

The invertase from X. dendrorhous has been purified, deglycosylated and crystallized and diffraction data have been collected to 2.3 Å resolution.
Keywords: yeast invertase, β-fructofuranosidases, glycoside hydrolase family 32
Abstract
Xanthophyllomyces dendrorhous invertase is an extracellular enzyme that releases β-fructose from the nonreducing termini of various β-d-fructofuranoside substrates. Its ability to produce neokestose by transglycosylation makes this enzyme an interesting research target for applications in industrial biotechnology. The native enzyme, which is highly glycosylated, failed to crystallize. Therefore, it was submitted to EndoH deglycosylating treatment and crystals were grown by vapour-diffusion methods. The crystals belonged to space group P21212, with unit-cell parameters a = 75.29, b = 204.93, c = 146.25 Å. Several diffraction data sets were collected using a synchrotron source. Self-rotation function and gel-filtration experiments suggested that the enzyme is a dimer with twofold symmetry.
1. Introduction
In recent years, great efforts have been focused on providing information about the domains implicated in the substrate binding and catalytic mechanisms of the glycosyl hydrolase (GH) families; to date, three-dimensional structures of at least a representative member of 52 of the 115 GH families have been reported. This information is continuously updated in the CAZy (Carbohydrate-Active Enzymes) database (Cantarel et al., 2009 ▶).
The GH32 family (EC 3.2.1.–) includes enzymes such as invertases or β-fructofuranosidases that catalyze the release of β-fructose from the nonreducing termini of various β-d-fructofuranoside substrates. These proteins contain two acidic residues, Asp or Glu, that act as the nucleophile and the acid/base catalyst responsible for the cleavage of glycosidic bonds and are included in the GH-J clan together with the GH68 (inulosucrase) family. To date, the three-dimensional structures of the β-fructofuranosidases from the bacterium Thermotoga maritima (Alberto et al., 2004 ▶) and the yeast Schwanniomyces occidentalis (Polo et al., 2009 ▶; Álvaro-Benito et al., 2010 ▶), as well as of an exoinulinase from the fungus Aspergillus niger (Nagem et al., 2004 ▶) and of a fructan exohydrolase and a cell-wall invertase from the plants Chicorium intybus (Verhaest et al., 2005 ▶) and Arabidopsis thaliana (Lammens et al., 2008 ▶), respectively, which are all members of the GH32 family, have been solved. Very recently, the structure of the fructosyltransferase from Aspergillus japonicus has also been reported (Chuankhayan et al., 2010 ▶). These studies have shown a common bimodular folding for the GH32 family, with an N-terminal fivefold β-propeller catalytic domain and a C-terminal β-sandwich domain, the function of which is still not well known. Interestingly, this β-sandwich domain was shown to be involved in dimerization of the S. occidentalis β-fructofuranosidase, being directly implicated in shaping its active site (Álvaro-Benito et al., 2010 ▶).
In addition to releasing β-fructose from the nonreducing termini of various β-d-fructofuranoside substrates, microbial β-fructofuranosidases may catalyze the synthesis of short-chain fructooligosaccharides (FOS), in which one to three fructosyl moieties are linked to the oligosaccharide used as the biosynthetic reaction substrate, which is generally sucrose, by different glycosidic bonds depending on the enzyme source (Sangeetha et al., 2005 ▶). FOS act as prebiotics, which are nondigestible food ingredients that improve the health of the consumer by stimulation of the growth of beneficial bifid bacteria in the digestive tract (Gibson & Roberfroid, 1995 ▶). At present, Aspergillus enzymes are the main industrial producers of FOS (Ghazi et al., 2007 ▶), giving a mixture of FOS with an inulin-type structure containing β-(2→1)-linked fructose oligomers (1F-FOS; 1-kestose, nystose or 1F-fructofuranosylnystose). However, there is an interest in the development of novel molecules with improved prebiotic and physiological properties. In this context, β-(2→6)-linked FOS, in which the link exists between two fructose units (6F-FOS; 6-kestose) or between fructose and the glucosyl moiety (6G-FOS; neokestose, neonystose and neofructofuranosylnystose), have enhanced prebiotic properties compared with commercial FOS (Marx et al., 2000 ▶; Kilian et al., 2002 ▶). The basidiomycetous yeast Xanthophyllomyces dendrorhous produces an extracellular β-fructofuranosidase (XdINV) which shows broad substrate specificity and hydrolyzes sucrose, 1-kestose and nystose. Unlike other microbial β-fructofuranosidases, it produces neokestose as the main transglycosylation product (Linde et al., 2009 ▶). Therefore, structural analysis of XdINV is necessary in order to fully understand its particular biological specificity and to improve its biotechnological potential. In this study, we describe an abbreviated purification protocol to overproduce the enzyme, its crystallization and a preliminary X-ray crystallographic analysis.
2. Experimental
2.1. Protein purification and quantification
X. dendrorhous ATCC MYA-131 was grown on maltose-based medium [0.7%(w/v) yeast nitrogen base (Difco), 2%(w/v) maltose]. The purification protocol was performed basically as described previously (Linde et al., 2009 ▶) with the following exceptions: the extracellular β-fructofuranosidase (invertase) activity produced (7.2 U ml−1) by 1 l yeast culture (A 660 = 2.4) was concentrated by tangential filtration through a 30 000 molecular-weight cutoff polyethersulfone membrane using a VivaFlow 50 system (Vivascience). The active fraction (40 ml; 37 U ml−1) was dialyzed in 20 mM sodium phosphate pH 7 (buffer A) and applied onto a DEAE-Sephacel chromatography column (10 ml) equilibrated with buffer A until the pH of the effluent was the same as that of the ingoing solution (about ten column volumes). The protein was eluted with a discontinuous gradient of 0, 0.05, 0.1 and 0.2 M NaCl (ten column volumes for each salt concentration at 1 ml min−1). The active fractions eluted at 0.1 M NaCl were pooled (45 ml; 44.5 U ml−1) and concentrated to 8 mg ml−1 using an Amicon Ultra-15 50K centrifugal filter (Millipore, Ireland). All procedures were carried out at room temperature (∼293 K). The protein profiles were determined by column chromatography, measuring the absorbance of the eluate at 280 nm. SDS–PAGE (8% polyacrylamide) of the samples confirmed the purity of XdINV (Fig. 1 ▶).
Figure 1.

SDS–PAGE analysis of XdINV. Lanes 1 and 2, the culture filtrate expressing fructofuranosidase activity before (lane 1) and after (lane 2) DEAE-Sephacel chromatography. Lane 3, deglycosylated XdINV. Lane 4, molecular-weight markers (indicated in kDa on the left).
β-Fructofuranosidase activity was determined by measuring the amount of glucose liberated from sucrose [0.5%(w/v) in 50 mM sodium phosphate buffer pH 5.5] over 10–20 min at 333 K. The mixture was boiled for 10 min and the glucose was measured using a glucose oxidase–peroxidase assay (Sigma Technical Bulletin No. 510). A calibration curve was established with 100 µg ml−1 glucose solution. One unit of activity (U) was defined as that corresponding to the release of 1 µmol of glucose per minute under the conditions described above.
2.2. Determination of the oligomeric state
The native molecular weight of purified β-fructofuranosidase was estimated by size-exclusion chromatography using a BioSep-SEC-S 2000 HPLC system (Phenomenex) equilibrated with 50 mM sodium phosphate pH 6.8 at 1 ml min−1 at room temperature (about 293 K). 40 µl of protein at a concentration of 0.35 mg ml−1 was used. Ferritin (450 kDa), aldolase (158 kDa), ovoalbumin (44 kDa) and cytochrome c (12.5 kDa) were used for column calibration, with elution volumes of 5.4, 6.3, 7.2 and 8.4 ml, respectively.
2.3. Deglycosylation
After purification of XdINV, deglycosylation was performed using endoglycosidase H (Endo H; New England BioLabs). Treatment with Endo H was accomplished following the manufacturer’s recommendations at 310 K during 17 h incubation. The deglycosylated protein was subjected to 1 min ultrafiltration using an Amicon Ultra-4 50K device (Millipore, UFC 803024) to remove the Endo H present in the sample (molecular mass 37 kDa) and was subsequently concentrated to 8 mg ml−1 with a 10 kDa cutoff membrane. The molecular weight of the deglycosylated form was determined by SDS–PAGE (8%). Molecular-weight markers from Sigma–Aldrich (S8320) that covered the range 45–200 kDa were used as standards.
2.4. Crystallization
Initial crystallization conditions for the purified and deglycosylated protein samples were investigated by high-throughput techniques with a NanoDrop robot (Innovadyne Technologies Inc.) using the commercially available screens Index and SaltRx from Hampton Research and PACT Suite and JCSG+ Suite from Qiagen. The assays were carried out using the sitting-drop vapour-diffusion method at 291 K in Innovaplate SD-2 microplates (Innovadyne Technologies Inc.) by mixing 250 nl protein solution with 250 nl precipitant solution and equilibrating against 60 µl well solution. Many conditions gave crystals from the deglycosylated sample and these conditions were optimized through further sitting-drop experiments by mixing 1 µl protein solution with 1 µl precipitant solution and equilibrating against 500 µl well solution in Cryschem plates (Hampton Research).
2.5. Data collection and processing
All crystals were soaked in precipitant solution containing an additional 30% glycerol before being flash-cooled to 100 K. Crystals were tested using in-house facilities (a rotating-anode generator with Cu Kα radiation) and synchrotron radiation. Diffraction data sets were collected at the ESRF (Grenoble, France). The data sets were processed using the program MOSFLM (Leslie, 1992 ▶) and the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
X. dendrorhous β-fructofuranosidase is a highly glycosylated protein with a monomer molecular weight of 160 kDa, which decreases to 66 kDa when it is deglycosylated (Fig. 1 ▶). The size of the native enzyme estimated by size-exclusion chromatography is 327 kDa and therefore the active form of this enzyme may be composed of two identical subunits.
A preliminary search for crystallization conditions using purified XdINV led to drops that were mostly clear in salt-containing solutions and phase-separated when PEG was used as a precipitant. In contrast, deglycosylated XdINV yielded many hits from initial screenings (Fig. 2 ▶). Plate-like crystals with various forms were obtained in sodium citrate and several ammonium salts, such as ammonium sulfate, phosphate, tartrate and citrate. Long needles also grew from PEG 3350 and PEG 6000 solutions at various pH values. Optimization was performed by varying the protein and precipitant concentration and the pH and by adding different additives to the crystallization solution: MPD, glycerol, sucrose and low-weight PEGs. The best rod-shaped crystals were grown by mixing equal amounts of 8 mg ml−1 XdINV (in 100 mM NaCl, 20 mM Tris pH 7) and a solution consisting of 1.3 M sodium citrate, 2–3% MPD and 0.1 M Tris pH 8.5. The final optimized crystals were tested using a synchrotron-radiation source to obtain high-resolution data. Several native data sets were collected at 100 K on the ID29 beamline at the ESRF (Grenoble, France) at resolutions of up to 2.3 Å. The data-collection statistics are given in Table 1 ▶. The crystals belonged to space group P21212, with unit-cell parameters a = 75.3, b = 146.2, c = 204.9 Å. As seen from the SDS–PAGE analysis, the molecular weight of the monomer is 66 kDa (Fig. 1 ▶); assuming the presence of two molecules in the asymmetric unit, the Matthews coefficient (Matthews, 1968 ▶) of 4.21 Å3 Da−1 corresponds to a 70% solvent content within the cell. Alternatively, the presence of two dimers in the asymmetric unit would lead to a Matthews coefficient of 2.8 Å3 Da−1, corresponding to a 42% solvent content. We investigated the local symmetry relating the units in the asymmetric unit using POLARRFN (Kabsch, 1976 ▶) from the CCP4 package. Several self-rotation functions were computed in the resolution range 15–2.5 Å, with Patterson vectors from 25 to 40 Å radius of integration. The stereographic projection (κ = 180° section) of the self-rotation is shown in Fig. 3 ▶. Analysis of the self-rotation peaks revealed the presence of noncrystallographic twofold symmetry on the ab plane, which is compatible with the presence of one or two functional dimers in the asymmetric unit of the crystals. Structure determination was carried out using the fructosyltransferase from A. japonicus (PDB code 3lf7; Chuankhayan et al., 2010 ▶), which shows 34% sequence identity, as a model. Molecular replacement was performed with a corrected model using the XdINV sequence in the MOLREP program (Vagin & Teplyakov, 1997 ▶) with data to 2.5 Å resolution. The rotation and translation functions yielded two clear positions for the model, with a final correlation coefficient of 0.29 and an R factor of 0.57. Preliminary structural refinement of this dimer with REFMAC (Murshudov et al., 1997 ▶) lowered the R factor to 0.41 (R free = 0.46). Model building and further refinement are ongoing.
Figure 2.

Crystals of deglycosylated XdINV obtained from (a) 1.4 M ammonium tartrate, 0.1 M Tris pH 8.5 and (b) 1.2 M sodium citrate, 2% MPD, 0.1 M Tris pH 8.5. The best crystals grew in 1–2 weeks to maximum dimensions of 0.6 × 0.1 × 0.1 mm.
Table 1. Data-collection statistics for the XdINV crystal.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 0.9763 |
| Source | ESRF |
| Beamline | ID29 |
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 75.29, b = 204.93, c = 146.25 |
| Resolution limits (Å) | 68.9–2.3 (2.43–2.3) |
| Unique reflections | 95649 |
| Rmerge† | 0.09 (0.36) |
| Completeness (%) | 98.3 (93.7) |
| Mean multiplicity | 6.7 (6.1) |
| Mean I/σ(I) | 13.9 (4.3) |
| Wilson B factor (Å2) | 33.1 |
R
merge = 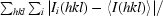
 , where Ii(hkl) is the ith observed amplitude of reflection hkl and 〈I(hkl)〉 is the mean amplitude for measurements of reflection hkl.
, where Ii(hkl) is the ith observed amplitude of reflection hkl and 〈I(hkl)〉 is the mean amplitude for measurements of reflection hkl.
Figure 3.

Plot of the self-rotation function of XdINV crystals using data between 15 and 2.5 Å resolution and a 30 Å radius of integration in the κ = 180° section. The view is down the c axis. ϕ = 0° and ϕ = 90° correspond to the a and b axes, respectively.
Acknowledgments
This work was supported by grants BIO2007-67708-C04-04 and BIO2007-67708-C04-03 from Dirección General de Investigación and by an institutional grant from the Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa. This is a product of the Project ‘Factoría Española de Cristalización’ Ingenio/Consolider 2010.
References
- Alberto, F., Bignon, C., Sulzenbacher, G., Henrissat, B. & Czjzek, M. (2004). J. Biol. Chem.279, 18903–18910. [DOI] [PubMed]
- Álvaro-Benito, M., Polo, A., González, B., Fernández-Lobato, M. & Sanz-Aparicio, J. (2010). J. Biol. Chem.285, 13930–13941. [DOI] [PMC free article] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res.37, D233–D238. [DOI] [PMC free article] [PubMed]
- Chuankhayan, P., Hsieh, C.-Y., Huang, Y.-C., Hsieh, Y.-Y, Guan, H.-H., Hsieh, Y.-C., Tien, Y.-C., Chen, C.-D., Chiang, C.-M. & Chen, C.-J. (2010). J. Biol. Chem.285, 23251–23264. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Ghazi, I., Fernández-Arrojo, L., Garcia-Arellano, H., Plou, F. J. & Ballesteros, A. (2007). J. Biotechnol.128, 204–211. [DOI] [PubMed]
- Gibson, G. R. & Roberfroid, M. B. (1995). J. Nutr.125, 1401–1412. [DOI] [PubMed]
- Kabsch, W. (1976). Acta Cryst. A32, 922–923.
- Kilian, S., Kritzinger, S., Rycroft, C., Gibson, G. & du Preez, J. (2002). World J. Microbiol. Biotechnol.18, 637–644.
- Lammens, W., Le Roy, K., Van Laere, A., Rabijins, A. & Van den Ende, W. (2008). J. Mol. Biol.377, 378–385. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Crystallographic Computing 5: From Chemistry to Biology, edited by D. Moras, A. D. Podjarny & J. C. Thierry, pp. 50–61. Oxford University Press.
- Linde, D., Macias, I., Fernández-Arrojo, L., Plou, F. J., Jiménez, A. & Fernández-Lobato, M. (2009). Appl. Environ. Microbiol.75, 1065–1073. [DOI] [PMC free article] [PubMed]
- Marx, S. P., Winkler, S. & Hartmeier, W. (2000). FEMS Microbiol. Lett.182, 163–169. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nagem, R. A. P., Rojas, A. L., Golubev, A. M., Korneeva, O. S., Eneyskaya, E. V., Kulminskaya, A. A., Neustroev, K. N. & Polikarpov, I. (2004). J. Mol. Biol.120, 621–623. [DOI] [PubMed]
- Polo, A., Álvaro-Benito, M., Fernández-Lobato, M. & Sanz-Aparicio, J. (2009). Acta Cryst. F65, 1162–1165. [DOI] [PMC free article] [PubMed]
- Sangeetha, P. T., Ramesh, M. N. & Prapulla, S. G. (2005). Process Biochem.40, 1085–1088.
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
- Verhaest, M., Van den Ende, W., Le Roy, K., De Ranter, C. J., Van Laere, A. & Rabijns, A. (2005). Plant J.41, 401–411. [DOI] [PubMed]