

Abstract
Harmful algal blooms (HABs) are a serious public health risk in coastal waters. As the intensity and frequency of HABs continue to rise, new methods of detection are needed for reliable identification. Herein, we developed a high-throughput, multiplex, bead array technique for the detection of the dinoflagellates Karenia brevis and Karenia mikimotoi. The method combined the Luminex detection system with two novel technologies: locked nucleic acid–modified oligonucleotides (LNA) and Mirus Label IT® nucleic acid technology. To study the feasibility of the method, we evaluated the performance of modified and unmodified LNA probes with amplicon targets that were biotin labeled with two different strategies: direct chemical labeling (Mirus Label IT) versus enzymatic end-labeling (single biotinylated primer). The results illustrated that LNA probes hybridized to complementary single-stranded DNA with better affinity and displayed higher fluorescence intensities than unmodified oligonucleotide DNA probes. The latter effect was more pronounced when the assay was carried out at temperatures above 53°C degree. As opposed to the enzymatic 5′ terminal labeling technique, the chemical-labeling method enhanced the level of fluorescence by as much as ~83%. The detection limits of the assay, which were established with LNA probes and Mirus Label IT system, ranged from 0.05 to 46 copies of rRNA. This high-throughput method, which represents the first molecular detection strategy to integrate Luminex technology with LNA probes and Mirus Label IT, can be adapted for the detection of other HABs and is well suited for the monitoring of red tides at pre-blooming and blooming conditions.
Harmful algal bloom (HABs) events are a threat to public health, marine life, commercial fisheries, tourism, recreation, and the environment. These outbreaks are known to decrease water quality by increasing the biological O2 demand, which can cause die-offs of marine life (Figley et al. 1979). Expanded spatial distribution of HABs can cover over a thousand kilometers and are triggered by the effects of winds, currents, storms, and ship ballast activities (Tester and Steidinger 1997; Doblin et al. 2004; Bolch and Salas 2007). Increases in HAB frequency and duration are probably fueled by changes in sea surface temperature, salinity, grazing, turbulence, irradiance, or increases in nutrient loads derived from run off from agricultural and urban areas (Reynolds 1989; Sterner 1989; Thomas and Gibson 1990; Anderson et al. 2002; Xiaonan and Wei 2004).
HABs are formed by species of phytoplankton or cyanobacteria, which can produce toxins that can be transferred through marine food webs, affecting consumers at higher trophic levels including fish, shellfish, marine mammals, and humans (Sellner et al. 2003; Smayda 1997). Exposure to HABs toxins in water, aerosols, or seafood can cause a myriad of poisoning syndromes in humans, i.e., paralytic shellfish poisoning (PSP), diarrhetic shellfish poisoning (DSP), neurotoxic shellfish poisoning (NSP), and azaspiracid shellfish poisoning (AZSP). Some of these syndromes, like NSP, can result in neurological disorders associated to a group of polyethers called brevetoxins, which are produced by the unarmored dinoflagellate, Karenia brevis (Anderson 1989; Anderson 1997; Hackett et al. 2004). Symptoms of NSP include paresthesia (reversal of hot-cold temperature sensation), myalgia (muscle pain), vertigo, ataxia (lack of coordination of muscle movements), abdominal pain, diarrhea, headache, and bradycardia (slow heart rate). When sufficient cell density and wind conditions are present (>1000 cells−L), aerosolized brevetoxin can cause respiratory distress (Kirkpatrick et al. 2003).
Karenia brevis outbreaks have been associated with mass mortalities of fish, marine mammals, birds, and sea turtles in the Gulf of Mexico and occasional blooms off the southeastern coast of the United States (Tester and Steidinger 1997; Steidinger et al. 1998; Landsberg 2002). For example, during 1946–1947, catastrophic mortalities of bottlenose dolphins, sea turtles, and numerous fish species were reported along a 150-mile stretch from Tarpon Springs to Key West, FL. This unprecedented event is to date one of the largest K. brevis red tide events in Florida waters (Gunter et al. 1948; Galtsoff 1948).
HABs blooms have caused substantial economic losses to coastal communities and commercial fisheries, with an estimated loss of $50 million dollars per year in the United States (Turgeon et al. 1998; Hoagland et al. 2002). Economic effects of single outbreaks have ranged between $7-$25 million dollars and have closed more than 400 km coastline (Shumway 1988; Tester et al. 1991).
Monitoring techniques for HAB communities are primarily based on microscopic identification, which is rather tedious and requires taxonomic expertise, because it relies on morphological characters that, in some cases, are insufficient for species level identification (Culverhouse et al. 2003; Taylor 1993). Photopigment analysis also has been used to monitor these events but, as in microscopy, the method lacks the capability to differentiate between closely related taxa (Millie et al. 1997). Current monitoring programs are based on the microscopic counts of K. brevis in the water sample along with measurement of shellfish toxicity. Although, this synergistic approach has proven to be successful, restricted sampling, and sometimes inaccurate identification have led to a limited view of the extent, composition, and dynamics of HABs. Thus, to improve our present early warning capabilities and to better protect human health and economic interests, new methods that allow detection before the onset of a bloom need to be explored. In the present study, we developed an array for the detection and identification of HABs, K. brevis and its closely related HABs species K. mikimotoi. The method combined xMAP multiplexing bead technology, a flow cytometer that allows simultaneous detection of up to 100 different analytes in a single reaction, and locked nucleic acid-modified capture probes (LNA). These nucleic acid probes contain specific nucleosides that bear a methylene bridge connecting the 2′-oxygen of the ribose with the 4′carbon. This bridge creates a locked 3′endo conformation that reduces the conformational flexibility of the ribose. The reduced flexibility promotes hybridization affinity of the oligonucleotides for their complementary targets (Simeonov and Nikiforov 2002) and significantly enhances the thermal stability (Koshkin et al. 1998). This study documented the performance of LNA-modified and unmodified probes when challenged with amplicon targets that were biotin labeled with two different strategies: direct chemical labeling (Mirus Label IT) versus enzymatic end-labeling (single biotinylated primer). The applicability of both type of probes and various labeling strategies were evaluated with a collection of field samples.
Materials and procedures
Strains studied and field samples
Voucher specimens (Table 1) were obtained from various culture collections: the Provasoli-Guillard Center for the Culture of Marine Phytoplankton (CCMP) and the Algae collection at Florida International University (FIU). Strains EPA-JR and JR1 were obtained from the United States Environmental Protection Agency Gulf Ecology Division Laboratory in Gulf Breeze, Florida. Cells were pelleted or filtered onto 5 μm, 47 mm mixed esters of cellulose (MEC) membrane filters (Millipore) and DNA was extracted using the FastDNA SPIN Kit for Soil (Qbiogene) and FastPrep Instrument (Qbiogene).
Table 1.
List of the strains used in this study.
| Organism | Strain | Origin |
|---|---|---|
| Alexandrium tamarense | CCMP 1493 | Da Ya Bay, China |
| Amphidinium carterae | CCMP 1314 | Falmouth, Massachusetts, USA |
| Amphidinium operculatum | CCMP 1344 | Knight Key, Florida, USA |
| Chattonella subsalsa | CCMP 1770 | West Boothbay Harbor, Maine, USA |
| Fragilidium sp. | KRl 1205 | Florida, USA |
| Gymnodinium sp. | KR 57 (FIU) | Florida USA |
| Gonyaulax cochlea | CCMP 1592 | Narragansett Bay, Rhode Island, USA |
| Heterocapsa triquetra | CCMP 1344 | Knight Key, Florida, USA |
| Heterosigma akashivo | KR 24 (FIU) | Florida, USA |
| Karenia brevis | CCMP 430 | Unknown |
| Karenia brevis | EPA-JR | Florida, USA |
| Karenia brevis | JR-1 | Pensacola, Florida, USA |
| Karenia brevis | Wilson | John’s Pass, Florida, USA |
| Karenia brevis | KR 28 (FIU) | Florida, USA |
| Karenia mikimotoi | CCMP 429 | Sutton Harbour, Plymouth, England |
| Karlodinium micrum | KR 41 (FIU)/CCMP 2778 | Mary Lake, Minnnesota, USA |
| Prorocentrum hoffmannianum | CCMP 683 | Knight Key, Florida, USA |
| Prorocentrum lima | CCMP 1368 | Knight Key, Florida, USA |
| Prorocentrum lima | PMIRGNA | Unknown |
| Prorocentrum mexicanum | CCMP 687 | Knight Key, Florida, USA |
| Prorocentrum micans | CCMP 1591 | Narragansett Bay, Rhode Island, USA |
| Prorocentrum minimum | CCMP 695 | Everglades, Florida, USA |
| Protoceratium reticulatum | KR 30 (FIU)/CCMP 2810 | Marquesas Keys, Florida, USA |
| Protoceratium reticulatum | KR 38 (FIU) | Florida, USA |
| Prorocentrum rhathymum | KR 9 (FIU) | Florida, USA |
| Pyrmnesium parvum | CCMP 708 | Millport, Isle of Cumbrae, Scotland |
| Scrippsiella sp. | KR 30/ CCMP 2810 (FIU) | Marquesas Keys, Florida, USA |
| Symbiodinium sp. | CCMP 831 | Great Barrier Reef, Australia |
Field sample analyses were conducted with an archived collection of samples derived from previous studies (Goodwin et al. 2005; LaGier et al. 2007). The samples were obtained from the Rookery Bay National Estuarine Research Reserve in Naples, Florida, and included the collection sites: Caxambas Pass, Hendersen’s Creek and Marco Pass. These water samples were collected from February 2002 through October 2003. Other field samples were collected during a 2002 red tide event in Tampa Bay area. The collection sites from this “blackwater” event included New Pass and Siesta Beach. The water samples were collected in sterile whirlpacks and were vacuum-filtered onto mixed esters of MEC filters. The filters were shipped to the National Oceanic and Atmospheric Administration (NOAA) Atlantic Oceanographic and Meteorological Laboratories (AOML) and were frozen at −20°C until used. DNA extraction used BIO 101 (Qbiogene) or FastDNA SPIN kit (Qbiogene). Detailed information about sampling and filter-handling procedures can be found in Goodwin et al. (2005). The samples were characterized and enumerated following the parameters established by the Fish and Wildlife Research Institute (FWRI). The FWRI classification system characterizes K. brevis pre-blooming and blooming conditions based on microscopy counts: not present, present (≤1,000 cells L−1), very low a (>1,000 to <5,000 cells L−1), very low b (5,000 to 10,000 cells L−1), low a (>10,000 to <50,000 cells L−1), low b (50,000 to <100,000 cells L−1), medium (100,000 to <106 cells L−1), and high (≥106 cells L−1).
Probe design and probe coupling
A sequence alignment of D1/D2 domains of the large-subunit rDNA (DNAstar Megalign) was constructed to facilitate probe design for the dinoflagellates, K. brevis and K. mikimotoi. This database, which comprises in-house and public domain sequences, was expanded to include over 120 dinoflagellate species. Areas displaying sequence divergence among the species were selected for the design of locked nucleic acid (LNA) and their nonmodified probe versions. A list of probe sequences is shown in Table 2. All LNA and nonmodified conventional probes were synthesized by Integrated DNA Technologies. LNA probes were synthesized with 3 LNA residues, which were strategically located in central areas displaying polymorphic sites. An exception was Pkmikilna2, which contained two consecutive interspersed residues flanked by two single interspersed LNA bases. LNA probes were designed to be 19 nucleotides long with melting temperature (Tm) values ranging from 57 to 68°C. Unmodified, conventional probes displayed similar sequence content as LNA probes but without LNA residues. Unmodified probe versions displayed Tm values ranging from 52.6 to 61°C. Thermodynamics of the unmodified probes were established using the software program OligoTM (Molecular Biology Insights), whereas LNA probes used the software tool obtained from Exiqon (http://www.exiqon.com). The specificity of the probes was performed with GenBank BLAST. The probes were synthesized with a 5′end amino C12 modification with standard desalting purification (IDT). The amino modification facilitated the coupling of the oligonucleotide probe sequence to the carboxylated surface of the microspheres. Coupling of the probes to unique sets of 5.6 μm polystyrene carboxylated microspheres followed the carbodiimide method (Fulton et al. 1997) with slight modifications (Diaz and Fell 2004). Coupling reactions were undertaken at a concentration of 0.2 nmol.
Table 2.
List of the probe sequences designed for the study. LNA-modified bases are represented in lower case letters.
| Target | Probe | Sequence |
|---|---|---|
| K. brevis | Pkb2lna | 5′-TGCTTCCGGgcgACTGAAT-3′ |
| K. brevis | Pkb658lna | 5′-GTCTGGtcgCACTGCTCCG-3′ |
| K. mikimotoi | Pkmikilna | 5′-CGCTTCCgagTGACTGAAT-3′ |
| K. mikimotoi | Pkmikilna2 | 5′-CGCTTcCgaGtGACTGAAT-3′ |
| K. brevis | Pkb2 | 5′-TGCTTCCGGGCGACTGAAT-3′ |
| K. brevis | Pkb658 | 5′-GTCTGGTCGCACTGCTCCG-3′ |
| K. mikimotoi | Pkmiki* | 5′-CGCTTCCGAGTGACTGAAT-3′ |
| K. brevis | Pkb* | 5′-TGTTGTCTAAGGTGATAGCTTGC-3′ |
Probes designed by Scorzetti et al. 2008.
PCR reactions
Amplified products from the large sub unit D1/D2 (LrRNA) region (~670 bp) were obtained using the universal forward primer D1R, (5′-ACCGCTGAATTTAAGCATA-3′) and the universal reverse primer, D2C (5′-CCTTGGTCCG TGTTTCAAGA-3′). Additionally, we designed a specific forward primer, Kr130 (5′-CTTGAATTGTAGTCTTGAGATGTGT TA-3′) to selectively amplify species within the Karenia cluster group. Kr130 was located 130 bp downstream from DIR, and generated amplicon sizes of ~540 bp. Enzymatic end-labeling reactions used a 5′end biotin-labeled D2C reverse primer, whereas chemical-labeling reactions (Mirus Label IT) used nonbiotinylated, D2C primer. Targets were amplified by standard PCR reaction and used Qiagen HotStarTaq Master Mix (QIAGEN Inc) in final volumes of 25 or 50 μL. The master mix contained template DNA ranging from 50 ng to 1 fg, 1.5 mM MgCl2, 0.4 μM of forward and reverse primer pairs, 2.5 units of HotStarTaq polymerase, dNTPs containing 200 μM each of dGTP, dCTP, dTTP, and dATP. Field samples used 1 μL to 2 μL of environmental genomic DNA suspension in a 50 μL reaction. PCR reactions were performed with an MJ Research PTC 100 thermocycler and used cycle numbers ranging from 30 to 40 cycles. The PCR program used 15 min of initial activation at 95°C, 30 s denaturating at 95°C, 30 s annealing at 50°C, and 1 min extension at 69°C. A final extension step was carried out at 69°C for 25 min. Amplicons were confirmed by agarose gel electrophoresis. Each run included two sets of negative controls: a blank (all chemicals except PCR amplicon) and a PCR negative control (water and PCR reagents).
Biotinylation of amplicons using Mirus Label IT
Nonbiotinylated amplicons were cleaned with QIAquick PCR purification kit (Qiagen) and DNA concentration was determined with a NanoDrop ND-1000 spectrophotometer with the absorbance at 260 nm. After purification, the amplicons were biotin labeled with the Mirus Label IT kit (Mirus, Madison, WI).
The labeling reactions were incubated in the dark at 37°C for 3 h and used a final volume of 20 μL and a 1:2 ratio of nucleic acid to labeling reagent. Pure cultures used 50 to 100 ng of PCR amplicon. Field samples used 2.5 to 5 μL of unlabeled amplicons (10 ng to 275 ng). After the labeling reaction, the samples were denatured using the manufacturer’s protocol.
To adjust the labeling density, several optimization steps that involved different ratios of Label IT biotin reagent to nucleic acid (1 μL to 4 μL of a 1:10 dilution of the Label IT reagent to 100 ng DNA) as well as different incubation times (1.5 h to 4 h) were assessed. To determine the optimal labeling efficiency under the described labeling conditions, 100 ng biotinylated targets were hybridized for 15 min at 55°C with their complementary probe sequence. The labeling efficiency was measured using the median fluorescence intensity (MFI) signals generated following the hybridization.
Hybridization assay
Hybridization reactions carried out in 96 well plates, used a 3M TMAC (tetramethyl ammonium chloride, 50 mM Tris, pH 8.0, 4 mM EDTA, pH 8.0, 0.1% sarkosyl) solution (Sigma). The hybridization reactions typically used 50 ng of biotinylated PCR followed by 12 μL of 1 × TE buffer (pH 8) and 33 μL of each of the labeled beads diluted in 1.5 × TMAC solution. Unless otherwise specified, field samples used 5 to 15 μL of labeled amplicon. The bead mixtures consisted of ~2500 microspheres for each set of probes. Prior to hybridization, the samples were denatured for 5 min at 95°C with a PTC-100 Thermocycler (MJ Research). After denaturing, the reaction mixture was incubated for 1.5 h at temperatures ranging from 50 to 56°C. The beads were pelleted by centrifugation at 2250g (3,700 rpm) for 3 min, and the unbound PCR products were carefully removed. The hybridized amplicons were incubated for 7 min at the specified hybridization temperature (50° to 56°C) with 300 ng of freshly made streptavidin-R-phycoerythrin (SAPE). The samples were centrifuged and washed with 75 μL of 1 × TMAC. The microsphere fluorescence was analyzed on the Luminex xMAP flow cytometer. Each assay was run twice. A blank and a set of positive and negative controls were included in each assay.
To test the detection limits of the Luminex assay, several tests were conducted with serial dilutions of genomic DNA (1 ng to 0.0001 pg) and amplicons (50 ng to 0.01 ng) labeled by the two methods. Detection limits were also determined at different temperature ranges (50 to 56°C) and were established with both LNA and nonmodified probes.
Detection analysis
Each sample was run on the Luminex xMAP flow cytometer, which was interfaced to a PC. Individual sets of microspheres were analyzed by a red laser (636 nm) and a green laser (532 nm). The red laser allowed classification of the microsphere sets according to spectral address, whereas the green laser allowed quantification of the capture probe reaction on the surface of each microsphere set. MFI values were calculated by Luminex 1.7 proprietary software (on a PC interfacing the Luminex analyzer). A blank (no target DNA) and a set of positive (K. brevis or K. mikimotoi) and negative controls were included in each assay. All samples were run in duplicates. The signal obtained from a blank sample was subtracted from the MFI value. A response was considered positive when the corrected MFI value (MFI value after subtraction of the blank value) was at least twice the blank value (Diaz and Fell 2004).
Results
Probe Signal Comparison of LNA versus nonmodified probes—
To determine the efficacy of LNA probes in signal enhancement, we assessed the performance of conventional, unmodified probes (Pkmiki, Pkb2, Pkb658) and compared with modified versions (Pkmkilna, Pkmikilna2; Pkb2lna, Pkb658lna) of the probe sequences. LNA probes were designed by introducing three centrally located LNA bases (Table 2). For this particular assay, we assessed the performance of the probes with both enzymatically labeled and chemically labeled amplicons. The results showed that incorporation of LNA residues to probe sequences did not significantly enhance the fluorescence signal intensity when the assay was undertaken at 50°C and used Mirus Label IT amplicons (Fig. 1). However, a moderate enhancement in fluorescence signal intensity ranging from 11.5% to 33%, was documented for Pkmikilna and Pkb2lna, when these probes were challenged with enzymatically labeled amplicons (data not shown).
Fig. 1.

Probe signal comparison of LNA modified and unmodified probes at different hybridization temperatures. The assay used 50 ng chemically labeled amplicon (Mirus Label IT). The error bars depict the standard deviation. The samples were run in duplicate, and the experiments were run twice.
To evaluate if an extra LNA residue could have an impact on specificity and/or signal intensity, we redesigned Pkmikilna by including an extra LNA residue into the probe sequence and interspacing two of the centrally located modifications (Pkmikilna2). This redesign strategy did not enhance the sensitivity of the probe when the assay was carried out at 50°C or 53°C since similar fluorescence intensities were attained when the probe was challenged with Mirus Label IT amplicons (Fig. 1). In contrast, at 56°C, Pkmikilna2 displayed signal intensities that were 15% higher than Pkmikilna (Fig. 1).
Effect of temperature on probe signal intensity
To determine the effect of temperature on hybridization, nonmodified and LNA probes were tested at different temperatures ranging from 50 to 56°C (Fig. 1). This particular assay was carried out in a multiplex format and employed 50 ng of amplicon labeled by two different methods. The results demonstrated that as the temperature of the assay was increased a gradual decrease in intensity was documented for all probes, with lowest signals recorded at 56°C. At that temperature, nonmodified probes Pkb658 and Pkb2 displayed fluorescent signal reductions as high as 72% when tested with Mirus Label IT amplicons (Fig. 1) and up to 81% with PCR biotinylated amplicons (data not shown). Among the probes, Pkb2 had the least tolerance to high temperatures as this probe showed the highest reduction in signal intensity. In contrast, LNA probes were more resilient to increase in temperatures. For instance, when LNA probes were hybridized at 56°C and were challenged with Mirus Label IT amplicons, Pk658lna and Pkb2lna showed a signal reduction of 36% and 47%, respectively (Fig. 1). Similar levels of signal reduction, ranging from 32% to 42% were also noted when LNA probes were hybridized to PCR biotinylated amplicons (data not shown). An exception was Pkb2lna, which displayed 65.8% signal reduction.
Overall, all LNA probes displayed higher signal intensities than conventional probes when the assay was carried out beyond 50°C and when challenged with Mirus Label IT amplicons. For instance at 56°C, the signals intensities of Pkb658lna, Pkb2lna, and Pkmikilna2 were 54%, 46%, and 54% higher than their nonmodified versions. Despite high temperatures, Pkb658lna, Pkb2lna, Pkmikilna, and Pkmikilna2 displayed relatively robust MFI signals (~1500 to 2900) (Fig. 1). In a similar fashion, LNA probes outperformed their conventional probe version when tested with enzymatically labeled amplicons (data not shown). At 53°C, Pkb658lna and Pkb2lna displayed signal intensities that ranged from 24% to 50% higher than conventional probes, whereas at 56°C, Pkb658lna and Pkb2lna displayed signal intensities that were 35% and 66% higher than that of their nonmodified counterparts, respectively.
Comparison of two labeling methods: chemical (Mirus Label IT) versus enzymatic labeling (end-labeling with a biotinylated primer)
Chemical-labeling biotinylation required various optimization steps that involved titration analysis of Label IT reagent as well as different incubation times. Toward this end, we conducted preliminary labeling experiments with K. brevis amplicon (~670 bp). Initial experiments were undertaken with 100 ng of amplicons. The amplicons were labeled using different amounts of a 1:10 dilution of Label IT reagent. Typically, a 1:1 ratio of Label IT biotin reagent to nucleic acid (one microgram of DNA) is suggested for an effective labeling (manufacturer’s instructions). However, based on our preliminary results, which showed a linear increment in fluorescence intensity as more Label IT volume was added to the reaction, a labeling ratio of 1:2 (100 ng of DNA: 2 μL of a 1:10 dilution of Label IT reagent) appeared to suffice to attain good labeling density. The reaction reached a plateau when 4 μL Label IT reagent were employed (data not shown). These results indicate that at these levels the reaction was nearly saturated.
To determine the optimum incubation time, the assay was monitored at over 1.5 to 4 h time intervals. As expected, higher MFI values were obtained when the labeling incubation time was lengthened (data not shown). For instance, 3 h incubation resulted in a 55% increase in signal intensity. Overall, optimal labeling conditions were reached with 3 h incubation at 37°C and a labeling ratio of 1:2 (100 ng DNA: 2 μL of a 1:10 dilution of Label IT reagent).
Once the optimum labeling parameters were established, we compared Mirus Label IT technology with the enzymatic 5′terminal labeling method. The experiment, which was carried out with PCR amplicons, showed that the chemical Label IT system outperformed the enzymatic 5′terminal labeling technique (Fig. 2). All eight probes showed significantly higher signal fluorescent intensities when challenged with amplicons labeled with Mirus Label IT technique (Fig. 2). For instance, at 50°C, the enhancement in signal intensity ranged from ~59% to 79% (Fig. 2). Similar levels of signal amplification (~63% to 83%) were documented at 53°C and 56°C (data not shown).
Fig. 2.
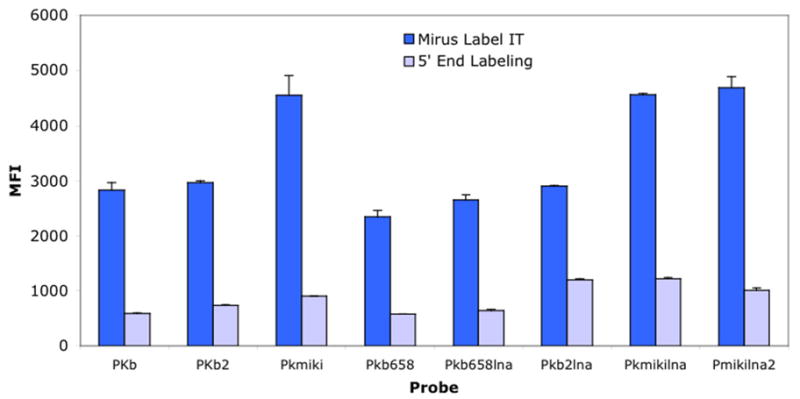
Comparison of Mirus Label IT and 5′ end labeled amplicons. Fifty nanograms of the labeled template was hybridized with different sets of K. brevis (Pkb, Pkb2, PKB658, Pkb2lna, PKB658lna) and K. mikimotoi probes (Pkmiklna and Pkmiklna2). Hybridization assay was undertaken at 50°C for 1.5 h. The error bars represent the standard deviation. The samples were run in duplicate, and the experiments were run twice.
Specificity
To determine the level of specificity of the hybridization assay, LNA-modified and unmodified probes (i.e., Pkmikilna2, Pkb2lna, Pkb658lna, Pkmiki, Pkb2, Pkb, and Pkb658) were challenged against the complementary target amplicon (perfect match) and a selective library of amplicons representing closely and more distantly related dinoflagellate species. These amplicons, which were derived from various culture collections (Table 1), were labeled with 5′end enzymatic labeling technique and Mirus Label IT. The selected species contained a variety of polymorphic sites representing various levels of mismatches (>2 bp). For instance, K. mikimotoi, which is a close relative of K. brevis, differs from probe sequences Pkb and Pkb658, by 2 and 3 bp base pairs, respectively. As expected, no significant cross reactivity was observed when LNA and nonmodified probes were challenged with Mirus Label IT amplicons representing closely and non–closely related dinoflagellate species (Fig. 3). An exception was Pkmiki, which displayed low cross reactivity with various dinoflagellate species. Although this cross reactivity was substantially diminished at 56°C, the probe continued to display some background noise (data not shown). This is in contrast to the counterpart LNA version of the probe, Pkmikilna2. The latter did not show any background noise or detectable cross reactivity with any of the species tested. Experiments undertaken with enzymatically labeled amplicons yielded similar results (data not shown).
Fig. 3.

Specificity of probes with Mirus Label IT D1/D2 amplicons. Probes were challenged with closely and non–closely related algal species. The hybridization was carried out for 1.5 h at 53°C and used 33 ng amplicon per well. The samples were run in duplicate and the experiments were run twice. The error bars represent the standard deviation.
Detection limits
Template detection limits: To determine the minimum amount of template DNA required in the PCR reaction, serial dilutions of template DNA (1 ng to 1 fg) were undertaken with Pkb2, Pkb2lna, Pkb658, Pkb658lna, Pkmikilna2, and Pkmiki (Fig. 4). The minimum detection limits that were established at 53°C were 10 fg (Pkmikilna2); 1 pg (Pkb2lna, Pkb658, Pkb658lna and Pkmiki), and 10 pg (Pkb2). Similar detection levels were observed with LNA probes when the assay was conducted at 56°C (data not shown). In contrast, the nonmodified probe versions were less sensitive as they showed a 5-fold (Pkb2) and 50-fold (Pkb658, Pkmiki) reduction in sensitivity at 56°C (data not shown).
Fig. 4.

Detection limits of genomic DNA. Mirus Label IT amplicon (10 μL) was hybridized to LNA and nonmodified probes. Hybridization conditions used 1.5 h at 53°C. The error bars depict the standard deviation. The samples were run in duplicate and the experiments were run twice.
Enzymatically labeled amplicons (5′ end biotin labeled amplicons): To determine the analytical sensitivity of the method, serial dilutions ranging from 50 ng to 10 pg were undertaken with enzymatically and chemically labeled amplicons. Based on our dilution series experiment, which was undertaken at 53°, the detection limits of the probes when challenged with enzymatically labeled amplicons were ~5 ng (Fig. 5). This detection level corresponds to ~21.1 fmol. When corrected to copy numbers and amplicon length, the sensitivity was estimated to be about 5.5 × 107 copies. Beyond 56°C, all K. brevis probes displayed similar detection levels, except for Pkb2, which failed to generate a fluorescent signal when employing 10 ng of enzymatically labeled amplicon (data not shown).
Fig. 5.

Analytic detection limits of chemically and enzymatically labeled amplicons. The amplicons were serially diluted and hybridized for 1.5 h at 53°C. The error bars represent the standard deviation. The samples were run in duplicate, and the experiments were run twice.
Chemically labeled amplicons (Mirus Label IT amplicons): For chemically labeled amplicons, Fig. 5 illustrates the detection levels of the assay conducted at 53°C. A steady decrease in fluorescence signal was documented as the amount of Mirus Label IT amplicon was decreased from 50 ng to 500 pg. Beyond 500 pg, the probe signals were close to background levels. No significant differences in detection levels were found between LNA and unmodified probes when the assay was conducted at 50°C (data not shown) or 53°C. At both temperatures LNA and unmodified probes displayed sensitivity levels of ~500 pg. However, at 56°C, a differential response between both set of probes was documented (data not shown). At that hybridization temperature, Pkb2, Pkb658, and Pkmiki exhibited a detection limit of ~5 ng, which corresponds to 5.52 × 107 copies for Pkb2 and Pkb658, and 5.38 × 107 copies for Pkmiki. In contrast, Pkb2lna, Pkmikilna2, and Pkb658lna showed detection limits of ~500 pg (data not shown). After correcting for PCR product length and assuming there are ~230 rRNA gene copies, the PCR product limit of detection for Pkb2lna, Pkb658lna, and Pkmikilna2 ranged from ~2.12 to ~2.05 fmol. This corresponded to detection limits ranging from 5.52 × 106 to 5.38 × 106 copies.
Assessment
A set of environmental samples representing a wide spectrum of FWRI categories was tested to determine the feasibility of the method for the detection of HABs. Testing field samples required a few optimization steps to Mirus Label IT and hybridization reactions. To this end, we titrated different amounts of amplicon (2.5–8 μL) in the labeling reaction and explored the effect of SAPE at various concentration (300–600 ng). After appropriate adjustments to the protocol method, analysis of field collected environmental samples typically employed 5 μL of unlabeled amplicon, a 1:2 ratio of the Label IT reagent to nucleic acid, 15 μL of biotinylated amplicon in the hybridization reaction, and 600 ng of SAPE.
Using the above-mentioned method, which included 1.5 h of incubation at 53°C, robust signal intensities were documented when LNA probes were challenged with field samples classified as high, medium, low b, low a, very low b, and very low a concentrations. For instance, Pkb2lna displayed signal intensities ranging from ~3255 (high) to ~383–469 MFI (very low A). As previously reported, non-LNA probes consistently displayed lower signal intensities than LNA probes. For example, Pkb658 signal intensities ranged from 1468 (High) to ~88–118 MFI (very low A), whereas Pkb658lna signal intensities ranged from ~3555 (High) to 327–405 MFI (very low A) (Fig. 6). As expected, the signal intensities generated by samples containing cell concentrations below 1,000 cells L−1 were significantly lower. At those cell levels, the signal intensity values for Pkb658lna ranged from 132 to 279 MFI. Similar ranges of values were also documented for Pkb2lna. With the exception of Pkb, which displayed signal intensity values of 130–230 MFI, lower signal intensities were documented for the non-modified probe versions, Pkb2 and Pkb658. In contrast to the robust signal intensities of the K. brevis probes, Pkmikilna2 and Pkmiki displayed lower signal intensities, which could be indicative of a low abundance of K. mikimotoi cells at the sites of field. However, this is difficult to confirm since the FWRI field sample data did not provide the concentration of K. miki-motoi. Samples classified as “not present” (i.e., #19 and #22) did not yield any significant signal that would indicate the presence of K. brevis or K. mikimotoi.
Fig. 6.

Performance of species-specific probes with field samples. Hybridization assay conditions used 15 μL chemical-labeled IT amplicon. The amplicons were generated using 40 cycles, and amplification was done with the universal primer set D2C and DIRF. The assay used 1.5 h incubations at 53°C. High (≥106 cells L−1); medium (100,000 to <106 cells L−1); low b (50,000 to <100,000 cells L−1); low a (>10,000 to <50,000 cells L−1); very low b (5000 to 10,000 cells L−1); very low a (>1000 to <5000 cells L−1); and not present (<1000 cell L−1). The samples were run in duplicate, and the experiments were run twice. The error bars represent the standard deviation. Inset legend represents sample IDs.
To make the assay more specific and to further enhance detection levels, a new forward primer (Kr130) was designed to selectively amplify species within the Karenia cluster group. The assay, which used the same conditions as those described above, proved to be more sensitive. All the probes displayed significantly higher MFI values than those previously documented with the universal primer set: D2C and DIRF. For instance, up to a 20-fold increase in signal enhancement was documented when Pkb658lna and Pkb2lna were challenged with a field sample (#17) containing below 1,000 cells L−1 (data not shown). Despite the enhancement of K. brevis detection, the data showed that under the chosen PCR and hybridization assay conditions, the system reached a plateau since the MFI signals did not follow a quantitative trend that correlated to the FWRI classification. Decreasing the amount of amplicon in the hybridization ameliorated but did not abolish the documented saturation levels (data not shown).
Because the observed DNA oversaturation appeared to be associated with PCR amplification effects (i.e., products reaching saturating concentrations), a series of amplification reactions were carried out with a lower numbers of cycles (i.e., 30 and 33 cycles). Upon reducing the number of cycles to 33, the DNA saturation effect was reduced while maintaining adequate detection levels (Fig. 7). For instance, good detection levels were documented for all K. brevis probes when tested with field samples classified as “present.” The documented MFI values for samples classified as “present” were as follows: Pkb658lna (1228-633 MFI); Pkb2lna (1395-739 MFI); Pkb2 (1143-442 MFI); Pkb (703-382 MFI); Pkb658 (489-205 MFI). In addition, all K. brevis LNA probes indicated the presence of K. brevis in two samples classified as “not present” (i.e., #30, #19 g), consistent with the increased detection sensitivity of the D2C and Kr130 primer set (Fig. 7). Overall, a nonsignificant reduction in fluorescence signal intensity was observed when the number of cycles was reduced to 30 (data not shown). However, when Pkb658lna and Pkb2lna were challenged with samples containing less than 1,000 cells L−1, the effect of reduction of signal became more apparent. For instance, when Pkb2lna was challenged with sample #17, an ~79% reduction in signal was documented (data not shown).
Fig. 7.

Probe performance with a collection of field samples. Amplifications were generated using 33 cycles with the universal primer D2C and the forward clade primer Kr130. Hybridization was carried out at 53°C for 1.5 h and used 5 μL of Mirus Label IT amplicon per well. FWRI classification: High (≥106 cells L−1); medium (100,000 to <106 cells L−1); low b (50,000 to <100,000 cells L−1); low a (>10,000 to <50,000 cells L−1); very low b (5,000 to 10,000 cells L−1); very low a (>1,000 to <5,000 cells L−1); and not present (<1,000 cell L−1). The error bars represent the standard deviation. The samples were run in duplicate, and the experiments were run twice. Inset legend represents sample IDs.
Discussion and Comments
Current traditional methods of HABs detection rely on morphological characteristics based on traditional light and electron microscopy. Light microscopy has been widely used as a method for cell enumeration but is tedious, not amenable to high-throughput analysis, and does not always provide enough resolution for accurate identification to the species level (Culverhouse et al. 2003). Electron microscopy allows precise species identification but is difficult for long-term monitoring, and does not always allow accurate cell density calculation (Lim et al. 1996). Optical signature analysis based on pigment methods is another attractive method for red tide detection. Although useful for monitoring and sketching the distribution map of red tides, this method is not sensitive enough for early detection of red tides and lacks the capability to differentiate between closely related taxa (Millie et al. 1997; Lauria et al. 1999). Traditional isolation and cultivation methods can take months, and cell sorting can cause morphological changes that preclude species identification (particularly for naked dinoflagellates such as K. brevis).
Numerous molecular methods based on nucleic acids have been formulated and shown to be a promising avenue to improve detection of HABs communities in coastal environments (Bowers et al. 2000; Connell 2002; Godhe et al. 2001). Many of these molecular methods have focused on the ribosomal DNA gene (rDNA) regions because they contain conserved and nonconserved regions that are phylogenetically informative (Goodwin et al. 2005; LaGier et al. 2007; Scorzetti et al. 2008). Other genes, i.e., the ribulose 1,5-bisphosphate carboxylase-oxygenase (RuBisCO) large subunit gene (rbcL) (Gray et al. 2003; Casper et al. 2004); the chloroplast large subunit (cp23S)-rDNA gene (Santos et al. 2002); the mitochondrial cytochrome B (Zhang and Lin 2002; Zhang et al. 2005); and the encoded form II Rubisco gene (Zhang and Lin 2003), have been used for the development of molecular assays for dinoflagellate species. The utility of a region is based on the level of phylogenetic variability of the target species. In this study, we found the D1/D2 region of the large subunit (LSU, 28S) provide adequate discriminatory power.
Some of the molecular methods used to study dinoflagellates have included the following: real time PCR (Galluzi et al. 2004; Park et al. 2007); denaturing gradient gel electrophoresis (DGGE) (Coyne et al. 2001); restriction fragment length polymorphism (Adachi et al 1994); fluorescent fragment detection PCR (Coyne et al. 2001); heteroduplex mobility assay (HMA) (Oldach et al. 2000); LSU rRNA FISH probe (Mikulski et al. 2005); fluorescent in situ hybridization (Allen 2000); quantitative real time PCR (Popels et al. 2003), real time reverse transcription (RT) PCR (Gray et al. 2003); nucleic acid sequence-based amplification (NASBA) (Casper et al. 2004); TaqMan probe-based RT PCR assay (Gray et al. 2003); sandwich hybridization assay (Tyrrell et al. 2002; Haywood et al. 2007); and sandwich hybridization assay nuclease protection (NPA-SH) (Cai et al. 2004).
In addition, several alternative detection strategies have been proposed. Although innovative, some of them had limited detection capabilities, sensitivity issues or need further development to be fully adopted. These included the following: quartz crystal microbalance biosensor (Nakanishi et al 1996); chemiluminescence biosensor (Asai et al. 1999; Asai et al. 2003); antibodies and lectins as cell surface markers (Scholin et al. 2003); surface plasmon resonance (Asai et al 2003); electrochemical assay (Litaker et al. 2001, LaGier et al. 2007); and target mediated aggregation detection technology (Costanzo et al. 2005). Most recently, novel platforms have been developed (Ellison and Burton 2005; Scorzetti et al. 2008). These platforms, which are based on bead-based technology, provided a versatile approach and basis for future development of tools for the simultaneous detection of multiple targets.
Herein, we describe a bead suspension assay that combines the high-throughput capability of Luminex techonology, the specificity of LNA probes, and a signal amplification system that used a non-enzymatic direct chemical labeling that allowed the covalent attachment of multiple biotin molecules. Merging these technologies, we developed a sensitive detection method that enabled the detection of K. brevis and K. mikimotoi at pre-blooming conditions. The method, which used a stringent hybridization assay format combined with Luminex 100 technology, provided sufficient specificity to permit accurate identification of the target species.
The sensitivity of the assay, which was determined with LNA and nonmodified probes, showed that the method can detect between 1 to 10 pg of genomic DNA template in the PCR reaction. Notably, for probe Pkmikilna2, we detected as little as 10 fg of genomic DNA template in the PCR reaction. Among all the probes, Pkb2 was the least sensitive, with a detection limit of 10 pg (Fig. 4). Assuming that K. mikimotoi has a minimum cell DNA quota similar as the one reported for K. brevis (~50 pg/cell) and the number of rRNA gene copies per dinoflagellate cell is ~230 copies (Kim and Martin 1974; Walker 1982; Rollo et al 1995; Kamykowski et al 1998; Yoon et al. 2005), 10 fg of genomic DNA corresponded to 0.05 copies of rRNA gene. Following the above calculation, 1 pg and 10 pg of genomic DNA correspond to 4.6 and 46 gene copies, respectively. When converted to cell numbers, these detection levels represented 0.02 and 0.2 cells, respectively. LNA probes displayed a similar range of detection levels when the assay was conducted at 53 and 56°C. In contrast, the nonmodified probe versions ie, Pkmiki, Pkb658 and Pkb2 were less sensitive at 56°C with detection limits of only 50 pg. Others studies, such as a real-time reverse transcription-PCR method (Gray et al. 2003), hybrid PCR (La Gier et al. 2007) and real-time nucleic acid sequence-based amplification (Casper et al. 2004), have reported detection levels of 1 to 10 cells of K. brevis. Except for the Pkb2 probe, our detection levels were more sensitive than those, including a report by Galluzi et al. (2004), who documented 10 copies of 5.8S rRNA (≤0.2 cell equivalent of algal lysate) when employing a real time PCR assay for the detection of the marine dinoflagellate, Alexandrium minimum.
LNA probes displayed similar hybridization efficiency as nonmodified probes when the assay was performed at 50°C (Fig. 1). Although the latter observation applied for enzymatically and chemically labeled amplicons, a difference in probe performance was observed when Pkb2lna was challenged with enzymatically labeled amplicon. This particular probe displayed MFI values that were 33% higher than its nonmodified version, Pkb2. The utility of the LNA probes was demonstrated as the temperature of the assay increased. At higher temperature, LNA probes consistently displayed higher fluorescent intensities and better sensitivity levels than their nonmodified probe versions. This effect was documented for both types of labeled amplicons. As opposed to conventional probes, we found that LNA probe sensitivity levels were not affected when the hybridization temperature was increased to 56°C. The observed differential response in probe performance and sensitivity level can be attributed to differences in the melting behavior of LNA and conventional probes. LNA probes are known to be resilient and stable under high temperatures due to the presence of LNA residues, which confers a high degree of thermodynamic stability (You et al. 2006). The thermal stability of the LNA/DNA duplex has been estimated to increase 3 to 8°C per modified base (Koshkin et al. 1998) and is known to increase the affinity of the oligonucleotide with its complementary target (Simeonov and Nikiforov 2002). Thus, by introducing LNA residues to the probe sequence, a desired melting temperature can be achieved without having to modify the sequence context of the probe or altered a pre-set hybridization temperature. The unique thermal property of LNA probes permits the assay to be run at high temperatures, which promote high stringent conditions. Overall, this study shows the enhanced level of thermal stability of LNA probes allows for a robust and efficient detection system that surpasses the specificity and sensitivity of conventional probes.
Although higher signal intensities have been reported with LNA probes compared with conventional and chemically modified probes [e.g., minor groove binder (MGB) probe and peptide nucleic acid (PNA) (Costa et al. 2004; Simeonov and Nikiforov 2002)]; others have reported no significant differences in signal strength or sensitivity levels (Johnson et al. 2004; Letertre et al. 2003; Laschi et al. 2009). For example, in a recent study using an enzyme amplified electrochemical hybridization assay format with micromagnetic beads, the levels of rRNA detection for DNA, PNA, and LNA probes were found to be 51, 60, and 78 pM, respectively (Laschi et al. 2009). Conversely, other studies have shown that depending upon the chosen assay platform or method, LNA probe performance can vary. For example, LNA probes displayed higher sensitivity levels and signal enhancement when challenged on a DNA microarray platform, but no signal enhancement was documented when tested on biosensors using a sandwich hybridization format (Diercks et al. 2009). Thus, parameters such as hybridization temperature, sequence context, LNA length, identity of the mismatch, number of LNA modifications, location of LNA substitutions, and assay platform can all have an intrinsic effect on the performance and utility of a LNA probe in hybridization assays (You et al. 2006).
Except for the Pkmiki probe, which showed nonspecific binding with some nontarget DNA, we found that LNA probe specificity was comparable to that observed with conventional DNA probes (Fig. 3). Moreover, the introduction of four centrally located interspersed LNA residues to Pkmiki enhanced the discriminatory power since no significant cross-reactivity was documented with any of the tested dinoflagellate species. Similarly, others have reported enhanced specificity when using LNA probes as genotyping tools (Simeonov and Nikiforov 2002; Johnson et al. 2004; den Buijsch et al. 2005; You et al. 2006). For instance, Simeonov and Nikiforov (2002) reported excellent specificity levels at single base pair mismatch when using hexamer and heptamer fluorescently labeled LNA probes. Moreover, a remarkable specificity has also been reported by the introduction of a single LNA residue in a sensor probe of a FRET assay (den Buijsch et al. 2005). Because of the presence of the additional methylene bridge, LNA probes display better base stacking hence conferring higher stability and affinity toward their complementary target. The conformational change that is introduced by the methylene bridge increases the melting temperature of the probe Tm and allows for better mismatch discrimination (Kauppinen et al. 2003; You et al. 2006).
In this study, we also investigated the labeling efficiency of Mirus Label IT technology versus the enzymatic 5′terminal labeling method. In contrast to enzymatic labeling methods like nick translation or random priming, this non-enzymatic direct chemical labeling system allows the covalent attachment of multiple biotin molecules to guanine residues or any other reactive heteroatom (e.g., N3 of adenine and N3 of cytosine) in the polynucleotide of DNA. After a series of assay optimizations that involved titrations of labeling reagent and different incubation times, our results illustrated that the chemical Label IT system appeared to be a more effective labeling system since it consistently outperformed the enzymatic 5′ terminal labeling technique with documented probe signal enhancement as much as 83%. This is not surprising as labeling efficiencies with this method have been estimated to be ~1 labeled molecule for every 20–60 base pairs of double-stranded DNA (Mirus BIO LLC). Thus, we demonstrated that adopting this chemical labeling system resulted in better analytical detection levels. For example at 53°C, LNA and unmodified probes displayed detection limits of ~500 pg when challenged with chemically labeled amplicons. In contrast, inferior levels of detection (~5 ng) were documented for both sets of probes when challenged with enzymatic end labeled amplicons. After correcting for PCR product length and rRNA gene copies, the detection limits of 500 pg and 5 ng correspond to 5.5 × 106 and 5.5 × 107 copies, respectively (Rollo et al 1995).
Analysis on field samples indicated that the method was sensitive at low cell numbers and over a broad concentration range as it allowed the detection of K. brevis in field samples classified as “present” with robust signal intensities. The method also allowed the detection of K. brevis in various samples that were below the detection limit of traditional microscopy (<1,000 cells L−1), and produced MFI values of sufficient strength to allow differentiation of positive versus negative signals. Although this discrepancy could potentially indicate false-positive results, the positive signal responses of three probes bearing different sequence composition confirmed the presence of K. brevis in those samples. Similar results were reported by Goodwin et al. (2005). In that case, the presence of K. brevis in a sample classified as “not present” by microscopy was identified through a DNA hybridization assay in microplate format, with the results confirmed by both standard PCR and by sequencing. The enhanced level of detection herein observed was especially clear when the assay employed a single forward primer designed to target K. brevis within the Karenia cluster group. Our results suggested that our molecular detection method was more sensitive than light microscopy and than a bead array developed for the detection of various HABs (Scorzetti et al. 2008). For instance, using our array format, we documented detection signals that were up to 5× higher than those reported by Scorzetti et al. (2008).
Even though the use of universal primers allows the amplification of a wide variety of species, it can also be a liability because an excess of nontarget DNA can be generated. For instance, the universal primer combination DIR and R635 can amplify a wide range of diatoms, fungi, and plants (Goodwin et al. 2005). In this scenario, an excess of amplified nontarget DNA can cause a “dilution factor” associated with the target DNA. This imbalance in the ratios of nonspecific to specific target material cannot only bias the potential detection of under-represented species, but can add some noise to the reaction, making it more difficult to achieve good sensitivity. Therefore to overcome this potential effect, which in our case was more noticeable in field samples containing less than 1,000 cells L−1 (as determined by microscopy cell counts), we were able to detect K. brevis at low cell counts when using a more specific forward primer. Lack of detection of K. mikimotoi on any of the field samples may have been due to the apparent near absence or extreme underrepresentation of that particular species at the time of collection.
Even though the data followed a semi-quantitative distribution that appeared to correlate with cell densities, the results also included outlier samples that did not necessarily correlate with microscopic enumeration (Fig. 7). Several factors could have contributed to this outcome: a) differential amplification efficiencies, b) differential DNA extraction efficiencies; c) inaccurate microscopic cell counts; d) presence of inhibiting substances. Although PCR-based methods are known to suffer from differential amplification efficiencies, it is interesting to note that fairly similar amplification patterns were obtained with these samples despite repeated amplifications and use of varying numbers of PCR cycles. Furthermore, data obtained using a microplate array confirmed similar outlier patterns (Goodwin unpubl. data). Thus, the observed variability appeared to be more related to DNA extraction efficiencies that led to different DNA yields or to errors associated with microscopic cell counts. Errors associated with cell counts can easily be introduced by sample manipulation, which often requires multiple sample dilution(s) and back calculations.
As previously observed with samples derived from cell cultures, LNA probes consistently displayed higher fluorescent intensities than their non-LNA modified versions when the probes were challenged with environmental samples. This method represents the first molecular strategy to integrate Luminex technology, LNA probes, and Mirus Label IT technology for the detection of HABs. The method, which does not require taxonomic expertise, proved to be especially useful for the detection of low-density populations and may provide an easy and relatively rapid procedure (approx. 8 h) that might be adopted as an alternative strategy for monitoring red tide at pre-blooming and blooming conditions.
The high throughput capacity of the 96-well plate format would allow more rapid screening of water samples, of which only a subset might require microscopic confirmation. Future directions should continue to expand the bead array platform to include other target species.
Acknowledgments
We are indebted to Dr. Robert Brazas for providing technical expertise on Mirus labeling methods. Financial support is gratefully acknowledged from the Cooperative Institute of Estuarine and Environmental Technology (CICEET) through a NOAA grant (NA06N0S4190167). Field samples and financial assistance for publication costs were provided by the Rookery Bay NERR and through grants from the National Science Foundation (OCE 0432368/0911373) and the National Institute of Environmental and Health Science (P50 ES12736). The authors thank the ARCH core facility at Florida International University for providing various DNA samples from their culture collection (NIEHS -S11ES11181).
References
- Adachi M, Sako Y, Ishida Y. Restriction fragment length polymorphism of ribosomal DNA internal transcribed spacer and 5.8S region in Japanese Alexandrium species (dynophyceae) J Phycol. 1994;30:857–863. doi: 10.1111/ j.0022-3646.1994.00857.x. [DOI] [Google Scholar]
- Allen CI. MS thesis. University of North Carolina; Greensboro: 2000. Utilization of the polymerase chain reaction and fluorescent in situ hybridization to assess fine scale and global distribution patterns of Pfiesteria species. [Google Scholar]
- Anderson DM. Toxic algal blooms and red tides: A global perspective. In: Okaichi T, Anderson DM, Nemoto T, editors. Red tides: Biology, environmental science and toxicology. Elsevier; 1989. pp. 11–16. [Google Scholar]
- Anderson DM. Turning back the harmful red tide. Nature. 1997;388:513–514. doi: 10.1038/41415. [DOI] [Google Scholar]
- Anderson DM, Glibert PM, Burkholder JM. Harmful algal blooms and eutrophication: Nutrient sources, composition and consequences. Estuaries. 2002;25:562–584. doi: 10.1007/BF02804901. [DOI] [Google Scholar]
- Asai R, Matsukawa R, Ikebukuro K, Karube I. Highly sensitive chemiluminescence flow-injection detection of the red tide phytoplankton, Heterosigma carterae. Anal Chim Acta. 1999;390:237–244. doi: 10.1016/S0003-2670 (99)00145-2. [DOI] [Google Scholar]
- Asai R, Nakanishi K, Nakamura C, Ikebukuro K, Miyake J, Karube I. A polymerase chain reaction-based ribosomal DNA detection using surface plasmon resonance detector for a red tide causing microalga, Alexandrium affine. Phycol Res. 2003;51:118–125. [Google Scholar]
- Bolch JS, de Salas MF. A review of the molecular evidence for ballast water introduction of the toxic dinoflagellates Gymnodinium catenatum and the Alexan-drium “tamarensis complex” to Australasia. Harm Algae. 2007;6:465–485. doi: 10.1016/j.hal.2006.12.008. [DOI] [Google Scholar]
- Bowers HA, Tengs T, Glasgow HB, Burkholder JM, Rublee PA, Oldach DW. Development of real time PCR assays for rapid detection of Pfiesteria piscicida and related dinoflagellates. Appl Environ Microbiol. 2000;66:4641–4648. doi: 10.1128/AEM.66.11.4641-4648.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Li R, Zhen Y, Mi T, Yu Z. Detection of two Prorocentrum species using sandwich hybridization integrated with nuclease protection assay. Harm Algae. 2004;5:300–309. doi: 10.1016/j.hal.2005.08.002. [DOI] [Google Scholar]
- Casper ET, Paul JH, Smith MC, Gray M. Detection and quantification of the red tide dinoflagellate Karenia brevis by real time nucleic acid sequence based amplification. Appl Environ Microbiol. 2004;70:4727–4732. doi: 10.1128/ AEM.70.8.4727-4732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell L. Rapid identification of marine algae (Raphi-dophyceae) using three-primer PCR amplification of nuclear internal transcribed spacer (ITS) regions from fresh and archived material. Phycologia. 2002;41:15–21. [Google Scholar]
- Costa JM, Ernault P, Olivi M, Gaillon T, Arar K. Chimeric LNA/DNA probes as a detection system for real time PCR. Clin Biochem. 2004;37:930–932. doi: 10.1016/j.clin biochem.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Costanzo PJ, Liang E, Patten TE, Collins SD, Smith RL. Biomolecule detection via target mediated nanoparticle aggregation and dielectrophoretic impedance measurement. Lab Chip. 2005;5:606–610. doi: 10.1039/b417535b. [DOI] [PubMed] [Google Scholar]
- Coyne KJ, Hutchins DA, Hare CE, Cary SC. Assessing temporal and spatial variability in Pfiesteria piscicida distributions using molecular probing techniques. Aquat Microb Ecol. 2001;24:275–285. doi: 10.3354/ame024275. [DOI] [Google Scholar]
- Culverhouse PF, Williams R, Reguera B, Herry V, González-Gil S. Do experts make mistakes? A comparison of human and machine identification of dinoflagellates. Mar Ecol Prog Ser. 2003;247:17–25. doi: 10.3354/meps 247017. [DOI] [Google Scholar]
- den Buijsch RO, de Vries JE, Loots WJG, Landt O, Wijnen PAHM, van Dieijen-Visser MP, Bekers O. Genotyping of the Pregnane X receptor A11156C polymorphism with locked nucleic acid containing fluorogenic probes. Pharmacogenom J. 2005;5:72–74. doi: 10.1038/sj. tpj.6500299. [DOI] [PubMed] [Google Scholar]
- Diaz MR, Fell JW. High-throughput detection of pathogenic yeasts of the genus Trichosporon. J Clin Microbiol. 2004;42:3696–3706. doi: 10.1128/JCM.42.8.3696-3706.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diercks S, Gescher C, Metflies K, Medlin LK. Evaluation of locked nucleic acids for signal enhancement of oligonucleotide probes for microalgae immobilized on solid surface. J Appl Phycol. 2009;21:657–668. doi: 10.1007/ s10811-008-9399-0. [DOI] [Google Scholar]
- Doblin MA, Popels LC, Coyne KJ, Hutchins DA, Cary SC, Dobbs FC. Transport of harmful bloom alga Aureococcus anophagefferens by oceangoing ships and coastal boats. Appl Environ Microbiol. 2004;70:6495–6500. doi: 10.1128/AEM.70.11.6495-6500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison CK, Burton RS. Application of bead array technology to community dynamics of marine phytoplankton. Mar Ecol Prog Ser. 2005;288:75–85. doi: 10.3354/ meps288075. [DOI] [Google Scholar]
- Figley W, Pyle B, Halgren B. Oxygen depletion and associated benthic mortalities in New York Bight, 1976. NOAA, U.S. Department of Commerce; 1979. Socioeconomic impacts; pp. 315–322. [No authors] NOAA professional paper 11. [Google Scholar]
- Fulton R, McDade R, Smith P, Kienker L, Kettman J. Advanced multiplexed analysis with the FlowMetrix system. Clin Chem. 1997;43:1749–1756. [PubMed] [Google Scholar]
- Galluzi L, Penna A, Bertozzini E, Villa M, Garcés E, Magnani M. Development of a real-time PCR assay for rapid detection and quantification of Alexandrium minimum (a dinoflagellate) Appl Environ Microbiol. 2004;70:1199–1206. doi: 10.1128/AEM.70.2.1199-1206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtsoff PS. Red tide. U. S. Fish and Wildlife. Serv Sp Sci Rept. 1948;46:1–44. [Google Scholar]
- Gunter G, Williams RN, Davis CC, Smith FGW. Catastrophic mass mortality of marine animals and coincident phytoplankton bloom on the west coast of Florida, November 1946 to August 1947. Ecol Monogr. 1948;18:309–324. [Google Scholar]
- Godhe A, Otta SK, Rehnstam-Holm AS, Karunasagar In, Karunasagar I. Polymerase chain reaction in detection of Gymnodinium mikimotoi and Alexandrium minitum in field samples from southwest India. Mar Biotechnol. 2001;3:152–162. doi: 10.1007/s101260000052. [DOI] [PubMed] [Google Scholar]
- Goodwin KD, Cotton SA, Scorzetti G, Fell JW. A DNA hybridization assay to identify toxic dinoflagellates in coastal waters: detection of Karenia brevis in the Rookery Bay National Estuarine research Reserve. Harm Algae. 2005;4:411–422. doi: 10.1016/j.hal.2004.07.005. [DOI] [Google Scholar]
- Gray M, Wawrik B, Paul J, Casper E. Molecular detection and quantitation of the red tide dinoflagellate Karenia brevis in the marine environment. Appl Environ Microbiol. 2003;69:5726–5730. doi: 10.1128/AEM.69.9.5726-5730. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JD, Anderson DM, Erdner DL, Bhattacharya D. Dinoflagellates: A remarkable evolutionary experiment. Am J Bot. 2004;91:1523–1534. doi: 10.3732/ajb.91. 10.1523. [DOI] [PubMed] [Google Scholar]
- Haywood AJ, Scholin CA, Marin R, III, Steidinger KA, Heil C, Ray J. Molecular detection of the breve-toxin-producing dinoflagellate Karenia brevis and closely related species using rRNA-targeted probes and a semiautomated sandwich hybridization assay. J Phycol. 2007;43:1271–1286. doi: 10.1111/j.1529-8817.2007.00407.x. [DOI] [Google Scholar]
- Hoagland P, Handerson DM, Kaoru Y, White AW. The economic effects of harmful algal blooms in the United States: estimates, assessment issues, and information needs. Estuaries. 2002;25:819–837. doi: 10.1007/BF0280 4908. [DOI] [Google Scholar]
- Johnson MP, Haupt LM, Griffiths LR. Locked nucleic acid (LNA) single nucleotide polymorphism (SNP) genotype analysis and validation using real time PCR. Nucl Acids Res. 2004;32:e55. doi: 10.1093/nar/gnh046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen S, Nielsen PS, Mouritzen P, Nielsen AT, Vissing H, Moller S, Ramsing NB. LNA microarrays in genomics. Pharma Genomics. 2003;3:24–32. [Google Scholar]
- Kamykowski D, Milligan EJ, Reed RE. Biochemical relationships in the orientation of the autotrophic dinoflagellate Gymnodinium breve under nutrient replete conditions. Mar Ecol Prog Ser. 1998;167:105–117. doi: 10.3354/meps167105. [DOI] [Google Scholar]
- Kim YS, Martin DF. Effects of salinity on synthesis of DNA, acidic polysaccharide and ichthyotoxin in Gymnodinium breve. Phytochemistry. 1974;13:533–538. doi: 10.1016/ S0031-9422(00)91348-7. [DOI] [Google Scholar]
- Kirkpatrick B, et al. Literature review of Florida red tide: implications for human health effects. Harm Algae. 2003;3:99–115. doi: 10.1016/j.hal.2003.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshkin AA, Nielsen P, Meldgaard M, Rajwanshi VK, Singh SK, Wengel J. LNA locked nucleic acid: an RNA mimic forming exceedingly stable LNA: LNA duplexes. J Amer Chem Soc. 1998;120:13252–13253. doi: 10.1021/ja9822862. [DOI] [Google Scholar]
- LaGier MJ, Fell JW, Goodwin KD. Electrochemical detection of harmful algae and other microbial contaminants in coastal waters using hand-held biosensors. Mar Poll Bull. 2007;54:757–770. doi: 10.1016/j.marpolbul.2006. 12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberg JH. The effects of harmful algal blooms on aquatic organisms. Rev Fish Sci. 2002;10:113–390. doi: 10.1080/ 20026491051695. [DOI] [Google Scholar]
- Laschi S, Palchetti I, Marrazza G, Mascini M. Enzyme amplified electrochemical hybridization assay based on PNA, LNA and DNA probe-modified micromagnetic beads. Bioelectrochemistry. 2009;76:214–220. doi: 10.1016/j. bioelechem.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Lauria ML, Purdie DA, Sharples J. Contrasting phytoplankton distributions controlled by tidal turbulence in an estuary. J Mar Syst. 1999;21:189–197. doi: 10.1016/S0924-7963(99)00013-5. [DOI] [Google Scholar]
- Letertre C, Perelle S, Dilasser F, Arar K, Fach P. Evaluation of the performance of LNA and MGB probes in 5′-nuclease PCR assays. Mol Cell Probes. 2003;17:307–311. doi: 10.1016/j.mcp.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Lim EL, Caron DA, Delong EF. Development and field application of a quantitative method for examining natural assemblages of protists with oligonucleotide probes. Appl Environ Microbiol. 1996;62:1416–1423. doi: 10.1128/aem.62.4.1416-1423.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litaker W, Sundseth R, Wojciechowski W, Bonaventura C, Henkens R, Tester P. Electrochemical detection of DNA or RNA from harmful algal bloom species. In: Hallagraeff GM, Blackburn SI, Bolch CJ, Lewis RJ, editors. Harmful algal blooms. Intergovernmental Oceanographic Commission Unesco; Paris: 2001. pp. 242–245. [Google Scholar]
- Millie DF, Schofield OM, Vinyard BT. Detection of harmful algal blooms using photopigments and absorption signatures: A case study of the Florida red tide dinoflagellate, Gymnodinium breve. Limnol Oceanogr. 1997;42:1240–1251. [Google Scholar]
- Mikulski CM, Morton SL, Doucette GJ. Development and application of LSU rRNA probes for Karenia brevis in the Gulf of Mexico, USA. Harm Algae. 2005;4:49–60. doi: 10.1016/j.hal.2003.11.001. [DOI] [Google Scholar]
- Nakanishi K, Adachi M, Sako Y, Ishida Y, Muguruma H, Karube I. Detection of the red tide-causing plankton Alexandrium affine by a piezoelectric immunosensor using a novel method of immobilizing antibodies. Anal Lett. 1996;29:1247–1258. [Google Scholar]
- Oldach DW, et al. Heteroduplex mobility assay guided sequence discovery: elucidation of the small subunit (18S) rDNA sequence of Pfiesteria piscicida from complex algal culture and environmental sample DNA pools. Proc Natl Acad Sci USA. 2000;97:4303–4308. doi: 10.1073/pnas. 97.8.4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TG, de Salas MF, Bolsch CJS, Hallegraeff GM. Development of real-time PCR for quantification of the heterotrophic dinoflagellate Cryptoperidiniopsis brodyi (Dinophyceae) in environmental samples. Appl Environ Microbiol. 2007;73:2552–2560. doi: 10.1128/AEM.02389-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popels LC, Cary SC, Hutchins DA, Forbes R, Pustuzzi F, Gobler CJ, Coyne KJ. The use of quantitative polymerase chain reaction for the detection and enumeration of the harmful alga Aureococcus anophagefferens in environmental samples along the United States East Coast. Limnol Oceanogr Methods. 2003;1:91–102. [Google Scholar]
- Reynolds CS. Physical determinants of phytoplankton succession. In: Sommer U, editor. Plankton ecology. Springer-Verlag; 1989. pp. 9–56. [Google Scholar]
- Rollo F, Sassaroli S, Boni L, Marota I. Molecular typing of the red tide dinoflagellate Gonyaulax polyedra in phytoplankton suspensions. Aquat Microb Ecol. 1995;9:55–61. doi: 10.3354/ame009055. [DOI] [Google Scholar]
- Santos SR, Taylor DJ, Kinzie RA, III, Hidaka M, Sakai K, Coffroth MA. Molecular phylogeny of symbiotic dinoflagellates inferred from partial chloroplast large subunit (cp23S)-rDNA sequences. Mol Phyl Evol. 2002;23:97–111. doi: 10.1016/S1055-7903(02)00010-6. [DOI] [PubMed] [Google Scholar]
- Scholin CA, Vrieling E, Peperzak L, Rhodes L, Rublee P. Detection of HAB species using lectin, antibody and DNA probes. In: Hallegraeff GM, Anderson DM, Cembella AD, editors. Manual on harmful marine microalgae. UNESCO; 2003. pp. 131–164. [Google Scholar]
- Scorzetti G, Brand LE, Hitchcock GL, Rein KS, Sinigalliano CD, Fell JW. Multiple simultaneous detection of harmful algal blooms (HABs) through a high-throughput bead array technology, with potential use in phytoplankton community analysis. Harm Algae. 2008;8:196–211. doi: 10.1016/j.hal.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellner KG, Doucette GJ, Kirkpatrick G. Harmful algal blooms: causes, impacts and detection. J Ind Microbiol Biotechnol. 2003;30:383–406. doi: 10.1007/s10295-003-0074-9. [DOI] [PubMed] [Google Scholar]
- Shumway SE. A review of the effects of algal blooms on shellfish and aquaculture. J World Aquacult Soc. 1988;21:65–104. doi: 10.1111/j.1749-7345.1990.tb00529.x. [DOI] [Google Scholar]
- Simeonov A, Nikiforov TT. Single nucleotide polymorphism genotyping using short fluorescently labeled locked nucleic acid (LNA) probes and fluorescence polarization detection. Nucl Acids Res. 2002;30:e91. doi: 10.1093/nar/gnf090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smayda TE. What is a bloom? A commentary. Limnol Oceanogr. 1997;42:1132–1136. [Google Scholar]
- Steidinger KA, Landsberg H, Truby EW, Roberts BS. First report of Gymnodinium pulchellum (Dino-phyceae) in North America and associated fish kills in the Indian River, Florida. J Phycol. 1998;34:431–437. doi: 10.1046/j.1529-8817.1998.340431.x. [DOI] [Google Scholar]
- Sterner RW. The role of grazers in phytoplankton succession. In: Sommer U, editor. Plankton ecology. Springer-Verlag; 1989. pp. 107–170. [Google Scholar]
- Taylor FJR. The species problem and its impact on harmful phytoplankton studies. In: Smayda TJ, Shimizu Y, editors. Toxic marine dinoflagellates. Elsevier; 1993. pp. 81–86. [Google Scholar]
- Tester PA, Stumpf RP, Vukovich FM, Fowler PK, Turner JT. An expatriate red tide bloom: transport, distribution and persistence. Limnol Oceanogr. 1991;36:1053–1061. [Google Scholar]
- Tester PA, Steidinger KA. Gymnodinium breve red tide blooms: Initiation, transport, and consequences of surface circulation. Limnol Oceanogr. 1997;42:1039–1051. [Google Scholar]
- Thomas WH, Gibson CH. Effects of small-scale turbulence on microalgae. J Appl Phycol. 1990;2:71–77. doi: 10.1007/BF02179771. [DOI] [Google Scholar]
- Tyrrell JV, Connell LB, Sholin CA. Monitoring for Heterosigma akashiwo using a sandwich hybridization assay. Harm Algae. 2002;1:205–214. doi: 10.1016/S1568-9883(02) 00012-4. [DOI] [Google Scholar]
- Turgeon DD, Sellner KG, Scavia D, Anderson D. Status of US harmful algal blooms: Progress towards a national program. NOAA, National Ocean Service, Centers for Coastal Ocean Science, Center for Monitoring and Assessment; 1998. [Google Scholar]
- Walker LM. Evidence for a sexual cycle in the Florida red tide dinoflagellate Ptychodiscus brevis (=Gymnodinium breve) Trans Am Microbiol Soc. 1982;101:287–293. doi: 10.2307/ 3225818. [DOI] [Google Scholar]
- Xiaonan L, Wei W. A relationship between red tide outbreaks and urban development along the coasts of Guangdong province. J Geogr Sci. 2004;14:219–255. doi: 10.1007/BF02837537. [DOI] [Google Scholar]
- You Y, Moreira BG, Behlke MA, Owczarzy R. Design of LNA probes that improve mismatch discrimination. Nucl Acids Res. 2006;34:e60. doi: 10.1093/nar/gkl175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HS, Hackett JD, Lidie L, Van Dolah FM, Nosenko T, Bhattacharya D. Tertiary endosymbiosis driven genome evolution in dinoflagellate algae. Mol Biol Evol. 2005;22:1299–1308. doi: 10.1093/molbev/msi118. [DOI] [PubMed] [Google Scholar]
- Zhang H, Lin S. Detection and quantification of Pfiesteria piscicida by using the mitochondrial cytochrome b gene. Appl Environ Microbiol. 2002;68:989–994. doi: 10.1128/ AEM.68.2.989-994.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lin S. Complex gene structure of the form II rubisco in the dinoflagellate Prorocentrum minimum (Dinophyceae) J Phycol. 2003;39:1160–1171. doi: 10.1111/j. 0022-3646.2003.03-055.x. [DOI] [Google Scholar]
- Zhang H, Bhattacharyaand D, Lin S. Phylogeny of dinoflagellates based on mitochondrial cytochrome b and nuclear small subunit rDNA sequence comparisons. J Phycol. 2005;41:411–420. doi: 10.1111/j.1529-8817.2005.04168.x. [DOI] [Google Scholar]