [141]), which has an incidence of one in 5,000 in the general population. Hemophilia A is categorized as severe (<1%), moderate (1-5%), or mild (5-20%), according to the relative amount of FVIII activity in the patient's plasma. The classic symptoms of hemophilia include bleeding episodes that affect joints, muscles, internal organs, and the brain. Whereas approximately one third of all hemophilia A cases are due to intron 22 inversions [142,143], missense mutations have been observed to be the most frequent mutation type and are almost exclusively responsible for the mild/moderate hemophilia A phenotypes [144]. In addition, stop codons account for ∼10% of all hemophilia A mutations, and one third of the identified FVIII gene defects other than intron inversions represent new mutations [144]. Most reported mutations have been registered in the hemophilia A mutation database (http://hadb.org.uk), and a graphic representation of the number of missense mutations per FVIII region is given in Figure 4.
Figure 4. Graphic representation of missense mutations per FVIII.
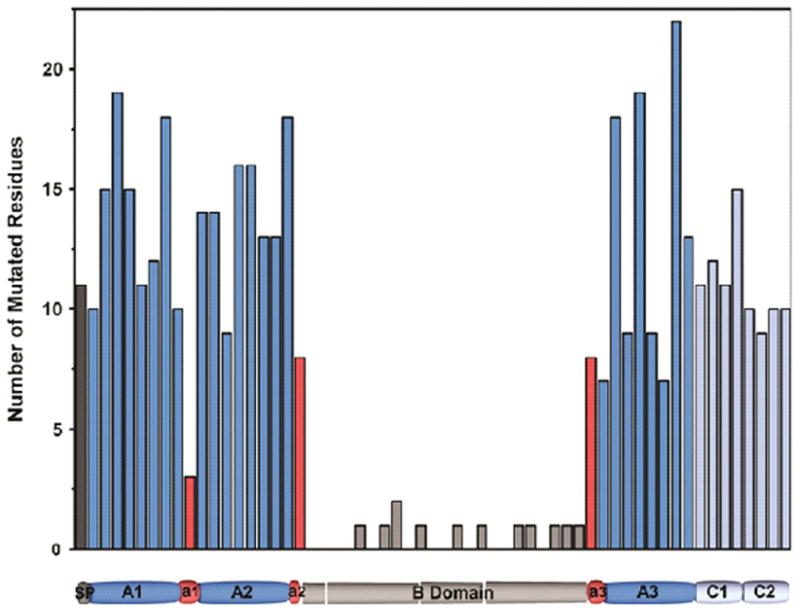
The number of hemophilia A causing mutations in FVIII was determined in 40 residue blocks, including the signal peptide. Mutations were derived from the Haemophilia A Mutation Database (http://hadb.org.uk), and mutations resulting in introduction of a stop codon were excluded. A schematic domain representation of FVIII is shown, indicating the location of the 40 residue stretches.
As expected, missense mutations at or near some of the thrombin activation sites have been reported to result in hemophilia A (Figure 4). Two different substitutions have been observed at position 1689 in a total of 35 mild to severe hemophilia A patients. Furthermore, there has been one report of a substitution at the P1′ position Ser1690 resulting in mild hemophilia. At position 372, four different missense mutations have been observed in a total of 22 reported cases of mild to severe hemophilia A, and missense mutations at P1′ Ser373 have been found in two mild hemophilia A patients. Remarkably, there have been no reports of missense mutations at or near position 740, which may suggest that mutations at this site do not result in hemophilia A.
Concluding remarks
The molecular process of maintaining FV and FVIII as inactive procofactors plays a critical regulatory role which has evolved to limit the expression of cofactor activity. Despite its significance, clear mechanistic insight by which the various proteolytic events lead to expression of FVa and FVIIIa procoagulant activity has proven difficult to pinpoint. Despite the similarities in structure and function of factors V and VIII, their molecular mechanisms of activation have been shown to differ substantially. For FV, the B domain plays a fundamentally important role as discrete conserved B domain sequences are involved in the mechanism by which FV persists as an inactive procofactor. The data suggest that the FV B domain serves an inhibitory function which, under normal physiological conditions, is efficiently removed upon proteolytic processing. In contrast, the FVIII B domain does not appear to be involved in regulating cofactor activity. Rather, cleavage between the A1 and A2 domains at position Arg372 is critical for the procoagulant activity of FVIII. The precise mechanism by which cleavage at Arg372 facilitates the transition to the active cofactor state remains to be determined. Collectively these studies lay the groundwork for further uncovering the precise molecular mechanism by which FV and FVIII transition from the procofactor to cofactor state.
Supplementary Material
Acknowledgments
Funding disclosure: Funding was provided by the National Institutes of Health (HL-88010 and HL-74124, Project 2; to RMC) and the National Hemophilia Foundation (Judith Graham Pool Postdoctoral Research Fellowship to MHAB).
Footnotes
Conflicts of Interest: RMC receives research support and royalties from Pfizer Pharmaceuticals for technology related to FXa. MHAB has no financial interest to disclose related to the contents of this article.
References
- 1.Khan AM, James MNG. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Prot Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Annual Rev Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 3.Kane W, Davie EW. Blood coagulation factors V and VIII: structural and functional similarities and their relationships to hemorrhagic and thrombotic disorders. Blood. 1988;71:539–555. [PubMed] [Google Scholar]
- 4.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor Proteins in the Assembly and Expression of Blood Clotting Enzyme Complexes. Ann Rev Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 5.Mann KG, Nesheim ME, Church WR, Haley P, Krishnaswamy S. Surface-Dependent Reactions of the Vitamin K-Dependent Enzyme Complexes. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 6.Jacquemin M, De Maeyer M, D'Oiron R, et al. Molecular mechanisms of mild and moderate hemophilia A. J Thromb Haemost. 2003;1:456–463. doi: 10.1046/j.1538-7836.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 7.Mannucci PM, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood. 2004;104:1243–1252. doi: 10.1182/blood-2004-02-0595. [DOI] [PubMed] [Google Scholar]
- 8.Ware AG, Guest MM, Seegers WH. Plasma Accelerator Factor and Purified Prothrombin Activation. Science. 1947;106(2741):41–42. doi: 10.1126/science.106.2741.41-a. [DOI] [PubMed] [Google Scholar]
- 9.Ware AG, Murphy RC, Seegers WH. The Function of Ac-Globulin in Blood Clotting. Science. 1947;106(2764):618–619. doi: 10.1126/science.106.2764.618-a. [DOI] [PubMed] [Google Scholar]
- 10.Owren PA. Parahaemophila: haemorrhagic diathesis due to absence of a previously unknown clotting factor. Lancet. 1947;249(6449):446–448. [PubMed] [Google Scholar]
- 11.Rapaport SI, Schiffman S, Patch MJ, Ames SB. The importance of activation of anti-haemophilic globulin and proaccelerin by traces of thrombin in the generation of intrinsic prothrombinase activity. Blood. 1963;21(2):221–236. [PubMed] [Google Scholar]
- 12.Mann KG, Kalafatis M. Factor V: A combination of Dr. Jekyll and Mr. Hyde. Blood. 2002;101:20–30. doi: 10.1182/blood-2002-01-0290. [DOI] [PubMed] [Google Scholar]
- 13.Fay PJ. Activation of factor VIII and mechanisms of cofactor action. Blood Reviews. 2004;18:1–15. doi: 10.1016/s0268-960x(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 14.Kane WH, Factor V. Hemostasis and Thrombosis. 5. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 177–92. [Google Scholar]
- 15.Tracy PB, Eide LL, Bowie EJW, Mann KG. Radioimmunoassay of factor V in human plasma and platelets. Blood. 1982;60:59–63. [PubMed] [Google Scholar]
- 16.Suzuki K, Dahlbäck B, Stenflo J. Thrombin-catalyzed activation of human coagulation factor V. J Biol Chem. 1982;257:6556–6564. [PubMed] [Google Scholar]
- 17.Esmon CT, Owen WG, Duiguid D, Jackson CM. The action of thrombin on blood clotting factor V: Conversion of factor V to a prothrombin-binding protein. Biochim Biophys Acta. 1973;310:289–294. doi: 10.1016/0005-2795(73)90034-2. [DOI] [PubMed] [Google Scholar]
- 18.Nesheim ME, Taswell JB, Mann KG. The contribution of bovine factor V and factor Va to the activity of prothrombinase. J Biol Chem. 1979;254:10952–10962. [PubMed] [Google Scholar]
- 19.Foster WB, Nesheim ME, Mann KG. The Factor Xa-catalyzed Activation of Factor V. J Biol Chem. 1983;258:13970–13977. [PubMed] [Google Scholar]
- 20.Jenny RJ, Pittman DD, Toole JJ, et al. Complete cDNA and derived amino acid sequence of human factor V. Proc Natl Acad Sci USA. 1987;84:4846–4850. doi: 10.1073/pnas.84.14.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Esmon CT. The subunit structure of thrombin-activated factor V. Isolation of activated factor V, separation of subunits and reconstitution of biological activity. J Biol Chem. 1979;254:964–973. [PubMed] [Google Scholar]
- 22.Nesheim ME, Mann KG. Thrombin-catalyzed activation of single chain bovine factor V. J Biol Chem. 1979;254:1326–1334. [PubMed] [Google Scholar]
- 23.Dharmawardana KR, Bock PE. Demonstration of exosite I-dependent interactions of thrombin with human factor V and factor Va involving the factor Va heavy chain: Analysis by affinity chromatography employing a novel method for active-site selective immobilization of serine proteinases. Biochemistry. 1998;37:13143–13152. doi: 10.1021/bi9812165. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki H, Shima M, Nogami K, et al. Factor V C2 domain contains a major thrombin-binding site responsible for thrombin-catalyzed factor V activation. J Thromb Haemost. 2006;4(6):1354–1360. doi: 10.1111/j.1538-7836.2006.01957.x. [DOI] [PubMed] [Google Scholar]
- 25.Krishnaswamy S, Russell GD, Mann KG. The reassociation of factor Va from its isolated subunits. J Biol Chem. 1989;264:3160–3168. [PubMed] [Google Scholar]
- 26.Nesheim ME, Foster WB, Hewick R, Mann KG. Characterization of factor V activation intermediates. J Biol Chem. 1984;259:3187–3196. [PubMed] [Google Scholar]
- 27.Monkovic DD, Tracy PB. Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J Biol Chem. 1990;265:17132–17140. [PubMed] [Google Scholar]
- 28.Thorelli E, Kaufman RJ, Dahlbäck B. Cleavage requirements for activation of factor V by factor Xa. Eur J Biochem. 1997;247:12–20. doi: 10.1111/j.1432-1033.1997.00012.x. [DOI] [PubMed] [Google Scholar]
- 29.Kane WH, Majerus PW. Purification and characterization of human coagulation factor V. J Biol Chem. 1981;256:1002–1007. [PubMed] [Google Scholar]
- 30.Segers K, Rosing J, Nicolaes GA. Structural models of the snake venom factor V activators from Daboia russelli and Daboia lebetina. Proteins. 2006;64(4):968–984. doi: 10.1002/prot.21051. [DOI] [PubMed] [Google Scholar]
- 31.Bakker HM, Tans G, Thomassen MCLGD, et al. Functional properties of human factor Va lacking the Asp683-Arg709 domain of the heavy chain. J Biol Chem. 1994;269:20662–20667. [PubMed] [Google Scholar]
- 32.Siigur J, Aaspollu A, Tonismagi K, et al. Proteases from Vipera lebetina venom affecting coagulation and fibrinolysis. Haemostasis. 2001;31(3-6):123–132. doi: 10.1159/000048055. [DOI] [PubMed] [Google Scholar]
- 33.Bradford HN, Annamalai A, Doshi K, Colman RW. Factor V is activated and cleaved by platelet calpain: Comparison with thrombin proteolysis. Blood. 1988;71:388–394. [PubMed] [Google Scholar]
- 34.Lee CD, Mann KG. Activation/Inactivation of human factor V by plasmin. Blood. 1989;73(1):185–190. [PubMed] [Google Scholar]
- 35.Kane WH, Mruk JS, Majerus PW. Activation of coagulation factor V by a platelet protease. J Clin Invest. 1982;70:1092–1100. doi: 10.1172/JCI110697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tracy PB, Nesheim ME, Mann KG. Proteolytic alteration of factor Va bound to platelets. J Biol Chem. 1983;258:662. [PubMed] [Google Scholar]
- 37.Tans G, Nicolaes GAF, Christella M, et al. Activation of human factor V by meizothrombin. J Biol Chem. 1994;269:15969–15972. [PubMed] [Google Scholar]
- 38.Oates AM, Salem HH. The regulation of human factor V by a neutrophil protease. Blood. 1987;70:846–851. [PubMed] [Google Scholar]
- 39.Allen DH, Tracy PB. Human coagulation factor V is activated to the functional cofactor by elastase and cathepsin G expressed at the monocyte surface. J Biol Chem. 1995;270:1408–1415. doi: 10.1074/jbc.270.3.1408. [DOI] [PubMed] [Google Scholar]
- 40.Samis JA, Garrett M, Manuel RP, Nesheim ME, Giles AR. Human neutrophil elastase activates human factor V but inactivates thrombin-activated human factor V. Blood. 1997;90:1065–1074. [PubMed] [Google Scholar]
- 41.Camire RM, Kalafatis M, Tracy PB. Proteolysis of factor V by cathepsin G and elastase indicates that cleavage at Arg1545 optimizes cofactor function by facilitating factor Xa binding. Biochemistry. 1998;37:11896–11906. doi: 10.1021/bi980520v. [DOI] [PubMed] [Google Scholar]
- 42.Esmon NL, Owen WG, Esmon CT. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of Protein C. J Biol Chem. 1982;257:859–864. [PubMed] [Google Scholar]
- 43.Keller FG, Ortel TL, Quinn-Allen MA, Kane WH. Thrombin-catalyzed activation of recombinant human factor V. Biochemistry. 1995;34:4118–4124. doi: 10.1021/bi00012a030. [DOI] [PubMed] [Google Scholar]
- 44.Steen M, Dahlbäck B. Thrombin-mediated proteolysis of factor V resulting in gradual B-domain release and exposure of the factor Xa-binding site. J Biol Chem. 2002;277:38424–38430. doi: 10.1074/jbc.M204972200. [DOI] [PubMed] [Google Scholar]
- 45.Smith CM, Hanahan DJ. The activation of factor V by factor Xa or α-chymotrypsin and comparison with thrombin and RVV-V action. An improved factor V isolation procedure. Biochemistry. 1976;15:1830–1837. doi: 10.1021/bi00654a007. [DOI] [PubMed] [Google Scholar]
- 46.Kane WH, Majerus PW. Purification and characterization of human coagulation factor V. J Biol Chem. 1981;256:1002–1007. [PubMed] [Google Scholar]
- 47.Kalafatis M, Beck DO, Mann KG. Structural requirements for expression of factor Va activity. J Biol Chem. 2004;278:33550–33561. doi: 10.1074/jbc.M303153200. [DOI] [PubMed] [Google Scholar]
- 48.Kane WH, Ichinose A, Hagen FS, Davie EW. Cloning of cDNAs coding for the heavy chain region and connecting region of human Factor V, a blood coagulation factor with four types of internal repeats. Biochemistry. 1987;26:6508. doi: 10.1021/bi00394a033. [DOI] [PubMed] [Google Scholar]
- 49.Mosesson MW, Church WR, DiOrio JP, et al. Structural model of factors V and Va based on scanning transmission electron microscope images and mass analysis. J Biol Chem. 1990;265(15):8863–8868. [PubMed] [Google Scholar]
- 50.Mosesson MW, Nesheim ME, DiOrio JP, et al. Studies on the structure of bovine factor V by scanning transmission electron microscopy. Blood. 1985;65(5):1158–1162. [PubMed] [Google Scholar]
- 51.Segers K, Dahlback B, Nicolaes GA. Coagulation factor V and thrombophilia: background and mechanisms. Thromb Haemost. 2007;98(3):530–542. [PubMed] [Google Scholar]
- 52.Kane WH, Devore-Carter D, Ortel TL. Expression and characterization of recombinant human factor V and a mutant lacking a major portion of the connecting region. Biochemistry. 1990;29:6762–6768. doi: 10.1021/bi00481a003. [DOI] [PubMed] [Google Scholar]
- 53.Toso R, Camire RM. Removal of B-domain sequences from factor V rather than specific proteolysis underlies the mechanism by which cofactor function is realized. J Biol Chem. 2004;279:21643–21650. doi: 10.1074/jbc.M402107200. [DOI] [PubMed] [Google Scholar]
- 54.Zhu H, Toso R, Camire RM. Inhibitory sequences within the B-domain stabilize circulating factor V in an inactive state. J Biol Chem. 2007;282(20):15033–15039. doi: 10.1074/jbc.M701315200. [DOI] [PubMed] [Google Scholar]
- 55.Camire RM, Bos MH. The molecular basis of factor V and VIII procofactor activation. J Thromb Haemost. 2009;7(12):1951–1961. doi: 10.1111/j.1538-7836.2009.03622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vos HL. Alignment of factor V sequences in twelve mammalian species shows several strongly conserved motifs in the B-domain. J Thromb Haemost. 2005;3(Supplement 1):P0041. [Google Scholar]
- 57.Vos HL, van Wijngaarden A. Variation and conservation of the B-domain of factor V. J Thromb Haemost. 2009;7(2):PP-MO-151. [Google Scholar]
- 58.Broad AJ, Sutherland SK, Coulter AR. The lethality in mice of dangerous Australian and other snake venom. Toxicon. 1979;17(6):661–664. doi: 10.1016/0041-0101(79)90245-9. [DOI] [PubMed] [Google Scholar]
- 59.Bos MH, Boltz M, St P L, et al. Venom factor V from the common brown snake escapes hemostatic regulation through procoagulant adaptations. Blood. 2009;114(3):686–692. doi: 10.1182/blood-2009-02-202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lavin MF, Masci PP. Prothrombinase complexes with different physiological roles. Thromb Haemost. 2009;102(3):421–423. doi: 10.1160/TH09-08-0050. [DOI] [PubMed] [Google Scholar]
- 61.Masci PP, Whitaker AN, de Jersey J. Purification and characterization of a prothrombin activator from the venom of the Australian brown snake, Pseudonaja textilis textilis. Biochem Int. 1988;17(5):825–835. [PubMed] [Google Scholar]
- 62.Rao VS, Kini RM. Pseutarin C, a prothrombin activator from Pseudonaja textilis venom: its structural and functional similarity to mammalian coagulation factor Xa-Va complex. Thromb Haemost. 2002;88(4):611–619. [PubMed] [Google Scholar]
- 63.Speijer H, Govers-Riemslag JW, Zwaal RF, Rosing J. Prothrombin activation by an activator from the venom of Oxyuranus scutellatus (Taipan snake) J Biol Chem. 1986;261(28):13258–13267. [PubMed] [Google Scholar]
- 64.Vos HL. Inherited defects of coagulation factor V: the thrombotic side. J Thromb Haemost. 2005;4:35–40. doi: 10.1111/j.1538-7836.2005.01572.x. [DOI] [PubMed] [Google Scholar]
- 65.Asselta R, Tenchini ML, Duga S. Inherited defects of coagulation factor V: the hemorrhagic side. J Thromb Haemost. 2006;4(1):26–34. doi: 10.1111/j.1538-7836.2005.01590.x. [DOI] [PubMed] [Google Scholar]
- 66.Vos HL. An online database of mutations and polymorphisms in and around the coagulation factor V gene. J Thromb Haemost. 2007;5(1):185–188. doi: 10.1111/j.1538-7836.2006.02258.x. [DOI] [PubMed] [Google Scholar]
- 67.Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 68.Greengard JS, Sun X, Xu X, et al. Activated protein C resistance caused by Arg506Gln mutation in factor Va. Lancet. 1994;343:1362–1363. doi: 10.1016/s0140-6736(94)92497-x. [DOI] [PubMed] [Google Scholar]
- 69.Sun X, Evatt B, Griffin JH. Blood coagulation factor Va abnormality associated with resistance to activated protein C in venous thrombophilia. Blood. 1994;83:3120–3125. [PubMed] [Google Scholar]
- 70.Voorberg J, Roelse J, Koopman R, et al. Association of idiopathic venous thromboembolism with single point-mutation at Arg506 of factor V. Lancet. 1994;343:1535–1538. doi: 10.1016/s0140-6736(94)92939-4. [DOI] [PubMed] [Google Scholar]
- 71.Zöller B, Svensson PJ, He X, Dahlbäck B. Identification of the same factor V gene mutation in 47 out of 50 thrombosis-prone families with inherited resistance to activated protein C. J Clin Invest. 1994;94:2521–2524. doi: 10.1172/JCI117623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koster T, Rosendaal FR, de Ronde H, et al. Venous thrombosis due to a poor anticoagulant response to activated protein C: Leiden Thrombophilia Study. Lancet. 1993;342:1503–1506. doi: 10.1016/s0140-6736(05)80081-9. [DOI] [PubMed] [Google Scholar]
- 73.Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk thrombosis in patients homozygous for factor V Leiden (activated protein C resistance) Blood. 1995;85:1504–1508. [PubMed] [Google Scholar]
- 74.Segers O, Castoldi E. Factor V Leiden and activated protein C resistance. Adv Clin Chem. 2009;49:121–157. doi: 10.1016/s0065-2423(09)49006-1. [DOI] [PubMed] [Google Scholar]
- 75.Duckers C, Simioni P, Rosing J, Castoldi E. Advances in understanding the bleeding diathesis in factor V deficiency. Br J Haematol. 2009;146(1):17–26. doi: 10.1111/j.1365-2141.2009.07708.x. [DOI] [PubMed] [Google Scholar]
- 76.Lunghi B, Pinotti M, Maestri I, Batorova A, Bernardi F. Evaluation of factor V mRNA to define the residual factor V expression levels in severe factor V deficiency. Haematologica. 2008;93(3):477–478. doi: 10.3324/haematol.11952. [DOI] [PubMed] [Google Scholar]
- 77.Ellestad SC, Zimmerman SA, Thornburg C, et al. Severe factor V deficiency presenting with intracranial haemorrhage during gestation. Haemophilia. 2007;13(4):432–434. doi: 10.1111/j.1365-2516.2007.01469.x. [DOI] [PubMed] [Google Scholar]
- 78.Guasch JF, Cannegieter S, Reitsma PH, van't Veer-Korthof E. Severe coagulation factor V deficiency caused by a 4 bp deletion in the factor V gene. Br J Haematol. 1998;101:32–39. doi: 10.1046/j.1365-2141.1998.00664.x. [DOI] [PubMed] [Google Scholar]
- 79.Lak M, Sharifian R, Peyvandi F, Mannucci PM. Symptoms of inherited factor V deficiency in 35 Iranian patients. Br J Haematol. 1998;103(4):1067–1069. doi: 10.1046/j.1365-2141.1998.01077.x. [DOI] [PubMed] [Google Scholar]
- 80.Salooja N, Martin P, Khair K, Liesner R, Hann I. Severe factor V deficiency and neonatal intracranial haemorrhage: a case report. Haemophilia. 2000;6(1):44–46. doi: 10.1046/j.1365-2516.2000.00362.x. [DOI] [PubMed] [Google Scholar]
- 81.Van WR, Montefusco MC, Duga S, et al. Coexistence of a novel homozygous nonsense mutation in exon 13 of the factor V gene with the homozygous Leiden mutation in two unrelated patients with severe factor V deficiency. Br J Haematol. 2001;114(4):871–874. doi: 10.1046/j.1365-2141.2001.03016.x. [DOI] [PubMed] [Google Scholar]
- 82.Cui J, O'Shea KS, Purkayastha A, Saunders TL, Ginsburg D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature. 1996;384:66–68. doi: 10.1038/384066a0. [DOI] [PubMed] [Google Scholar]
- 83.Gewirtz AM, Keefer M, Doshi K, et al. Biology of human megakaryocyte Factor V. Blood. 1986;67:1639–1642. [PubMed] [Google Scholar]
- 84.Chiu HC, Schick P, Colman RW. Biosynthesis of Factor V in isolated guinea pig megakaryocytes. J Clin Invest. 1985;75:339–346. doi: 10.1172/JCI111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giampaolo A, Vulcano F, Macioce G, et al. Factor-V expression in platelets from human megakaryocytic culture. Br J Haematol. 2005;128(1):108–111. doi: 10.1111/j.1365-2141.2004.05279.x. [DOI] [PubMed] [Google Scholar]
- 86.Camire RM, Pollak ES, Kaushansky K, Tracy PB. Secretable human platelet-derived factor V originates from the plasma pool. Blood. 1998;92:3035–3041. [PubMed] [Google Scholar]
- 87.Christella M, Thomassen LG, Castoldi E, et al. Endogenous factor V synthesis in megakaryocytes contributes negligibly to the platelet factor V pool. Haematologica. 2003;88(10):1150–1156. [PubMed] [Google Scholar]
- 88.Gould WR, Simioni P, Silveira JR, et al. Megakaryocytes endocytose and subsequently modify human factor V in vivo to form the entire pool of a unique platelet-derived cofactor. J Thromb Haemost. 2005;3(3):450–456. doi: 10.1111/j.1538-7836.2005.01157.x. [DOI] [PubMed] [Google Scholar]
- 89.Bouchard BA, Williams JL, Meisler NT, Long MW, Tracy PB. Endocytosis of plasma-derived factor V by megakaryocytes occurs via a clathrin-dependent, specific membrane binding event. J Thromb Haemost. 2005;3(3):541–551. doi: 10.1111/j.1538-7836.2005.01190.x. [DOI] [PubMed] [Google Scholar]
- 90.Bouchard BA, Meisler NT, Nesheim ME, et al. A unique function for LRP-1: a component of a two-receptor system mediating specific endocytosis of plasma-derived factor V by megakaryocytes. J Thromb Haemost. 2008;6(4):638–644. doi: 10.1111/j.1538-7836.2008.02894.x. [DOI] [PubMed] [Google Scholar]
- 91.Gould WR, Silveira JR, Tracy PB. Unique in vivo modifications of coagulation factor V produce a physically and functionally distinct platelet-derived cofactor: characterization of purified platelet-derived factor V/Va. J Biol Chem. 2004;279(4):2383–2393. doi: 10.1074/jbc.M308600200. [DOI] [PubMed] [Google Scholar]
- 92.Duckers C, Simioni P, Spiezia L, et al. Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood. 2009 doi: 10.1182/blood-2009-08-237719. [DOI] [PubMed] [Google Scholar]
- 93.Duckers C, Simioni P, Spiezia L, et al. Low plasma levels of tissue factor pathway inhibitor in patients with congenital factor V deficiency. Blood. 2008;112(9):3615–3623. doi: 10.1182/blood-2008-06-162453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cripe LD, Moore D, Kane WH. Structure of the gene for human coagulation factor V. Biochemistry. 1992;31:3777–3785. doi: 10.1021/bi00130a007. [DOI] [PubMed] [Google Scholar]
- 95.Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. Nature. 1984;312:326–330. doi: 10.1038/312326a0. [DOI] [PubMed] [Google Scholar]
- 96.Koschinsky ML, Funk WD, van Oost BA, MacGillivray RT. Complete cDNA sequence of human preceruloplasmin. Proc Natl Acad Sci U S A. 1986;83(14):5086–5090. doi: 10.1073/pnas.83.14.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davidson CJ, Tuddenham EG, McVey JH. 450 million years of hemostasis. J Thromb Haemost. 2003;1(7):1487–1494. doi: 10.1046/j.1538-7836.2003.00334.x. [DOI] [PubMed] [Google Scholar]
- 98.Jiang Y, Doolittle RF. The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes. Proc Natl Acad Sci U S A. 2003;100(13):7527–7532. doi: 10.1073/pnas.0932632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Newell JL, Fay PJ. Acidic residues C-terminal to the A2 domain facilitate thrombin-catalyzed activation of factor VIII. Biochemistry. 2008;47(33):8786–8795. doi: 10.1021/bi8007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nogami K, Zhou Q, Myles T, et al. Exosite-interactive regions in the A1 and A2 domains of factor VIII facilitate thrombin-catalyzed cleavage of heavy chain. J Biol Chem. 2005;280(18):18476–18487. doi: 10.1074/jbc.M412778200. [DOI] [PubMed] [Google Scholar]
- 101.Jacquemin M, Lavend'homme R, Benhida A, et al. A novel cause of mild/moderate hemophilia A: mutations scattered in the factor VIII C1 domain reduce factor VIII binding to von Willebrand factor. Blood. 2000;96(3):958–965. [PubMed] [Google Scholar]
- 102.Leyte A, Verbeet MP, Brodniewicz-Proba T, Van Mourik JA, Mertens K. The interaction between human blood-coagulation factor VIII and von Willebrand factor. Characterization of a high-affinity binding site on factor VIII. Biochem J. 1989;257(3):679–683. doi: 10.1042/bj2570679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lollar P, Hill-Eubanks DC, Parker CG. Association of the factor VIII light chain with von Willebrand factor. J Biol Chem. 1988;263:10451–10455. [PubMed] [Google Scholar]
- 104.Saenko EL, Scandella D. The acidic region of the factor VIII light chain and the C2 domain together form the high affinity binding site for von willebrand factor. J Biol Chem. 1997;272(29):18007–18014. doi: 10.1074/jbc.272.29.18007. [DOI] [PubMed] [Google Scholar]
- 105.Lenting PJ, Van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92(11):3983–3996. [PubMed] [Google Scholar]
- 106.Vehar GA, Davie EW. Preparation and properties of bovine factor VIII (antihemophilic factor) Biochemistry. 1980;19(3):401–410. doi: 10.1021/bi00544a001. [DOI] [PubMed] [Google Scholar]
- 107.Fass DN, Knutson GJ, Katzmann JA. Monoclonal antibodies to porcine factor VIII coagulant and their use in the isolation of active coagulant protein. Blood. 1982;59:594–600. [PubMed] [Google Scholar]
- 108.Fulcher CA, Roberts J, Zimmerman TS. Thrombin proteolysis of purified FVIII procoagulant protein: Correlation of activation with generation of specific polypeptides. Blood. 1983;60:807. [PubMed] [Google Scholar]
- 109.Lollar P, Knutson G, Fass D. Stabilisation of thrombin activated porcine Factor VIII:C by Factor IXa and phospholipid. Blood. 1984;63:1303–1308. [PubMed] [Google Scholar]
- 110.Toole JJ, Knopf JL, Wozney JM, et al. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312:342–347. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- 111.Wood WI, Capon DJ, Simonsen CC, et al. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312:330–336. doi: 10.1038/312330a0. [DOI] [PubMed] [Google Scholar]
- 112.Eaton D, Rodriguez H, Vehar G. Proteolytic processing of human FVIII. Correlation of specific cleavages by thrombin FXa and activated protein C with activation and inactivation of Factor VIII coagulant activity. Biochemistry. 1986;25:505–512. doi: 10.1021/bi00350a035. [DOI] [PubMed] [Google Scholar]
- 113.Vehar G, Keyt B, Eaton D, et al. Structure of human factor VIII. Nature. 1984;312:337–342. doi: 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]
- 114.Lollar P, Parker C. pH-dependent denaturation of thrombin-activated porcine factor VIII. J Biol Chem. 1990;265:1688–1692. [PubMed] [Google Scholar]
- 115.Fay PJ, Haidari PJ, Smudzin TM. Human factor VIIIa subunit structure. Reconstitution of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem. 1991;266:8957–8962. [PubMed] [Google Scholar]
- 116.Lollar P, Parker ET. Structural basis for the decreased procoagulant activity of human factor VIII compared to the porcine homolog. J Biol Chem. 1991;266(19):12481–12486. [PubMed] [Google Scholar]
- 117.Pittman DD, Kaufman RJ. Proteolytic requirements for thrombin activation of anti-hemophilic factor (factor VIII) Proc Natl Acad Sci USA. 1988;85:2429–2433. doi: 10.1073/pnas.85.8.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Newell JL, Fay PJ. Proteolysis at Arg740 facilitates subsequent bond cleavages during thrombin-catalyzed activation of factor VIII. J Biol Chem. 2007;282(35):25367–25375. doi: 10.1074/jbc.M703433200. [DOI] [PubMed] [Google Scholar]
- 119.Hamer RJ, Koedam JA, Beeser-Visser NH, et al. Factor VIII binds to von Willebrand factor via its Mr-80,000 light chain. Eur J Biochem. 1987;166:37–43. doi: 10.1111/j.1432-1033.1987.tb13480.x. [DOI] [PubMed] [Google Scholar]
- 120.Hill-Eubanks DC, Parker CG, Lollar P. Differential proteolytic activation of factor VIII-von Willebrand factor complex by thrombin. Proc Natl Acad Sci USA. 1989;86:6508–6512. doi: 10.1073/pnas.86.17.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Regan LM, Fay PJ. Cleavage of factor VIII light chain is required for maximal generation of factor VIIIa activity. J Biol Chem. 1995;270:8546–8552. doi: 10.1074/jbc.270.15.8546. [DOI] [PubMed] [Google Scholar]
- 122.Donath MJSH, Lenting PJ, Van Mourik JA, Mertens K. The role of cleavage of the light chain at positions Arg1689 or Arg1721 in subunit interactions and activation of human blood coagulation factor VIII. J Biol Chem. 1995;270:3648–3655. doi: 10.1074/jbc.270.8.3648. [DOI] [PubMed] [Google Scholar]
- 123.Pipe SW, Kaufman RJ. Characterization of genetically engineered inactivation-resistant coagulation factor VIIIa. Proc Natl Acad Sci USA. 1997;94:11851–11856. doi: 10.1073/pnas.94.22.11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shima M, Ware J, Yoshioka A, Fukui H, Fulcher CA. An arginine to cysteine amino acid substitution at a critical thrombin cleavage site in dysfunctional factor VIII molecule. Blood. 1989;74:1612–1617. [PubMed] [Google Scholar]
- 125.Arai M, Inaba H, Higuchi M, et al. Direct characterization of factor VIII in plasma: Detection of a mutation altering a thrombin cleavage site (arginine-372 to histidine) Proc Natl Acad Sci USA. 1989;86:4277–4281. doi: 10.1073/pnas.86.11.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.O'Brien DP, Pattinson JK, Tuddenham EGD. Purification and characterization of factor VIII 372-Cys: A hypofunctional cofactor from a patient with moderately severe haemophilia A. Blood. 1990;75:1664–1672. [PubMed] [Google Scholar]
- 127.Nogami K, Zhou Q, Wakabayashi H, Fay PJ. Thrombin-catalyzed activation of factor VIII with His substituted for Arg372 at the P1 site. Blood. 2005;105(11):4362–4368. doi: 10.1182/blood-2004-10-3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fay PJ, Mastri M, Koszelak ME, Wakabayashi H. Cleavage of factor VIII heavy chain is required for the functional interaction of a2 subunit with factor IXa. J Biol Chem. 2001;276(15):12434–12439. doi: 10.1074/jbc.M009539200. [DOI] [PubMed] [Google Scholar]
- 129.Shen BW, Spiegel PC, Chang CH, et al. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111(3):1240–1247. doi: 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ngo JC, Huang M, Roth DA, Furie BC, Furie B. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure. 2008;16(4):597–606. doi: 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 131.Curtis JE, Helgerson SL, Parker ET, Lollar P. Isolation and characterization of thrombin-activated human factor VIII. J Biol Chem. 1994;269(8):6246–6251. [PubMed] [Google Scholar]
- 132.O'Brien LM, Huggins CF, Fay PJ. Interacting regions in the A1 and A2 subunits of factor VIIIa identified by zero-length cross-linking. Blood. 1997;90(10):3943–3950. [PubMed] [Google Scholar]
- 133.Sudhakar K, Fay PJ. Exposed hydrophobic sites in factor VIII and isolated subunits. J Biol Chem. 1996;271(38):23015–23021. doi: 10.1074/jbc.271.38.23015. [DOI] [PubMed] [Google Scholar]
- 134.Toole JJ, Pittman DD, Orr EC, et al. A large region (∼95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci USA. 1986;83:5939–5942. doi: 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eaton DL, Wood WI, Eaton D, et al. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry. 1986;25:8343–8347. doi: 10.1021/bi00374a001. [DOI] [PubMed] [Google Scholar]
- 136.Pittman DD, Alderman EM, Tomkinson KN, et al. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted FVIII. Blood. 1993;81(11):2925–2935. [PubMed] [Google Scholar]
- 137.Lind P, Larsson K, Spira J, et al. Novel forms of B-domain deleted recombinant factor VIII molecules. Construction and biochemical characterization. Eur J Biochem. 1995;232:19–27. doi: 10.1111/j.1432-1033.1995.tb20776.x. [DOI] [PubMed] [Google Scholar]
- 138.Roelse JC, Koopman MM, Buller HR, et al. Absence of mutations at the activated protein C cleavage sites of factor VIII in 125 patients with venous thrombosis. Br J Haematol. 1996;92(3):740–743. doi: 10.1046/j.1365-2141.1996.349885.x. [DOI] [PubMed] [Google Scholar]
- 139.Amano K, Michnick DA, Moussalli M, Kaufman RJ. Mutation at either Arg336 or Arg562 in factor VIII is insufficient for complete resistance to activated protein C-mediated inactivation: Implications for the APC resistance test. Thromb Haemost. 1998;79:557–563. [PubMed] [Google Scholar]
- 140.Kamphuisen PW, Eikenboom JC, Bertina RM. Elevated factor VIII levels and the risk of thrombosis. Arterioscler Thromb Vasc Biol. 2001;21(5):731–738. doi: 10.1161/01.atv.21.5.731. [DOI] [PubMed] [Google Scholar]
- 141.Graw J, Brackmann HH, Oldenburg J, et al. Haemophilia A: from mutation analysis to new therapies. Nat Rev Genet. 2005;6(6):488–501. doi: 10.1038/nrg1617. [DOI] [PubMed] [Google Scholar]
- 142.Lakich D, Kazazian HH, Antonarakis SE, Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nature Genet. 1993;5:236–241. doi: 10.1038/ng1193-236. [DOI] [PubMed] [Google Scholar]
- 143.Naylor J, Brinke A, Hassock S, Green PM, Giannelli F. Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet. 1993;2(11):1773–1778. doi: 10.1093/hmg/2.11.1773. [DOI] [PubMed] [Google Scholar]
- 144.Oldenburg J, Ananyeva NM, Saenko EL. Molecular basis of haemophilia A. Haemophilia. 2004;10 4:133–139. doi: 10.1111/j.1365-2516.2004.01005.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.