

The carbohydrate-binding region of GspB from S. gordonii strain M99 was crystallized in space group P212121 and data were collected to 1.3 Å resolution.
Keywords: GspB, glycoproteins, Streptococcus gordonii, sialic acid, adhesins, endocarditis, lectins
Abstract
The carbohydrate-binding region of the bacterial adhesin GspB from Streptococcus gordonii strain M99 (GspBBR) was expressed in Escherichia coli and purified using affinity and size-exclusion chromatography. Separate sparse-matrix screening of GspBBR buffered in either 20 mM Tris pH 7.4 or 20 mM HEPES pH 7.5 resulted in different crystallographic behavior such that different precipitants, salts and additives supported crystallization of GspBBR in each buffer. While both sets of conditions supported crystal growth in space group P212121, the crystals had distinct unit-cell parameters of a = 33.3, b = 86.7, c = 117.9 Å for crystal form 1 and a = 34.6, b = 98.3, c = 99.0 Å for crystal form 2. Additive screening improved the crystals grown in both conditions such that diffraction extended to beyond 2 Å resolution. A complete data set has been collected to 1.3 Å resolution with an overall R merge value of 0.04 and an R merge value of 0.33 in the highest resolution shell.
1. Introduction
GspB is a 3072-residue cell-wall-anchored glycoprotein from Streptococcus gordonii that mediates the binding of this bacterium to salivary glycoproteins and human platelets (Bensing & Sullam, 2002 ▶). The former interaction is important for colonization of the oral cavity, while the latter is a key event in the pathogenesis of infective endocarditis (Xiong et al., 2008 ▶). This unusual adhesin is a member of an expanding family of serine-rich glycoproteins that are found in most pathogenic streptococci and staphylococci (Takahashi et al., 2006 ▶; Siboo et al., 2005 ▶; Rose et al., 2008 ▶; Seifert et al., 2006 ▶) and are believed to mediate pathogen host attachment and promote bacterial biofilm formation (Sanchez et al., 2010 ▶; Wu et al., 2007 ▶). GspB contains an atypical 90-amino-acid N-terminal signal peptide, a serine-rich region, a basic region, a second serine-rich region and a C-terminal cell-wall-anchoring domain (Fig. 1 ▶). The basic region (GspBBR) mediates the binding of GspB to sialylated carbohydrate moieties on platelet glycoprotein Ibα (Takamatsu et al., 2005 ▶), salivary mucin MG2 and salivary agglutinin (Takamatsu et al., 2006 ▶) through its high-affinity interaction with NeuAcα(2–3)Galβ(1–3)GalNAc (sialyl-T antigen). Of note, the binding properties of GspB homologs vary considerably, with some homologs (e.g. Hsa of S. gordonii strain Challis and SrpA of S. sanguinis) binding to a broader or different range of carbohydrate motifs, while others (such as PsrP of S. pneumoniae) have no apparent lectin-like activity (Shivshankar et al., 2009 ▶; Takamatsu et al., 2005 ▶; Yajima et al., 2005 ▶).
Figure 1.

Overall architecture of GspB. GspB is comprised of a signal peptide (SP), a short serine-rich region (SRR1), a unique basic region (BR) that is responsible for carbohydrate binding, a second, longer, serine-rich region (SRR2) and a cell-wall-anchoring domain (CWAD). A recently published structural study of the Fap1 adhesin from S. parasanguinis identified the structural elements of both the serine-rich repeats and the unique region in that protein (Ramboarina et al., 2010 ▶). The repeat region of GspB is likely to form a super-helical fibril like that observed in the repeat region of Fap1.
These studies will ultimately identify the molecular details of carbohydrate selectivity by GspB and related serine-rich repeat adhesins. This is a critical starting point for understanding how carbohydrate binding by GspB and related lectins affects pathogen infectivity. Furthermore, a crystal structure may allow the design of small-molecular inhibitors to disrupt carbohydrate binding, which offers a new route for the design of therapeutics. Here, we report the expression, purification and crystallization of the carbohydrate-binding domain GspBBR from S. gordonii strain M99.
2. Materials and methods
2.1. Expression and purification
GspBBR comprises residues 233–615 of GspB and retains lectin behavior (Takamatsu et al., 2005 ▶). The gene encoding GspBBR was amplified from a patient isolate of S. gordonii strain M99 and was cloned into the pGEX plasmid encoding an N-terminal GST fusion tag as previously described (Takamatsu et al., 2005 ▶). This plasmid was transformed into electrocompetent Escherichia coli BL21 Gold (DE3) cells (Stratagene) and grown in LB medium with 100 µg ml−1 ampicillin at 310 K until the optical density at 600 nm (OD600) was between 0.4 and 0.5. The cells were then cold-shocked by incubation in an ice–water bath for 20 min. GspBBR expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM and the cells were incubated with shaking at 291 K overnight. The cells were harvested by centrifugation at 4000g for 15 min at 277 K and the pellets were washed with ice-cold phosphate-buffered saline (PBS) and spun down at 4500g for 15 min at 277 K.
The cell pellets were resuspended in ice-cold PBS supplemented with 1 mM dithiothreitol, 1 µg ml−1 leupeptin, 1 µg ml−1 pepstatin A, 0.17 mg ml−1 phenylmethylsulfonyl fluoride, 1 µg ml−1 DNaseI and 10 µg ml−1 RNase. The cells were disrupted by sonication for 20 min with 0.7 s pulses at 277 K. The lysate was clarified by centrifugation at 27 000g for 30 min. The supernatant was passed through a 0.45 µm filter and loaded onto a 5 ml GSTrap column (GE Healthcare) at 1 ml min−1. The column was washed with PBS supplemented with 1 mM dithiothreitol until the absorbance at 280 nm (A 280) returned to 0 and was then eluted with buffer containing 50 mM Tris pH 8.0 and 10 mM l-glutathione. The eluted protein was concentrated using a 50 kDa molecular-weight cutoff spin concentrator (Millipore). The GST tag was cleaved using 1 U factor Xa (NEB) in a buffer consisting of 100 mM NaCl, 2 mM CaCl2 and 50 mM Tris pH 8.0 for 36 h at room temperature (296 K). The cleavage reaction was passed through a 0.2 µm filter (Costar, Spin-X) and separated at 0.3 ml min−1 on a 24 ml Hi-Load Superdex 200 10/300 size-exclusion column (GE Healthcare) equilibrated in 20 mM Tris pH 7.4 or 20 mM HEPES pH 7.5. Fractions were analyzed using SDS–PAGE (Fig. 2 ▶) and concentrated using a 30 kDa molecular-weight cutoff concentrator (Millipore). The protein concentration was determined using the Quickstart Bradford Assay Kit (Bio-Rad).
Figure 2.

Purification of GspBBR for crystallization. The gel shows two separate concentrations of the GST-GspBBR fusion protein after affinity purification and two concentrations of purified isolated GspBBR(246–601) after size-exclusion chromatography. Lane 1, Kaleidoscope Ladder (Bio-Rad; labeled in kDa); lanes 2 and 3, 4 µg GST-GspBBR; lane 4, 1 µg GST-GspBBR; lane 5, 4 µg of GST and GspBBR after factor Xa cleavage; lane 6, 4 µg GspBBR after size-exclusion chromatography; lane 7, 1 µg GspBBR after size-exclusion chromatography; lane 8, dissolved crystals grown in 26% Jeffamine-ED 2001 and 0.05 M HEPES pH 7.5.
2.2. Crystallization
GspBBR formed diffraction-quality crystals (Fig. 3 ▶) in two chemically distinct sets of conditions that resulted from sparse-matrix screening of the protein in two different buffers: 20 mM Tris pH 7.4 and 20 mM HEPES pH 7.5. Sparse-matrix screening of GspBBR in either buffer was performed using a Mosquito crystallization robot (TTP LabTech) and sampled conditions from Crystal Screen, Crystal Screen 2 and Index Screen (Hampton Research) and Wizard I and II (Emerald BioSystems).
Figure 3.

Crystals of GspBBR from S. gordonii strain M99 grown using the hanging-drop vapor-diffusion method. (a) Crystals of GspBBR grown from 25% PEG 3350, 0.15 M ammonium acetate, 0.1 M HEPES pH 7.5 and 10 mM spermidine. (b) Crystals of GspBBR grown from 34% Jeffamine ED-2001, 0.15 M KCl and 0.1 M HEPES pH 7.5.
The first crystal form was grown using the hanging-drop vapor-diffusion method with drops consisting of 1 µl 10 mg ml−1 GspBBR in 20 mM HEPES pH 7.5 mixed with 1 µl reservoir solution (25% PEG 3350, 0.15 M ammonium acetate, 0.1 M HEPES pH 7.5) and equilibrated against 1 ml reservoir solution. Crystals grew within 3 d and were flash-cooled in liquid nitrogen without the addition of an additional cryopreservation agent. The initial crystals diffracted to 3 Å resolution. Optimization was performed using grid screening together with the Additive Screen kit from Hampton Research. The inclusion of 10 mM spermidine in the crystallization conditions increased the crystal size and improved the diffraction limit to 1.9 Å resolution (Fig. 4 ▶ a).
Figure 4.

Typical diffraction for each of the crystal forms. (a) Diffraction image for crystals grown using PEG 3350 as the precipitant. This diffraction image was collected on the SSRL 11-1 beamline and diffraction to 1.8 Å resolution can be observed. (b) Typical diffraction image for crystals grown using Jeffamine ED-2001 as the precipitant. This diffraction image was collected on the LS-CAT ID-21-G beamline and diffraction to 1.3 Å resolution can be observed.
A second set of chemically distinct crystallization conditions was identified for GspBBR after repeating sparse-matrix screening with 6 mg ml−1 GspBBR in 20 mM Tris pH 7.4. Crystals were grown using the hanging-drop vapor-diffusion method with drops consisting of 1 µl GspBBR and 1 µl reservoir solution [33% Jeffamine ED-2001 (Hampton Research) and 0.1 M HEPES pH 7.5] equilibrated against 1 ml reservoir solution. Crystals appeared within 2 d, but required three weeks to grow to full size. Prior to flash-cooling in liquid nitrogen, crystals were cryoprotected in a solution containing all of the components of the reservoir solution with the addition of 15% glycerol. Additive screening identified that the inclusion of 0.15 M KCl improved the diffraction quality of these crystals from 1.8 to 1.3 Å resolution (Fig. 4 ▶ b).
2.3. Data collection and processing
Crystal quality was assessed on Stanford Synchrotron Radiation Lightsource (SSRL) beamlines 9-2, 11-1 and 12-2 and the Life Sciences Collaborative Access Team (LS-CAT) ID-21-D/F/G beamlines. Although the two crystallization conditions are chemically distinct, both resulted in the formation of orthorhombic crystals in space group P212121 (Table 1 ▶). Diffraction data for the crystals obtained using PEG 3350 as the precipitant were collected on beamline 11-1 at SSRL using a MAR 325 CCD detector with a distance of 250 mm and a wavelength of 1.03034 Å. A data set consisting of 90 frames was collected with a rotation angle of 90° and an exposure time of 20 s per frame. Diffraction data for the crystals obtained using Jeffamine ED-2001 as the precipitant were collected on beamline ID-21-G at the Advanced Photon Source using a MAR 325 CCD detector with a distance of 165 mm and a wavelength of 0.97856 Å. A data set consisting of 240 frames was collected with a rotation angle of 90° and an exposure time of 2 s per frame. All data were processed and scaled using the HKL-2000 program package (Otwinowski & Minor, 1997 ▶).
Table 1. X-ray data-collection statistics for crystals scaled in space group P212121 .
Values in parentheses are for the highest resolution shell.
| Crystals obtained using | ||
|---|---|---|
| PEG 3350 | Jeffamine ED-2001 | |
| Space group | P212121 | P212121 |
| Wavelength (Å) | 1.03034 | 0.97856 |
| Beamline | SSRL 11-1 | LS-CAT ID-21-G |
| Resolution (Å) | 50–1.90 | 50–1.29 |
| Unit-cell parameters (Å, °) | a = 33.3, b = 86.7, c = 117.9, α = β = γ = 90 | a = 34.6, b = 98.3, c = 99.0, α = β = γ = 90 |
| No. of measured reflections | 84235 | 380613 |
| No. of unique reflections | 26193 | 83230 |
| Multiplicity | 3.2 (2.9) | 4.6 (2.8) |
| 〈I/σ(I)〉 | 16.8 (3.9) | 21.3 (2.6) |
| Completeness (%) | 93.8 (92.4) | 96.7 (73.6) |
| Rmerge† (%) | 6.8 (35.3) | 4.0 (33.2) |
| No. of molecules in asymmetric unit | 1 | 1 |
| Matthews coefficient (Å3 Da−1) | 2.2 | 2.1 |
| Solvent content (%) | 43.2 | 42.5 |
R
merge = 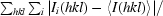
 , where I
i(hkl) is the ith instance of the intensity at position hkl and 〈I(hkl)〉 is the average of all instances of the reflections at position hkl.
, where I
i(hkl) is the ith instance of the intensity at position hkl and 〈I(hkl)〉 is the average of all instances of the reflections at position hkl.
3. Results and discussion
3.1. Crystallization and data collection
Sparse-matrix screening of the GspBBR protein in different buffers resulted in the identification of dramatically different crystallization conditions (Fig. 3 ▶). This suggests that parallel sparse-matrix screening of proteins in different buffers may be a general method to increase the probability of growing diffraction-quality crystals of a protein. In this case, orthorhombic crystals of GspBBR with 20 mM HEPES pH 7.5 as the buffer formed from conditions that used PEG 3350 as the precipitant, while crystals of GspBBR with 20 mM Tris pH 7.4 as the buffer formed from conditions using Jeffamine ED-2001 as the precipitant. Crystals of GspBBR grown using Jeffamine ED-2001 as the precipitant displayed superior diffraction quality (Fig. 4 ▶) and reproducibility. As a result, this crystal form was used exclusively for structure determination.
The unit-cell parameters for the crystals grown from the conditions containing PEG 3350 were a = 33.3, b = 86.7, c = 117.9 Å, α = β = γ = 90°, while the unit-cell parameters for the crystals grown from the conditions containing Jeffamine ED-2001 as the precipitant were a = 34.6, b = 98.3, c = 99.0 Å, α = β = γ = 90° (Table 1 ▶). Since the b and c unit-cell parameters are similar in length for the crystals grown from Jeffamine ED-2001, the crystals were originally assumed to be tetragonal; however, scaling in SCALEPACK (Otwinowski & Minor, 1997 ▶) resulted in unreasonable R merge values and an unreasonable number of rejected reflections. This strongly suggested that the crystals were orthorhombic. Specific volume calculations (Matthews, 1968 ▶) suggested the presence of one molecule of GspBBR per asymmetric unit and a solvent content of 43% for both crystal forms (Table 1 ▶).
3.2. Identification of heavy-atom derivatives
GspBBR does not exhibit significant sequence similarity to any protein of known structure; it contains only two methionines and no cysteines. As a result, heavy-atom derivatives were prepared for phasing. Two heavy-atom derivatives, Dy3+ and Ho3+, were prepared by soaking GspBBR crystals with either 1 mM DyCl3 for 3 d or 10 mM HoCl3 for 3 d. Data were collected using the wavelengths and beamlines listed in Table 2 ▶ and were processed and scaled using the HKL (Otwinowski & Minor, 1997 ▶) and CCP4 (Collaborative Computational Project, Number 4, 1994 ▶) suites of programs. The location of the positions of both Dy3+ and Ho3+ were independently identified as the same site using the SHELXD (Sheldrick, 2008 ▶) subroutine in the program SHARP (de La Fortelle & Bricogne, 1997 ▶). The extreme non-isomorphism between all data sets suggested that the inclusion of multiple crystals in the phasing calculation would be detrimental. As a result, three-wavelength multiple-wavelength anomalous dispersion data sets for both the Dy3+ and Ho3+ derivatives were carefully collected and used for phasing in the absence of a native reference data set. Phases were calculated using SHARP (de La Fortelle & Bricogne, 1997 ▶) and were further improved by solvent flattening using DM (Collaborative Computational Project, Number 4, 1994 ▶; Cowtan, 1994 ▶). Details of the structure determination and analysis will be published elsewhere.
Table 2. Data-collection statistics for the DyCl3 and HoCl3 MAD data sets.
Values in parentheses are for the highest resolution shell.
| DyCl3 | HoCl3 | |||||
|---|---|---|---|---|---|---|
| Peak | Inflection | Remote | Peak | Inflection | Remote | |
| Wavelength (Å) | 1.59083 | 1.59122 | 1.02463 | 1.53523 | 1.53583 | 1.01623 |
| Beamline | SSRL 9-2 | SSRL 9-2 | SSRL 9-2 | SSRL 9-2 | SSRL 9-2 | SSRL 9-2 |
| Resolution (Å) | 50–1.98 | 50–1.98 | 50–1.55 | 50–2.90 | 50–2.80 | 50–2.75 |
| Unit-cell parameters (Å, °) | a = 35.5, b = 98.5, c = 99.1, α = β = γ = 90 | a = 34.5, b = 98.5, c = 99.1, α = β = γ = 90 | a = 34.5, b = 98.4, c = 99.0, α = β = γ = 90 | a = 34.6, b = 99.0, c = 98.3, α = β = γ = 90 | a = 34.6, b = 99.0, c = 99.0, α = β = γ = 90 | a = 34.7, b = 99.1, c = 98.8, α = β = γ = 90 |
| No. of measured reflections | 203951 | 206064 | 431338 | 45488 | 48993 | 53679 |
| No. of unique reflections | 23918 | 23929 | 48691 | 6172 | 6842 | 6889 |
| Multiplicity | 8.5 (8.1) | 8.6 (8.1) | 8.9 (8.3) | 7.4 (6.0) | 7.2 (5.1) | 7.8 (6.6) |
| I/σ(I) | 31.2 (19.3) | 31.2 (18.4) | 27.2 (6.2) | 12.4 (3.6) | 11.1 (2.7) | 10.4 (3.1) |
| Completeness (%) | 98.0 (96.2) | 98.0 (95.6) | 97.2 (99.6) | 77.1 (80.6) | 76.9 (75.6) | 73.1 (77.7) |
| Rmerge† (%) | 8.7 (17.9) | 8.0 (17.7) | 6.1 (35.9) | 14.5 (32.6) | 13.9 (32.0) | 12.8 (36.2) |
R
merge =
, where I
i(hkl) is the ith instance of the intensity at position hkl and 〈I(hkl)〉 is the average of all instances of the reflection at position hkl.
Acknowledgments
This work was supported by American Heart Association grant 09GRNT2220122 (TMI), Pilot Project funds from the Vanderbilt Institute of Chemical Biology (TMI), Pilot funds from the VICTR CTSA UL1 RR024975 from NCRR/NIH (TMI), the Department of Veterans Affairs (PMS and GC) and grants AI041513 and AI057433 (PMS) and GM61606 (GC) from the National Institutes of Health. Portions of this research used facilities supported by Vanderbilt Core Grant in Vision Research P30EY008126. Portions of this research were carried out on beamlines 9-2, 11-1 and 12-2 at Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and the National Institute of General Medical Sciences. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor in support of this research program (grant 085P1000817). We thank Thomas Tomasiak, Timothy Panosian, Dr Mikio Tanabe, Dr Jessica Vey and Tarjani Thaker for assistance with data collection.
References
- Bensing, B. A. & Sullam, P. M. (2002). Mol. Microbiol.44, 1081–1094. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cowtan, K. (1994). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.31, 34–38.
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Ramboarina, S. et al. (2010). J. Biol. Chem.285, 32446–32457. [DOI] [PMC free article] [PubMed]
- Rose, L., Shivshankar, P., Hinojosa, E., Rodriguez, A., Sanchez, C. J. & Orihuela, C. J. (2008). J. Infect. Dis.198, 375–383. [DOI] [PubMed]
- Sanchez, C. J., Shivshankar, P., Stol, K., Trakhtenbroit, S., Sullam, P. M., Sauer, K., Hermans, P. W. & Orihuela, C. J. (2010). PLoS Pathog.6, e1001044. [DOI] [PMC free article] [PubMed]
- Seifert, K. N., Adderson, E. E., Whiting, A. A., Bohnsack, J. F., Crowley, P. J. & Brady, L. J. (2006). Microbiology, 152, 1029–1040. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shivshankar, P., Sanchez, C., Rose, L. F. & Orihuela, C. J. (2009). Mol. Microbiol.73, 663–679. [DOI] [PMC free article] [PubMed]
- Siboo, I. R., Chambers, H. F. & Sullam, P. M. (2005). Infect. Immun.73, 2273–2280. [DOI] [PMC free article] [PubMed]
- Takahashi, Y., Takashima, E., Shimazu, K., Yagishita, H., Aoba, T. & Konishi, K. (2006). Infect. Immun.74, 740–743. [DOI] [PMC free article] [PubMed]
- Takamatsu, D., Bensing, B. A., Cheng, H., Jarvis, G. A., Siboo, I. R., Lopez, J. A., Griffiss, J. M. & Sullam, P. M. (2005). Mol. Microbiol.58, 380–392. [DOI] [PubMed]
- Takamatsu, D., Bensing, B. A., Prakobphol, A., Fisher, S. J. & Sullam, P. M. (2006). Infect. Immun.74, 1933–1940. [DOI] [PMC free article] [PubMed]
- Wu, H., Zeng, M. & Fives-Taylor, P. (2007). Infect. Immun.75, 2181–2188. [DOI] [PMC free article] [PubMed]
- Xiong, Y. Q., Bensing, B. A., Bayer, A. S., Chambers, H. F. & Sullam, P. M. (2008). Microb. Pathog.45, 297–301. [DOI] [PMC free article] [PubMed]
- Yajima, A., Takahashi, Y. & Konishi, K. (2005). Microbiol. Immunol.49, 795–800. [DOI] [PubMed]