

A comparison is made between the structures of dihydroorotase from four different organisms, including B. anthracis dihydroorotase, and reveals substantial variations in the active site, dimer interface and overall tertiary structure.
Keywords: dihydroorotatse, Bacillus anthracis, pyrimidine biosynthesis
Abstract
Dihydroorotase (EC 3.5.2.3) catalyzes the reversible cyclization of N-carbamoyl-l-aspartate to l-dihydroorotate in the third step of the pyrimidine-biosynthesis pathway in Bacillus anthracis. A comparison is made between the structures of dihydroorotase from four different organisms, including B. anthracis dihydroorotase, and reveals substantial variations in the active site, dimer interface and overall tertiary structure. These differences demonstrate the utility of exploring multiple structures of a molecular target as expressed from different organisms and how these differences can be exploited for structure-based drug discovery.
1. Introduction
The pyrimidine-biosynthesis pathway is a unique and generally crucial pathway in all organisms and affords a novel opportunity for the development of therapeutics against various bacterial agents. Dihydroorotase (DHOase) is the enzyme that catalyzes a key step involving the cyclical dehydration of N-carbamoyl-l-aspartate (CA-asp) to l-dihydroorotate (DHO). The forward reaction is optimally favored at low pH (below pH 6) and the reverse reaction is favored at high pH (above pH 8). The dihydroorotase gene, pyrC, has been identified as an essential gene in several organisms, including Bacillus subtilis (Samant et al., 2008 ▶), Staphylococcus aureus (Forsyth et al., 2002 ▶) and Mycobacterium tuberculosis (Sassetti et al., 2003 ▶). The mammalian form of DHOase is substantially different, both functionally and structurally, since it is part of a trifunctional enzyme channeling glutamine to DHO and further supports the choice of the gene product as a viable molecular target for antibacterial drug discovery.
Over a dozen wild-type, mutant and ligand-bound structures, coupled with kinetic data, of Escherichia coli DHOase (EC-DHOase) have been reported (Thoden et al., 2001 ▶; Lee et al., 2005 ▶; Lee, Maher, Christopherson et al., 2007 ▶; Lee, Chan et al., 2007 ▶; Lee, Mayer & Guss, 2007 ▶), in addition to structures of DHOase from other organisms, including a recent report on the structure of S. aureas DHOase (SA-DHOase). We take advantage of the E. coli structures to examine variations in the active site, dimer interface and tertiary structural interactions with substrate, product and other ligands compared with the structure of B. anthracis DHOase (BA-DHOase). However, since E. coli is a Gram-negative bacterium, we also include a comparison of the structure of BA-DHOase with SA-DHOase since these are both Gram-positive bacteria and have a relatively high sequence identity. We also include a fourth comparison to the structure of Thermus thermophilus DHOase (TT-DHOase) since it also shares a similar sequence identity with BA-DHOase.
2. Experimental methods
2.1. Protein expression and purification
The pyrC gene was PCR-amplified from genomic DNA isolated from B. anthracis Sterne strain. Techniques for cloning and protein expression have been described previously (May et al., 2007 ▶). The cell lysate was purified by affinity and size-exclusion chromatography (SEC). The position of the eluate fraction from SEC suggests that the protein is a dimer in solution.
2.2. Crystallization, data collection and processing
BA-DHOase was crystallized using hanging-drop vapor diffusion at a protein concentration of 10 mg ml−1 at 288 K. Protein:reservoir volume ratios of 1:1, 1:2 and 2:1 were employed with 1–2 µl drop volumes. Five conditions, ranging in pH from 5.1 to 8.5, yielded small crystals from these coarse screening trials using Hampton Research Index and PEG/Ion screens. A crystallization condition consisting of 200 mM MgCl2 pH 6 and 20% PEG 3350 yielded large thin plates that were suitable for data collection.
Native crystals were flash-cooled directly from the crystallization drop. A series of ten data sets with successive rotations were collected from different portions of a single crystal on the Advanced Photon Source NE-CAT beamline with a 20 µm X-ray beam cross-section. A combination of 242 images yielded a data set that was 99% complete to 2.6 Å resolution (Table 1 ▶). The intensities were integrated with HKL-2000 (Otwinowski & Minor, 1997 ▶) and reduced to structure factors with CCP4 (Collaborative Computational Project, Number 4, 1994 ▶).
Table 1. Data-collection, phasing and refinement information.
Values in parentheses are for the highest resolution shell.
| Unit-cell parameters (Å, °) | a = 50.36, b = 80.78, c = 104.77, β = 100.21 |
| Space group | P21 (No. 4) |
| Data collection | |
| Wavelength (Å) | 0.97918 |
| No. of images | 242 |
| Resolution (Å) | 20–2.55 (2.59–2.55) |
| No. of unique reflections | 26859 (1205) |
| Completeness (%) | 98.6 (89.9) |
| Mean I/σ(I) | 31.1 (3.3) |
| Redundancy | 4.3 (2.5) |
| Rmerge† | 0.088 (0.263) |
| Refinement | |
| Resolution range (Å) | 20–2.60 |
| No. of reflections (work/test) | 24041/1224 |
| Rcryst/Rfree‡ | 0.217/0.287 |
| R.m.s.d. (bond lengths) (Å) | 0.0041 |
| R.m.s.d. (bond angles) (Å) | 1.09 |
| Average B (chain A/chain B) (Å2) | 45.7/52.9 |
| No. of non-H protein atoms | 6512 |
| No. of Zn atoms | 4 |
| No. of water molecules | 180 |
R
merge = 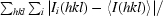
 , where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean for that set of observations.
, where I
i(hkl) is the intensity of the ith observation and 〈I(hkl)〉 is the mean for that set of observations.
R
cryst = 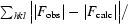
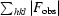 , where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes for reflection hkl in the working set; R
free is the same for the test set.
, where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes for reflection hkl in the working set; R
free is the same for the test set.
2.3. Structure determination and refinement
A search model was constructed from the structure of TT-DHOase from T. thermophilus (PDB entry 2z00; M. Kanagawa, S. Baba, S. Kuramitsu, S. Yokoyama, G. Kawai & G. Sampei, unpublished work) and sequence alignment using ClustalW (Larkin et al., 2007 ▶). The sequence identity (41%) was the highest among a comparison of BA-DHOase with known DHOase structures from seven other organisms. (The structure of SA-DHOase was not known or available at this time.) Molecular replacement using Phaser (McCoy et al., 2007 ▶) resulted in an initial model with two chains in the asymmetric unit in space group P21. Substantial manual rebuilding and refinement using CNS (Brünger et al., 1998 ▶) and O (Jones, 2001 ▶) yielded a final model which included 426 residues and two Zn atoms in each chain and a total of 180 water molecules. The secondary structure of the two chains is identical (see below) and the coordinates of chain A were restrained to chain B by noncrystallographic symmetry during the initial stages of refinement and were released during the final stages of refinement and rebuilding. Atomic coordinates have been deposited in the Protein Data Bank (PDB code 3mpg) and validated by MolProbity (Chen et al., 2010 ▶).
3. Results and discussion
3.1. Overall structure of DHO from B. anthracis
The BA-DHOase structure is a homodimer with the two chains related by a noncrystallographic twofold and centered in the middle of the interface between the two chains. Superposition of the two chains using SuperPose (Maiti et al., 2004 ▶) resulted in root-mean-square differences (r.m.s.d.s) of 0.37 Å for the 426 Cα atoms and of 0.75 Å for all 3256 atoms in a single chain. Thus, the secondary structure of one monomer is virtually identical to that of the other. The overall tertiary structure formed from the TIM-barrel secondary-structure motif is conical, with the active site residing at the base of the cone (Fig. 1 ▶). The B factors are fairly uniform throughout the chain, with the exception of the N-terminus and C-terminus and two large loops spanning residues 20–46. PyMOL (DeLano, 2006 ▶) was used to draw all figures.
Figure 1.

Depiction of the homodimer of BA-DHOase. The N-termini are shown in dark blue, the C-termini in red and the Zn atoms as orange spheres. The dimer interface is at the center of the figure.
The active site includes a binuclear Zn center, with two histidine (His59 and His61) and two asparagine (Asp151 and Asp304) residues bound to the more buried ‘α’ Zn atom and two histidine (178 and 231) residues and Asp151 bound to the ‘β’ Zn atom (Fig. 2 ▶). The carboxylate side group of Asp151 forms a bridge between the two Zn atoms, which are separated by 3.3 Å. The active site is exposed to solvent and the β-Zn atom can be further coordinated to solvent and ligand molecules. No ligand is bound in the active site of either chain in this structure. Several attempts to cocrystallize the enzyme with substrate or product were unsuccessful and further cocrystallization and soaking experiments are ongoing.
Figure 2.

The active site, with α-Zn on the left and β-Zn on the right. Electron density from a 2F o − F c map is drawn at 6.5σ.
The crystal structure reveals a large interface between monomers involving 18 residues from chain A and 17 residues from chain B (Fig. 3 ▶). The total interface accessible surface area (ASA) is 866 Å2 with a total of four hydrogen bonds and 77 nonbonded contacts as determined using PDBSUM (Laskowski, 2009 ▶).
Figure 3.

Dimer-interface region, with corresponding regions colored similarly in each chain. Water molecules at the interface are shown as brown spheres. The side groups of residues involved in hydrogen bonding and nonbonded contacts are shown as sticks from the cartoon trace.
3.2. Sequence homology of DHOase
The alignment of DHOase sequences from 39 bacterial sources revealed 17 conserved residues (Porter et al., 2004 ▶). All of these residues are near the active site and seven coordinate directly to the Zn centers. The remaining ten residues are in contact with active-site residues and serve to orient the active-site residues for optimal metal binding and ligand contact, e.g. the conserved triad Met91-Pro92-Asn93 forms a wall against active-site residues His59 and His61 by hydrophobic contact against Met91 and through hydrogen bonding to Asn93. Fig. 4 ▶ shows the alignment of DHOase from B. anthracis with those from three other bacteria: S. aureus, T. thermophilus and E. coli; they share sequence identities with BA-DHOase of 61, 41 and 15%, respectively. The large C-terminal segment, residues 315–428, contains no conserved residues and is distant from the active site.
Figure 4.

Sequence alignment of dihydroorotase from four organisms: B. anthracis, S. aureus, T. thermophilus and E. coli.
3.3. Comparison of the crystal structures of DHOase
There are two substantial differences in a comparison of the active sites of various DHOases. Firstly, there is only one Zn center in SA-DHOase (PDB entry 3gri; J. S. Brunzelle, Z. Wawrzak, T. Skarina, O. Onopriyenko, A. Savchenko & W. F. Anderson, unpublished work) compared with two Zn centers in the majority of DHOase structures. An examination of the 22 DHOase crystal structures in the PDB reveals that DHOase from Aquifex aeolicus (AA-DHOase; PDB entries 1xrf, 1xrt and 3d6n; Martin et al., 2005 ▶; Zhang et al., 2009 ▶) is the only other entry with a single Zn center in the active site. In AA-DHOase, residue His179 (active-site residue His178 in BA-DHOase) rotates away and Cys180 rotates towards and occupies the site of the missing β-Zn center. In SA-DHOase, the active site is similar to that in BA-DHOase, but His177 and Asp150 (His178 and Asp151 in BA-DHOase) are also rotated and shifted in this region without a second metal ion for alignment.
Secondly, there is a post-translationally modified carboxylated Lys coordinated to the Zn atom in all EC-DHOases compared with Asp (residue 151 in BA-DHOase) in many of the other sequences. Both the carboxylated lysine and aspartate residues bind to one or both of the Zn centers through their carboxylate side group. As noted above, there is substantial flexibility in the loop that contains Asp151, which can accommodate differences in carboxylate binding. Asp304 (Asp250 in EC-DHOase) is also singularly important in responding to a change in pH and initiating deprotonation of the amide N atom of CA-asp in the forward reaction (Porter et al., 2004 ▶). Use of a carboxylate compared with carbamate in the active site affords subtle variation in the efficiency of deprotonation, especially in the aci-carbamate resonance form, with an additional opportunity for the protonated Nζ of the carboxylated lysine to form a hydrogen bond to Tyr104 and stabilize the long active-site loop in EC-DHOase; the shorter active-site loops in other bacteria may not require this loop stabilization.
We note a variation in the shape and accessibility of the active sites in these four DHOases. There are substantial differences in the shape of the active site and the accessibility of the Zn centers. The flexible loop near residue 153 is important in changing in the shape of the active site and allowing access of ligand for active-site binding.
All four bacterial enzymes examined here are dimers in the crystal structure. SA-DHOase has an interface ASA between the two monomer chains of 937 Å2, with 27 residues from chain A and 28 residues from chain B yielding 14 hydrogen bonds and ten salt bridges. In comparison, TT-DHOase (PDB entry 2z00) has a dimer interface ASA that is larger and more extensive (1067 Å2), with 22 hydrogen bonds and 36 salt bridges. EC-DHOase (PDB code 2eg6; Lee, Chan et al., 2007 ▶) has the largest ASA of 1117 Å2, with 22 hydrogen bonds and 17 salt bridges. With the exception of EC-DHOase, the enzymes function as a monomer. Substantial positive cooperatively between the two active sites has only been reported for EC-DHOase, with a Hill coefficient of 1.57 (Lee et al., 2005 ▶). The monomer–monomer contacts are substantially different in EC-DHOase in comparison to the other three bacterial DHOases and may have subtle influences on cooperativity and catalysis.
While there is a general secondary-structural similarity of the monomers, we also examined the superposition of dimers between these bacterial species. The r.m.s.d. of Cα atoms in BA-DHOase compared with SA-DHOase is 0.98 Å2 for a single chain (monomer) superposition and 1.05 Å2 when both chains of the dimer are included. (Some Cα atoms with large displacements were omitted from this superposition, leaving an overlap of 829 atoms.) Thus, the two structures are very similar to each other as both a monomer and a dimer. The greatest difference is in the region of a flexible loop flanking the active site: Thr149–Gln156 (in BA-DHOase). The flexible loop has been observed in different conformations for EC-DHOase depending upon substrate or product docking. The flexible loop is substantially shorter in BA-DHOase, TT-DHOase and SA-DHOase and may reduce the specificity of the enzyme to bind ligand, enabling the design of inhibitors that are more selective for B. anthracis, S. aureus and T. thermophilus than for E. coli.
Likewise, the r.m.s.d. between monomers of BA-DHOase and TT-DHOase is 1.36 Å2 and is 1.81 Å2 for dimers (superposition of 805 Cα atoms), suggesting a similar secondary structure for the monomers but a different orientation of the noncrystallographic twofold axis to generate the dimer. For EC-DHOase, the r.m.s.d. is 1.96 Å2 (282 Cα atoms) for superposition with the BA-DHOase monomer; there is no relevant overlap between BA-DHOase and EC-DHOase as dimers as different EC-DHOase residues are involved in the chain A–chain B interface.
Supplementary Material
PDB reference: dihydroorotase, 3mpg
Acknowledgments
We acknowledge the use of the Advanced Photon Source, which is supported by the US Department of Energy, Basic Energy Sciences, Office of Science under contract No. DE-AC02-06CH11357. NE-CAT Sector 24 is supported by the National Institutes of Health, National Center for Research Resources under grant No. RR-15301.
References
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2006). MacPyMOL v.0.99. DeLano Scientific LLC, Palo Alto, California, USA.
- Forsyth, R. A. et al. (2002). Mol. Microbiol.43, 1387–1400. [DOI] [PubMed]
- Jones, T. A. (2001). Methods in Macromolecular Crystallography, edited by D. Turk & L. Johnson, pp. 142–147. Amsterdam: IOS Press.
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibbons, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Laskowski, R. A. (2009). Nucleic Acids Res.37, D355–D359. [DOI] [PMC free article] [PubMed]
- Lee, M., Chan, C. W., Graham, S. C., Christopherson, R. I., Guss, J. M. & Maher, M. J. (2007). J. Mol. Biol.370, 812–815. [DOI] [PubMed]
- Lee, M., Chan, C. W., Guss, J. M., Chistopherson, R. I. & Maher, M. J. (2005). J. Mol. Biol.348, 523–533. [DOI] [PubMed]
- Lee, M., Maher, M. J., Christopherson, R. I. & Guss, J. M. (2007). Biochemistry, 46, 10538–10550. [DOI] [PubMed]
- Lee, M., Maher, M. J. & Guss, J. M. (2007). Acta Cryst. F63, 154–161. [DOI] [PMC free article] [PubMed]
- Maiti, R., Van Domselaar, G. H., Zhang, H. & Wishart, D. S. (2004). Nucleic Acids Res.32, W590–W594. [DOI] [PMC free article] [PubMed]
- Martin, P. D., Purcarea, C., Zhang, P., Vaishnav, A., Sadecki, S., Guy-Evans, H. I., Evans, D. R. & Edwards, B. F. (2005). J. Mol. Biol.348, 535–547. [DOI] [PubMed]
- May, M., Mehboob, S., Mulhearn, D. C., Wang, Z., Yu, H., Thatcher, G. R., Santarsiero, B. D., Johnson, M. E. & Mesecar, A. D. (2007). J. Mol. Biol.371, 1219–1237. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst.40, 658–674. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Porter, T. N., Li, Y. & Raushel, F. M. (2004). Biochemistry, 43, 16285–16292. [DOI] [PubMed]
- Samant, S., Lee, H., Ghassemi, M., Chen, J., Cook, J. L., Mankin, A. S. & Neyfakh, A. A. (2008). PLoS Pathog.4, 1–10. [DOI] [PMC free article] [PubMed]
- Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Mol. Microbiol.48, 77–84. [DOI] [PubMed]
- Thoden, J. B., Phillips, G. N. Jr, Neal, T. M., Raushel, F. M. & Holden, H. M. (2001). Biochemistry, 40, 6989–6997. [DOI] [PubMed]
- Zhang, P., Martin, P. D., Purcarea, C., Vaishnav, A., Brunzelle, J. S., Fernando, R., Guy-Evans, H. I., Evans, D. R. & Edwards, B. F. (2009). Biochemistry, 48, 766–778. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: dihydroorotase, 3mpg