

Abstract
Exposure of alcohols 1a-1i to butadiene in the presence of a cyclometallated iridium catalyzed derived from allyl acetate, 4-methoxy-3-nitrobenzoic acid and BIPHEP (2,2′-bis(diphenylphosphino)biphenyl) results in hydrogen transfer to generate aldehyde-allyliridium pairs, which engage in C-C coupling to form products of carbonyl crotylation. Under related conditions using 1,4-butanediol as hydrogen donor, butadiene reductively couples to aldehydes 2e-2g and 2i to furnish carbonyl crotylation products 3e-3g and 3i. Thus, butadiene mediated carbonyl crotylation occurs with equal facility from the alcohol or aldehyde oxidation level with complete levels of branched regioselectivity.
Keywords: iridium, butadiene, crotylation, green chemistry, byproduct-free
We have found that hydrogen exchange between alcohols and π-unsaturated reactants triggers generation of electrophile-nucleophile pairs en route to products of C-C coupling.1,2 Using ruthenium- catalysts, the “transfer hydrogenative coupling” of alcohols to dienes,3 enynes,4 alkynes,5 and allenes6 delivers products of carbinol C-H functionalization or “hydro-hydroxyalkylation”. Recently, related alcohol-enal couplings catalyzed by ruthenium were reported where C-C bond formation is followed by redox isomerization.7 Using ruthenium-based catalysts, diastereoselective hydro-hydroxyalkylation only has been achieved in one isolated case6b and, to date, enantioselective variants have proven elusive. In contrast, excellent levels of relative and absolute stereocontrol have been achieved in the iridium catalyzed C-C coupling of alcohols to π-unsaturated reactants, including allylic acetates,8,9 dimethylallene8e,g,10 and 1,3-cyclohexadiene.11 This fact prompted us to explore alcohol-butadiene C-C coupling under the conditions of iridium catalyzed transfer hydrogenation. Our efforts were further motivated by the fact that the most broadly utilized protocol for stereoselective carbonyl crotylation, the method reported by Brown,12 is attended by significant preactivation and the superstoichiometric generation of the secondary alcohol byproduct, isopinocampheol, has proven problematic.13 In contrast, butadiene is an abundant petrochemical feedstock that could potentially deliver products of carbonyl crotylation in the absence of stoichiometric byproducts, while bypassing discrete steps devoted to alcohol oxidation (Scheme 1).14
Scheme 1.

Metal catalyzed hydro-hydroxyalkylation of butadiene circumvents the preactivation and byproduct generation that attends “state-of-the-art” carbonyl crotylation methodology.
In an initial series of experiments, the coupling of butadiene to alcohol 1a was explored using the ortho-cyclometallated iridium complex derived from [Ir(cod)Cl]2, various 4-substituted 3-nitrobenzoic acids, allyl acetate and the chelating phosphine ligand BIPHEP (2,2′-bis(diphenylphosphino)biphenyl). Unlike other iridium catalyzed couplings developed in our laboratory,8–11 reactions attempted in THF produced only trace quantities of 3a. Improved conversion to 3a was observed in non-Lewis basic solvents, and toluene was best among the solvents screened (Table 1, entries 1–5). Additionally, a dramatic electronic effect involving the C,O-benzoate was evident: whereas the catalyst derived from 4-cyano-3-nitrobenzoic acid “BIPHEP-I-CN”provides only an 8% yield of C-C coupling product 3a, the catalyst derived from 4-methoxy-3-nitrobenzoic acid “BIPHEP-I-OMe” delivers 3a in 62% yield (Table 1, entries 5–7). Finally, mild basic additives, in particular sodium acetate, were found to enhance conversion (Table 1, entries 5, 8–11). Under optimal conditions employing “BIPHEP-I-OMe” as precatalyst in toluene solvent employing sodium acetate as base, butadiene and alcohol 1a are converted to the product of C-C coupling 3a in 80% isolated yield. Notably, a single regioisomer is formed (Table 1).
Table 1.
Selected experiments in the optimization of the iridium catalyzed hydro-hydroxyalkylation of butadiene.a
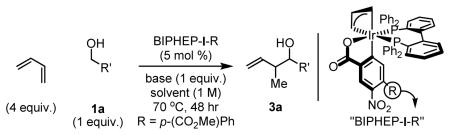 | ||||
|---|---|---|---|---|
| entry | solvent | base | R | yield 3a (syn:anti) |
| 1 | THF | NaHCO3 | OMe | trace |
| 2 | MeCN | NaHCO3 | OMe | trace |
| 3 | dioxane | NaHCO3 | OMe | 32% (1.4:1) |
| 4 | DCE | NaHCO3 | OMe | 49% (1.4:1) |
| 5 | PhMe | NaHCO3 | OMe | 62% (1.4:1) |
| 6 | PhMe | NaHCO3 | H | 57% (1.4:1) |
| 7 | PhMe | NaHCO3 | CN | 8% (1:1) |
| 8 | PhMe | No Base | OMe | 31% (1.4:1) |
| 9 | PhMe | KHCO3 | OMe | 57% (1.4:1) |
| 10 | PhMe | Li2CO3 | OMe | trace |
| 11 | PhMe | NaOAc | OMe | 80% (1.4:1) |
Reactions were performed in 13 × 100 mm pressure tubes. The cited yields are of material isolated by silica gel chromatography. See Supporting Information for experimental details.
Under these optimized conditions, butadiene was coupled to alcohols 1a-1i. Benzylic alcohols 1a-1f were converted to products of carbonyl crotylation 3a-3f in good yield. Allylic alcohols 1g and 1h also couple to butadiene to provide moderate yields homoallylic alcohols 3g and 3h. Finally, the unactivated aliphatic alcohol 1i combines with butadiene to provide a 52% yield of the hydro-hydroxyalkylation product 3i. Although products 3a-3i are obtained as mixtures of syn- and anti-diastereomers, only a single regioisomer is formed in each case (Table 2).
Table 2.
Iridium catalyzed hydro-hydroxyalkylation of butadiene employing alcohols 1a-1i.a
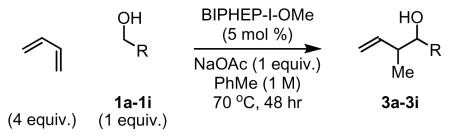 | ||
|---|---|---|
| 1a, R = p-(CO2Me)Ph | 1b, R = p-(COMe)Ph | 1c, R = p-CF3Ph |
| 1d, R = p-NO2Ph | 1e, R = p-BrPh | 1f, R = 2-Furyl |
| 1g, R = CH=CHPh | 1h, R = CH=CHCH2OBn | 1i, R = (CH2)7Me |
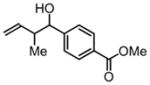 3a, 80% Yield 1.4:1 (syn: anti) |
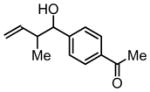 3b, 82% Yield 1.4:1 (syn: anti) |
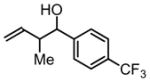 3c, 86% Yield 1.4:1 (syn: anti) |
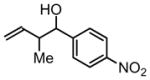 3d, 70% Yield 1:1.3 (syn: anti) |
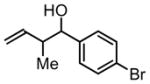 3e, 62% Yield 1.7:1 (syn: anti) |
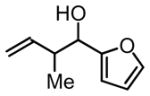 3f, 73% Yield 1.1:1 (syn: anti)b |
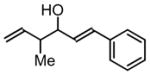 3g, 64% Yield 1.5:1 (syn: anti) |
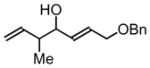 3h, 62% Yield 1.4:1 (syn: anti) |
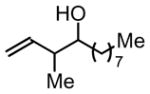 3i, 52% Yield 1.5:1 (syn: anti)c |
The modest levels of syn-diastereoselectivity suggests a kinetic preference for butadiene hydrometallation from the s-cis conformer to deliver the anti-π-allyl which, in turn, provides the (Z)-σ-allyl stereoisomer. It is likely that conversion of the kinetically preferred (Z)-σ-allyl stereoisomer to the thermodynamically preferred (E)-σ-allyl stereoisomer occurs at a rate comparable to carbonyl addition. Alternatively, a Curtin-Hammett scenario might be operative, wherein full equilibration between the (Z) and (E)-σ-allyl stereoisomers is achieved, yet there exists a slight kinetic preference for carbonyl addition from the thermodynamically less stable (Z)-σ-allyl stereoisomer (Scheme 2).
Scheme 2.

Proposed catalytic mechanism for iridium catalyzed hydro-hydroxyalkylation of butadiene.
For nearly all the C-C bond forming transfer hydrogenations we have developed,3–11 carbonyl addition may be achieved from both the alcohol or aldehyde oxidation levels. In the latter case, a stoichiometric reductant such as isopropanol or formic acid is required. Using BIPHEP-I-OMe as precatalyst, the reductive coupling of butadiene to aldehydes mediated by 1,4-butanediol14 occurs smoothly, as demonstrated by the conversion of aldehydes 2e-2g and 2i to crotylation products 3e-3g and 3i. Thus, butadiene-mediated carbonyl crotylation occurs with equal facility from the alcohol or aldehyde oxidation level (Scheme 3).
Scheme 3.

Iridium catalyzed reductive coupling of butadiene to aldehydes 2e-2g and 2i.
Having established favorable reactivity, a preliminary evaluation of chirally modified catalysts was undertaken. Toward this end, the ortho-cyclometallated iridium C,O-benzoate derived from (S)-SEGPHOS and (R)-WALPHOS (SL-W002-1) were prepared and assayed in the coupling of butadiene to alcohol 1a. Using (S)-SEGPHOS-I-OMe, the crotylation product 3a was obtained in 80% yield as a 1.4:1 mixture of syn- and anti-diastereomers, respectively. High levels of enantiomeric enrichment were observed for the syn-stereoisomer (87% ee). Using (R)-WALPHOS-I-OMe, the crotylation product 3a was obtained in 40% yield as a 1:2.2 mixture of syn- and anti-diastereomers, respectively. High levels of enantiomeric enrichment were observed for the major anti-diastereomer (88% ee) (Scheme 4).
Scheme 4.

Preliminary evaluation of chirally modified catalysts.
In summary, we report the iridium catalyzed hydro-hydroxyalkylation of butadiene employing alcohols 1a-1i to furnish products of carbonyl crotylation 3a-3i. Using 1,4-butanediol as hydrogen donor, butadiene couples to aldehydes 2e-2g and 2i to provide products of carbonyl crotylation 3e-3g and 3i. Thus, butadiene-mediated carbonyl crotylation occurs with equal facility from the alcohol or aldehyde oxidation level. Although products 3a-3i are obtained as diastereomeric mixtures, preliminary studies employing chiral iridium catalysts modified by (S)- SEGPHOS and (R)-WALPHOS (SL-W002-1) reveal promising levels of asymmetric induction and catalyst-directed diastereocontrol. Future studies will focus on the development of second-generation catalysts that promote butadiene-mediated carbonyl crotylation with control of relative and absolute stereochemistry.
Experimental Section
Procedure for the Synthesis of BIPHEP-I-OMe
To an oven-dried sealed tube under an atmosphere of nitrogen charged with [Ir(cod)Cl]2 (100 mg, 0.15 mmol, 100 mol%), BIPHEP (156 mg, 0.3 mmol, 200 mol%), Cs2CO3 (195 mg, 0.6 mmol, 400 mol%) and 4-methoxy-3-nitrobenzoic acid (100 mg, 0.6 mmol, 400 mol%) was added THF (3 mL, 0.05 M). The reaction mixture was heated at 80 °C for 30 min and was then allowed to cool to ambient temperature. Allyl acetate (75 mg, 0.75 mmol, 500 mol%) was added and the reaction mixture was allowed to stir for an additional 90 min at 80 °C, at which point the reaction mixture was allowed to cool to ambient temperature. The reaction mixture was filtered and washed with THF (15 mL) until all yellow residue was dissolved. The filtrate was concentrated in vacuo and hexanes (50 mL) was added. The resulting yellow precipitate was collected by filtration and dried under vacuum (253 mg, 0.266 mmol, 90% yield).
General Procedure for Iridium Catalyzed Hydro-hydroxyalkylation of Butadiene
To an oven-dried sealed tube under an atmosphere of nitrogen charged with alcohol 1a (50 mg, 0.30 mmol, 100 mol%), Ir-BIPHEP-1-OMe (14 mg, 0.015 mmol, 5 mol%), sodium acetate (25 mg, 0.30 mmol, 100 mol%) was added toluene (1.0 M, 0.3 mL) followed by 1,3-butadiene (0.1 ml, 1.2 mmol, 400 mol%, chilled at −78 °C). The reaction mixture was placed in a 70 °C oil batch and was allowed to stir for 48 hr, at which point the reaction mixture was concentrated in vacuo. Purification of the product by column chromatography (SiO2; ethyl acetate:hexanes, 1:10) provides the product of carbonyl crotylation 3a (53 mg) as a colorless oil in 80% yield as a single regioisomer and as a 1.4:1 mixture of syn/anti diastereomers.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation and the NIH-NIGMS (RO1-GM069445) for partial support of this research.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.200######.
Spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including scanned images of 1H and 13C NMR spectra. Scanned images of HPLC traces corresponding to racemic and enantiomerically enriched products.
References
- 1.For selected reviews, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063–1072. doi: 10.1021/jo061895m.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394–1401. doi: 10.1021/ar7001123.Shibahara F, Krische MJ. Chem Lett. 2008;37:1102–1107. doi: 10.1246/cl.2008.1102.Patman RL, Bower JF, Kim IS, Krische MJ. Aldrichim Acta. 2008;41:95–104.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem. 2009;121:36–48. doi: 10.1002/anie.200802938.Angew Chem Int Ed. 2009;48:34–46.
- 2.For related “hydrogen auto-transfer” reactions, alcohol dehydrogenation and nucleophile generation occur independently. Hence, conventional pre-activated nucleophiles are required. Such processes deliver products of formal alcohol substitution rather than carbonyl addition. For selected reviews, see: Guillena G, Ramón DJ, Yus M. Angew Chem. 2007;119:2410–2416. doi: 10.1002/anie.200603794.Angew Chem Int Ed. 2007;46:2358–2416.Hamid MHSA, Slatford PA, Williams JMJ. Adv Synth Catal. 2007;349:1555–1575.Nixon TD, Whittlesey MK, Williams JMJ. Dalton Trans. 2009:753–762. doi: 10.1039/b813383b.Dobereiner GE, Crabtree RH. Chem Rev. 2010;110:681–703. doi: 10.1021/cr900202j.Guillena G, Ramón DJ, Yus M. Chem Rev. 2010;110:1611–1641. doi: 10.1021/cr9002159.
- 3.a) Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:6338–6339. doi: 10.1021/ja801213x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shibahara F, Bower JF, Krische MJ. J Am Chem Soc. 2008;130:14120–14122. doi: 10.1021/ja805356j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Smejkal T, Han H, Breit B, Krische MJ. J Am Chem Soc. 2009;131:10366–10367. doi: 10.1021/ja904124b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Han H, Krische MJ. Org Lett. 2010;12:2844–2846. doi: 10.1021/ol101077v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patman RL, Williams VM, Bower JF, Krische MJ. Angew Chem. 2008;120:5298–5301. doi: 10.1002/anie.200801359. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:5220–5223. doi: 10.1002/anie.200801359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Patman RL, Chaulagain MR, Williams VM, Krische MJ. J Am Chem Soc. 2009;131:2066–2067. doi: 10.1021/ja809456u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Williams VM, Leung JC, Patman RL, Krische MJ. Tetrahedron. 2009;65:5024–5029. doi: 10.1016/j.tet.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Ngai MY, Skucas E, Krische MJ. Org Lett. 2008;10:2705–2708. doi: 10.1021/ol800836v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Skucas E, Zbieg JR, Krische MJ. J Am Chem Soc. 2009;131:5054–5055. doi: 10.1021/ja900827p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Grant CD, Krische MJ. Org Lett. 2009;11:4485–4487. doi: 10.1021/ol9018562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denichoux A, Fukuyama T, Doi T, Horiguchi J, Ryu I. Org Lett. 2010;12:1–3. doi: 10.1021/ol902289r. [DOI] [PubMed] [Google Scholar]
- 8.a) Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340–6341. doi: 10.1021/ja802001b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891–14899. doi: 10.1021/ja805722e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514–2520. doi: 10.1021/ja808857w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem. 2009;121:5118–5121. doi: 10.1002/anie.200901648. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2009;48:5018–5021. doi: 10.1002/anie.200901648. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Itoh J, Han SB, Krische MJ. Angew Chem. 2009;121:6431–6434. [Google Scholar]; Angew Chem, Int Ed. 2009;48:6316–6316. [Google Scholar]; f) Hassan A, Lu Y, Krische MJ. Org Lett. 2009;11:3112–3115. doi: 10.1021/ol901136w. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Bechem B, Patman RL, Hashmi S, Krische MJ. J Org Chem. 2010;75:1795–1798. doi: 10.1021/jo902697g. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Han SB, Han H, Krische MJ. J Am Chem Soc. 2010;132:1760–1761. doi: 10.1021/ja9097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For applications in target oriented synthesis, see: Lu Y, Krische MJ. Org Lett. 2009;11:3108–3111. doi: 10.1021/ol901096d.Harsh P, O’Doherty GA. Tetrahedron. 2009;65:5051–5055. doi: 10.1016/j.tet.2009.03.097.
- 10.Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916–6917. doi: 10.1021/ja902437k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bower JF, Patman RL, Krische MJ. Org Lett. 2008;10:1033–1035. doi: 10.1021/ol800159w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Brown HC, Bhat KS. J Am Chem Soc. 1986;108:293–294. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]; b) Brown HC, Bhat KS. J Am Chem Soc. 1986;108:5919–5923. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 13.Stoichiometric generation of isopinocampheol frequently complicates product isolation in Brown allylations and crotylations: Ireland RE, Armstrong JD, III, Lebreton J, Meissner RS, Rizzacasa MA. J Am Chem Soc. 1993;115:7152–7165.Burova SA, McDonald FE. J Am Chem Soc. 2004;126:2495–2500. doi: 10.1021/ja039618+.Ramachandran PV, Prabhudas B, Chandra JS, Reddy MVR. J Org Chem. 2004;69:6294–6304. doi: 10.1021/jo0492416.White JD, Hansen JD. J Org Chem. 2005;70:1963–1977. doi: 10.1021/jo0486387.Gao D, O’Doherty GA. Org Lett. 2005;7:1069–1072. doi: 10.1021/ol047322i.Gao D, O’Doherty GA. J Org Chem. 2005;70:9932–9939. doi: 10.1021/jo051681p.Liu D, Xue J, Xie Z, Wei L, Zhang X, Li Y. Synlett. 2008:1526–1528.
- 14.A single example of rhodium catalyzed butadiene-aldehyde reductive coupling to furnish products of crotylation is reported, see: Kimura M, Nojiri D, Fukushima M, Oi S, Sonoda Y, Inoue Y. Org Lett. 2009;11:3794–3797. doi: 10.1021/ol901527b.Also see reference 3a.
- 15.Maytum HC, Tavassoli B, Williams JMJ. Org Lett. 2007;9:4387–4389. doi: 10.1021/ol702029n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.