

Abstract
The reaction of 5,10,15-tritolylcorrole with EtMgBr opens the way for novel functionalizations of the corrole ring. DDQ oxidation of the macrocycle, followed by addition of the Grignard reagent, led to the formation of 5- and 10-alkyl substituted isocorroles in satisfying yields. Together with the one-pot formation of these isocorrole isomers, the use of such a nucleophile evidenced the competitive reactivity of the macrocycle β-positions, leading to the formation of 2-bromo- and 3-bromo-5,10,15-tritolylcorrole. While the formation of these monobromocorrole derivatives is not unprecedented, this is the first time the isomers have been separated and fully characterized. Furthermore, the higher yields of the 2-substituted species highlight a useful regioselectivity for the substitution reaction.
Keywords: corrole, isocorrole, oxidation, synthesis
INTRODUCTION
Modification of the porphyrin molecular framework has been intensively investigated in the past [1] with two principal aims: to probe structure-property relationships, and to modulate the properties of the macrocycle for potential applications. One of the many possible variations consists of the introducing sp3 hybridized carbon atoms in the porphyrin molecular core. Depending on the skeletal position where this modification is introduced, different hydroporphyrins can be obtained, both aromatic (chlorin) and non-aromatic (phlorin, isoporphyrin and porphodimethene) [2]. The importance of these hybrid materials has to be related not only to their role as intermediates in natural processes or in their occurrence in oxidative reactions involving porphyrins [3], but also to their distinctive electronic absorption and redox behavior, which make them promising as photosensitizers in PDT [4] or as receptors for anions and small molecules [5].
Inspired by the well-established chemistry of the porphyrinoids, researchers have made several attempts to prepare their corrole analogs. In particular, isocorroles have been reported wherein the sp3-carbon atom is located in a meso-position and their occurrence in many corrole-related reactions has already been documented [6]. For the isocorrole non-aromatic macrocycle, two isomers have been isolated, depending on the meso-position involved; these are the 10-isocorrole and the 5-isomer (Fig. 1). After the first publication of a nickel 10-isocor-rolate complex [7], other groups have devoted their attention to the synthesis of these macrocycles. Deahen and co-workers reported the preparation of a Ni-5-isocorrole complex by using the MacDonald [2+2] condensation of a 1,9-diformyl-5,5-dipropyldipyrromethane with a 5-aryldipyrromethane in the presence of Ni-acetate [8]. The isocorrole, called pseudocorrole to distinguish it from the (2.0.1.0)-type corrole analog, and reported in some articles also as isocorrole [9], was isolated in only trace amounts, along with calix[4]phyrins. Higher yields of isocorroles (10%) were obtained using 1-formyl-5,5-dipropyldipyrromethane in the condensation, instead of the diformyl derivative. More recently, the synthesis of the free-base isocorroles has been reported. In 2006, Setsune et al. [10] described the isolation of 10,10-dimethylisocorrole in 28% yield, and more recently Geier and co-workers investigated two complementary routes leading to the formation of 5,5-dimethylisocorroles (31%) [11].
Fig. 1.

Molecular structure of (a) 10-isocorrole and (b) the corresponding 5-isomer
A common feature of these approaches is the lengthy and difficult preparation of the pyrrolic precursors, and the formation of one of the two possible isomers. We have taken a different route for the preparation of isocorroles, using a 5,10,15-triarylcorrole as starting material [12]. Oxidation of the corrole ring by DDQ in the presence of methanol afforded the simultaneous preparation of both 5- and 10-methoxy isocorroles (74% overall yield). The scope of the reaction was quite general, being successful with a wide range of 5,10,15-triarylcorroles. It is worth mentioning that this is the only reported method for the preparation of triarylisocorrole derivatives and that these macrocycles can be further functionalized at the β-pyrrolic carbon atoms.
The availability of free-base isocorroles led us to investigate the coordination chemistry of this macro-cycle. The Ni and Cu complexes of isocorrole were obtained, but in the case of metal ions having formal oxidation states higher than +2 when coordinated to corroles, the metalation reaction induced the rearomatization of the macrocycle, affording the corresponding corrole metal complexes. Electrochemical studies showed that isocorroles were converted into the corresponding corroles upon reduction [13]. On the other hand, Setsune et al. reported the metalation of gem-dimethylisocorrole [14] to give stable four-, five- and six-coordinate complexes, with the exception of Zn(II) isocorrole that easily undergoes demetalation. Such a difference could confidently be attributed to the stabilization of the gem-dialkyl substitution in the starting isocorrole free bases, whereas the –OCH3 group is a better leaving group, which can allow rearomatization during the metalation process.
These results showed that the coordination chemistry of 5- and 10-alkoxy substituted isocorroles is limited and led us to explore the possible preparation of more stable species following the same one-pot procedure. We report here the preparation of 5- and 10-ethyl-triarylisocorroles together with the first characterization of monobromo-substituted corroles obtained using the same reaction pathway.
EXPERIMENTAL
General
Silica gel 60 (70–230 mesh, Carlo Erba) was used for column chromatography. PLC silica gel 60 (2 mm, Merck) was used for preparative TLC. Reagents and solvents (Aldrich, Merck or Fluka) were of the highest grade available and were used without further purification. CH2Cl2 used for the spectrophotometric measurements was stored over activated molecular sieves. 1H NMR spectra were recorded on a Bruker AV300 (300 MHz) spectrometer. Chemical shifts are given in ppm relative to residual CHCl3 (7.25 ppm). UV-vis spectra were measured on a Cary 50 spectrophotometer. Mass spectra (FAB mode) were recorded on a VG Quattro spectrometer in the positive-ion mode using m-nitrobenzyl alcohol (Aldrich) as a matrix. Elemental analysis (C, H, N) were obtained at the Microanalytical Laboratory of the University of Padova, Italy.
Synthesis of isocorroles
Preparation
A solution of TTCorH3 [15] (100 mg, 0.176 mmol) and DDQ (42 mg, 0.176 mmol) in anhydrous toluene (100 mL) was placed in a 250 mL round-bottomed flask and stirred at room temperature under nitrogen. After 5 min, a solution of EtMgBr 1 M in THF (352 μL, 0.352 mmol) was added. The reaction progress was monitored both by TLC (CH2Cl2/hexane, 1:1) and UV-vis spectrophotometry. After 30 min, although there still remained some starting corrole, hydrazine was added to quench the unreacted DDQ and the crude mixture was filtered through a Celite bed. The solvent was then evaporated and the products were purified by chromatography on silica gel using a mixture of CH2Cl2/hexane (1:1) as the eluent. The first green band (Rf = 0.83, 40% overall yield) was further purified by preparative TLC on silica gel plates (CH2Cl2/hexane 1:3, eluent) to afford a first fraction corresponding to the 10-isomer, and a second one corresponding to the 5-isomer. Both of the isomers were recrystallized from CH2Cl2/MeOH (1:2), affording 10-ethyl-5,10,15-tritolyl-isocorrole (Rf = 0.22) and 5-ethyl-5,10,15-tritolyl-isocor-role (Rf = 0.10) in 25% (26 mg; 0.044 mmol) and 13% (14 mg; 0.023 mmol) yields, respectively.
10-ethyl-5,10,15-tritolyl-isocorrole (1)
mp > 300 °C. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, M−1.cm−1) 358 (29.3), 432 (48.2), 648 (6.6), 692 (7.2). 1H NMR (300 MHz, CDCl3): δ, ppm 14.30 (s, 2 H, NH), 7.53 (d, 4 H, J = 7.38 Hz, 5- and 15-phenyl), 7.30 (m, 6 H, phenyl), 7.08 (d, 2 H, J = 7.92 Hz, 10-phenyl), 6.85 (s, 4 H, β-pyrrole), 6.75 (d, 2 H, J = 3.93 Hz, β-pyrrole), 6.15 (d, 2 H, J = 3.78 Hz, β-pyrrole), 2.88 (q, 2 H, -CH2CH3), 2.47 (s, 6 H, 5- and 15-phenyl-CH3), 2.31 (s, 3 H, 10-phenyl-CH3), 0.89 (t, 3 H, -CH2CH3). MS (FAB): m/z (%) 597 [M]+ (100). Anal. calcd. for C42H36N4: C, 84.53; H, 6.08; N, 9.39%. Found: C, 84.47; H, 6.10; N, 9.43.
5-ethyl-5,10,15-tritolyl-isocorrole (2)
mp > 300 °C. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, M−1.cm−1) 408 (37.2), 655 (5.8), 722 (4.7). 1H NMR (300 MHz, CDCl3): δ, ppm 14.76 (s, 1 H, NH), 14.39 (s, 1 H, NH), 7.50 (d, 2 H, J = 8.07 Hz, phenyl), 7.32 (m, 8 H, phenyl), 7.15 (d, 1 H, J = 5.13 β-pyrrole), 7.05 (m, 3 H, phenyl and β-pyrrole), 6.77 (m, 1 H,β-pyrrole), 6.73 (d, 1 H,J= 4.41 Hz,β-pyrrole), 6.54 (m, 1 H, β-pyrrole), 6.44 (d, 1 H, J = 4.41 Hz, β-pyrrole), 6.37 (m, 1 H, β-pyrrole), 5.99 (m, 1 H, β-pyrrole), 2.88 (q, 2 H, -CH2CH3), 2.48 (s, 3 H, phenyl-CH3), 2.46 (s, 3 H, phenyl-CH3), 2.30 (s, 3 H, phenyl-CH3), 0.70 (t, 3 H, -CH2CH3). MS (FAB): m/z (%) 597 [M]+ (100). Anal. calcd. for C42H36N4: C, 84.53; H, 6.08; N, 9.39%. Found: C, 84.55; H, 6.07; N, 9.36.
From the first chromatographic purification, two other fractions were isolated. Recrystallization from CH2Cl2/MeOH (1:2) afforded 2-bromoTTCorH3 (Rf = 0.72, 30% yield) (34 mg; 0.053 mmol) and the 3-bromoTTCorH3 (Rf = 0.63, 10% yield) (11 mg; 0.018 mmol).
2-bromo-5,10,15-tritolylcorrole (3)
mp > 300 °C. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, M−1.cm−1) 421 (122.5), 525 (sh), 564 (15.4), 612 (10.0), 657 (21.4). 1H NMR (300 MHz, CDCl3): δ, ppm 9.17 (d, 1 H, J = 3.87 Hz, β-pyrrole), 8.89 (d, 1 H, J = 4.62 Hz, β-pyrrole), 8.82 (d, 1 H, J = 4.50 Hz, β-pyrrole), 8.65 (s, 1 H, β-pyrrole), 8.54 (m, 3 H, β-pyrrole), 8.34 (d, 2 H, J = 7.53 Hz, phenyl), 8.15 (d, 2 H, J = 7.47 Hz, phenyl), 8.06 (d, 2 H, J = 7.50 Hz, phenyl), 7.62 (m, 6 H, phenyl), 2.71 (s, 9 H, phenyl-CH3). MS (FAB): m/z (%) 648 [M]+ (100). Anal. calcd. for C40H31BrN4: C, 74.19; H, 4.82; N, 8.65%. Found: C, 74.21; H, 4.83; N, 8.61.
3-bromo-5,10,15-tritolylcorrole (4)
mp > 300 °C. UV-vis (CH2Cl2): λmax, nm (ε × 10−3, M−1.cm−1) 417 (100.5), 524 (sh), 563 (13.9), 617 (9.7), 651 (15.2). 1H NMR (300 MHz, CDCl3): δ, ppm 8.92 (s, 1 H, β-pyrrole), 8.87 (d, 2 H, J = 4.25 Hz, β-pyrrole), 8.66 (d, 1 H, J = 4.93 Hz, β-pyrrole), 8.57 (d, 1 H, J = 4.22 Hz, β-pyrrole), 8.50 (d, 2 H, J = 4.64 Hz, β-pyrrole), 8.29 (d, 2 H, J = 7.83 Hz, phenyl), 8.04 (m, 4 H, phenyl), 7.65 (d, 2 H, J = 7.81 Hz, phenyl), 7.56 (m, 4 H, phenyl), 2.70 (s, 9 H, phenyl-CH3). MS (FAB): m/z (%) 648 [M]+ (100). Anal. calcd. for C40H31BrN4: C, 74.19; H, 4.82; N, 8.65%. Found: C, 74.17; H, 4.84; N, 8.64.
x-ray crystallography data
For 1. C42H36N4·CHCl3, monoclinic space group P21/n, a = 13.904(3), b = 10.158(2), c = 25.288(5) Å, = 93.148(15)°, V = 3566.2(13) Å3, Z = 4, calcd = 1.334 g.cm−3, (CuKα) = 2.614 mm−1, R = 0.123 for 1353 data with Fo2 > 2(Fo2) of 5169 unique data and 455 refined parameters. A total of 23,449 data was collected at T = 90.0(5) K to = 59.7° with CuKα radiation (= 1.54178 Å) on a Bruker Kappa Apex-II diffractometer, using a green lath crystal of dimensions 0.40 × 0.12 × 0.01 mm.
For 3. C40H31BrN4·2CHCl3, monoclinic space group P21/c, a = 16.0322(15), b = 14.6126(14), c = 16.8920(15) Å, = 103.071(5)°, V = 3854.8(6) Å3, Z = 4, calcd = 1.527 g.cm−3, (CuKα) = 5.575 mm−1, R = 0.048 for 5415 data with Fo2 > 2(Fo2) of 6895 unique data and 519 refined parameters. A total of 24,302 data was collected at T = 90.0(5) K to = 68.5° with CuKα radiation on a Bruker Kappa Apex-II diffractometer, using a dark green plate crystal of dimensions 0.31 × 0.22 × 0.06 mm. Absorption correction were by face-indexed numerical methods. For both compounds, refinement was by full-matrix least-squares using SHELXL, with H atoms in idealized positions. NH hydrogen atoms were placed in idealized positions, guided by difference maps, and their positions were refined for 3. There is a slight disorder in 3, in which the bromo substituent is located at the C18 position with approximately 15% occupancy, as well as disorder in one of the solvent molecules.
RESULTS AND DISCUSSION
With the aim to obtain stable isocorroles using the same one-pot approach developed for methoxy-substituted derivatives, TTCorH3 was used as the starting material. Since the plausible reaction mechanism for the isocorrole formation was the initial oxidation of corrole by DDQ, followed by the addition of methanol [12] (or water [6c]), it was decided to attempt the oxidation in toluene, followed by the addition of an opportune nucleophile. To obtain an alkyl-substituted isocorrole, the suitable reagents used were organolithium or Grignard reagents, both of which have been used in the case of porphyrins [16]. An organolithium compound, BuLi, was first investigated but no reaction occurred; oxidation of the corrole was observed, but after the quenching step, only the starting material was recovered. Attention was then focused on Grignard reagents and, in particular, on EtMgBr. The reaction was attempted without the addition of DDQ but no reactivity was observed other than the corrole anion formation. The reaction was performed in anhydrous toluene under inert atmosphere with the initial oxidation of corrole by addition of an equimolar amount of DDQ and then adding EtMgBr in a 1:2 molar ratio. The reaction monitoring by UV-vis and TLC showed, along with some unreacted material, the presence of three new green bands characterized by higher Rf than the starting TTCorH3. The excess of DDQ was quenched using hydrazine and, after filtering the solution through a bed of Celite, the solvent was evaporated under reduced pressure.
The crude mixture was purified by column chromatography using silica gel as stationary phase and a CH2Cl2/hexane solution (1:1) as the eluent. The unreacted corrole was separated and three fast-moving green fractions were collected. The most polar fraction (40% overall yield) showed a UV-vis spectrum having features characteristic of an isocorrole derivative. TLC analysis indicated the presence of two compounds, which were then separated by preparative TLC on silica gel (CH2Cl2/hexane, 1:3), leading to the isolation of 1 and 2, in an approximately 2:1 ratio, respectively (Chart 1).
Chart 1.

The 1H NMR spectrum of the first eluted band was consistent with the symmetrical structure of 1, with the presence of a single peak at 14.30 ppm, assigned to the inner NHs, and two signals (2:1 intensity ratio) for the -CH3 of the tolyl groups in the 5,15- and 10-positions respectively. The second eluted fraction was in accord with the less symmetrical structure of 2, presenting two different signals, at 14.79 and 14.39 ppm, for the inner core NHs, and three separate resonances for the above-mentioned -CH3 of the tolyl groups.
Slow diffusion of MeOH into a CHCl3 solution of 1 afforded a single crystal that allowed the characterization by X-ray crystallography (Fig. 2). Although the quality of the structure determination was not high, the identity of the compound was however confirmed.
Fig. 2.
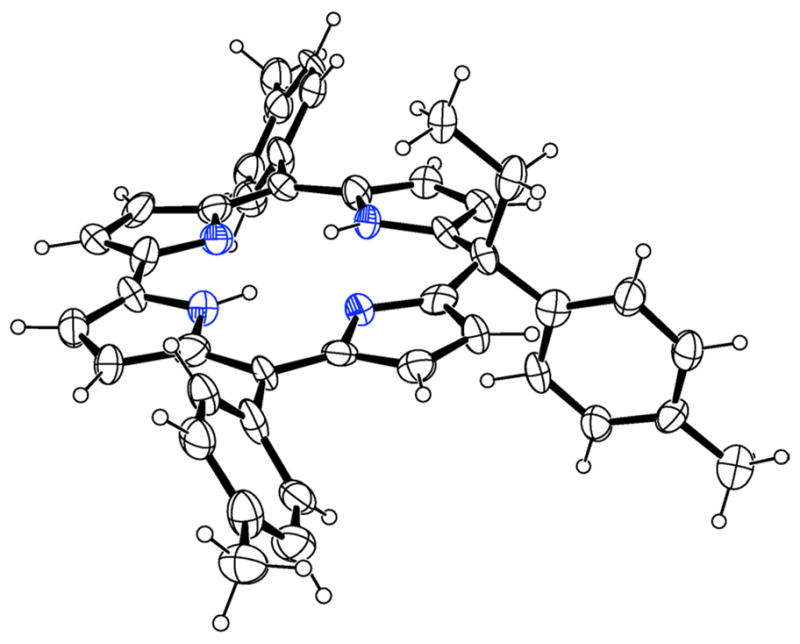
X-ray molecular structure of 1
The 23-atom core shows only minor deviations from coplanarity, with mean deviation 0.07 Å and maximum deviation 0.16(1) Å. The four N atoms are coplanar with 0.04(1) Å, and the two pyrrole rings in the background in Fig. 2 tilt slightly in the same direction, out of the N4 plane by 7° and 9°. The ethyl substituent at C10 is turned inward over the central core of the molecule and is coplanar with the tolyl group also at C10. The plane of the ethyl and tolyl substituents on C10 is approximately perpendicular to the main ring system, forming a dihedral angle of 88.3(2)° with the N4 plane. The other two tolyl planes form dihedral angles of 55.1(4) and 57.1(2)° with the N4 plane and a dihedral angle of 67.8(4)° with each other.
While these products indicated the successful formation of the desired isocorrole species, the other two chromatographic fractions showed UV-vis spectra, indicating the retention of the aromatic corrole structure. This result indicated the formation of β-substituted corroles, as confirmed by their 1H NMR spectra; the presence of a singlet at 8.65 ppm for the first isolated compound and at 8.92 ppm for the second fraction provided evidence of the monosubstitution (Fig. 3). However, no ethyl group resonances were present in either spectrum, thereby excluding the substitution by the ethyl group of the Grignard reaction at the β-pyrrolic position of the corrole ring. FAB mass spectra for both fractions, on the other hand, were compatible with the presence of a Br substituent. This result suggested that the nucleophile reacting at the pyrrolic position of the oxidized corrole was the bromide group of the Grignard reagent, leading to the monobromo-substituted corrole. In agreement with the data reported in the literature for the 1H NMR assignment of pyrrolic resonances of corrole [17], the two fractions were respectively characterized as the 2- and 3-Br substituted corrole (Fig. 3).
Fig. 3.

1H NMR spectra (aromatic region) of (a) TTCorH3, (b) 3, (c) 4
We were able to obtain single crystals of 3 that were characterized by X-ray crystallography (Fig. 4). While the four N atoms are reasonably coplanar, with mean deviation 0.013 Å; the 23-atom core is quite distorted from planarity, with mean deviation 0.19 Å and maximum deviation 0.496(3) Å. The distortion can be described as tipping of the pyrrole rings out of the N4 plane. The two directly-linked pyrroles tip toward the same side of the molecule, forming dihedral angles of 21.6(1) and 32.0(1)° with the N4 plane. One of the other pyrroles tips toward the opposite side of the molecule, forming a dihedral angle of 32.6(1)° with the N4 plane. The fourth pyrrole is more nearly coplanar with N4, tipping by only 6.1(1)°. The pyrrole tipping allows avoidance of extremely short intramolecular N-H···H-N contacts, the nearest being 2.18 Å. The C-Br distance is 1.874(3) Å, and the tipping of the brominated pyrrole causes the Br atom to be 1.586(1) Å out of the N4 plane.
Fig. 4.

X-ray molecular structure of 2-bromoTTCorH3
It is worth mentioning that even though the formation of a mixture of monobrominated corrole isomers has been previously reported in the literature [18], the separation of the regioisomers was not at that time possible. This is the first example where the regioisomers have been separated and fully characterized. Moreover, it is interesting to note that the regioselectivity of this reaction does not follow the usual pattern observed for corrole, where the reactivity order is C3 > C2 ≥ C8 > C7 [19, 20]. In the past, in fact, tribromo-derivatives [18, 21] have been isolated and unambiguously characterized by crystallography: the substitution pattern, as in the case of deuteration of other corrolic rings [22], suggested a higher reactivity for the C3 (or C17) position. On the other hand, in our case the first eluted isomer 3 represents the most abundant species (30%), in contrast with the hypothetical identification reported by Du et al. [18]. In this case the most reactive site for the A-pyrrole (D-pyrrole) is the one closer to the direct bond, namely the C2 position. One can hypothesize that the different reactivity suggests a different reaction mechanism in this case and further studies are now in progress in our laboratories to thoroughly investigate this topic; these results will be reported in due course.
CONCLUSION
The results obtained have demonstrated that stable isocorroles can be successfully synthesized using our new approach. This methodology can be of great importance since it is based on the use of different and readily available macrocycles as starting material that are still susceptible, after the isocorrole formation, to further functionalization; this can subsequently be used to tune the properties of the products for use in real applications.
The use of Grignard reagents illustrates the competitive reactivity of the β-pyrrole positions compared with the meso-positions. This allows, along with the formation of isocorroles, the isolation of previously unreported mono-bromocorrole isomers.
Supplementary Material
Acknowledgments
This work was supported by MIUR, Italy (PRIN project 2007C8RW53) and the US National Institutes of Health (K.M.S., Grant CA-132861).
Footnotes
Crystallographic data have been deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition numbers CCDC 780147 and 780148. Copies can be obtained on request, free-of-charge, via www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223-336-033 or deposit@ccdc.cam.ac.uk).
References
- 1.a) Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Vol. 2. Academic Press; San Diego, CA: 2000. [Google Scholar]; b) Sessler JL, Weghorn SJ. Expanded, Contracted & Isomeric Porphyrins. Pergamon Press; 2000. [Google Scholar]
- 2.a) Vicente MGH. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 1. Academic Press; San Diego, CA: 2000. pp. 149–199. [Google Scholar]; b) Li X, Chmielewski PJ, Xiang J, Xu J, Jiang L, Li Y, Liu H, Zhu D. J Org Chem. 2006;71:9739–9742. doi: 10.1021/jo0618268. [DOI] [PubMed] [Google Scholar]; c) Senge MO. Accounts of Chemical Research. 2005;38:733–743. doi: 10.1021/ar0500012. [DOI] [PubMed] [Google Scholar]; d) O’Brien AY, McGann JP, Geier GR., III J Org Chem. 2007;72:4084–4092. doi: 10.1021/jo070216k. [DOI] [PubMed] [Google Scholar]; e) Abhilash GJ, Bhuyan J, Singh P, Maji S, Pal K, Sarkar S. Inorg Chem. 2009;48:1790–1792. doi: 10.1021/ic800864w. [DOI] [PubMed] [Google Scholar]; f) Lash TD, El-Beck JA, Colby DA. J Org Chem. 2009;74:8830–8833. doi: 10.1021/jo901959k. [DOI] [PubMed] [Google Scholar]
- 3.a) Scheer H. Chlorophylls. CRC Press; Boca Raton, FL: 1991. [Google Scholar]; b) Balaban TS. Acc Chem Res. 2005;38:612–623. doi: 10.1021/ar040211z. [DOI] [PubMed] [Google Scholar]; c) Senge MO, Wiehe A, Ryppa C. In: In Advances in Photosynthesis and Respiration. Grimm B, Porra R, Rudiger W, Scheer H, editors. Vol. 25. Springer; Dordrecht: 2006. pp. 27–37. [Google Scholar]
- 4.Pandey RK, Zheng G. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 6. Academic Press; San Diego, CA: 2000. pp. 157–230. [Google Scholar]
- 5.a) Sessler JL, Gale PA. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Vol. 6. Academic Press; San Diego, CA: 2000. pp. 257–278. [Google Scholar]; b) Sessler JL, Zimmerman RS, Bucher C, Kral V, Andrioletti B. Pure Appl Chem. 2001;73:1041–1057. [Google Scholar]; c) Sessler JL, Gross DE, Chow S, Lynch VM, Schmidtchen FP, Bates GW, Light ME, Gale PA. J Am Chem Soc. 2006;128:12281–12288. doi: 10.1021/ja064012h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Finnigan EM, Giordani S, Senge OM, McCabe T. J Phys Chem A. 2010;114:2464–2470. doi: 10.1021/jp909573m. [DOI] [PubMed] [Google Scholar]
- 6.a) Mandoj F, Nardis S, Pomarico G, Stefanelli M, Schiaffino L, Ercolani G, Prodi L, Genovese D, Zaccheroni N, Fronczek FR, Smith KM, Xiao X, Shen J, Kadish KM, Paolesse R. Inorg Chem. 2009;48:10346–10357. doi: 10.1021/ic9014866. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stefanelli M, Shen J, Zhu W, Mastroianni M, Mandoj F, Nardis S, Ou Z, Kadish KM, Fronczek FR, Smith KM, Paolesse R. Inorg Chem. 2009;48:6879–6887. doi: 10.1021/ic900859a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mandoj F, Nardis S, Pomarico G, Paolesse R. J Porphyrins Phthalocyanines. 2008;12:19–26. doi: 10.1142/S1088424610002513. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Stefanelli M, Mastroianni M, Nardis S, Licoccia S, Fronczek FR, Smith KM, Zhu W, Ou Z, Kadish KM, Paolesse R. Inorg Chem. 2007;46:10791–10799. doi: 10.1021/ic7014572. [DOI] [PubMed] [Google Scholar]
- 7.Hohlneicher G, Bremm D, Wytko J, Bley-Escrich J, Gisselbrecht JP, Gross M, Michels M, Lex Johann, Vogel E. Chem Eur J. 2003;9:5636–5642. doi: 10.1002/chem.200305094. [DOI] [PubMed] [Google Scholar]
- 8.Orlewska C, Maes W, Toppet S, Dehaen W. Tetrahedron Lett. 2005;46:6067–6070. [Google Scholar]
- 9.a) Will S, Rahbar A, Schmickler H, Lex J, Vogel E. Angew Chem Int Ed Engl. 1990;29:1390–1393. [Google Scholar]; b) Vogel E, Binsack B, Hellwig Y, Erben C, Heger A, Lex J, Wu YD. Angew Chem Int Ed Engl. 1997;36:2612–2615. [Google Scholar]
- 10.Setsune J, Tsukajima A, Watanabe J. Tetrahedron Lett. 2006;47:1817–1820. [Google Scholar]
- 11.Flint DL, Fowler RL, LeSaulnier TD, Long AC, O’Brien AY, Geier GR., III J Org Chem. 2010;75:553–563. doi: 10.1021/jo902452c. [DOI] [PubMed] [Google Scholar]
- 12.Nardis S, Pomarico G, Fronczek FR, Vicente MGH, Paolesse R. Tetrahedron Lett. 2007;48:8643–8646. [Google Scholar]
- 13.Pomarico G, Xiao X, Nardis S, Paolesse R, Fronczek FR, Smith KM, Fang Y, Ou Z, Kadish KM. Inorg Chem. 2010;49:5766–5774. doi: 10.1021/ic100730j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Setsune J, Tsukajima A, Okazaki N. J Porphyrins Phthalocyanines. 2009;13:256–265. [Google Scholar]
- 15.Paolesse R, Marini A, Nardis S, Froiio A, Mandoj F, Nurco DJ, Prodi L, Montalti M, Smith KM. J Porphyrins Phthalocyanines. 2003;7:25–36. [Google Scholar]
- 16.a) Senge MO. Acc Chem Res. 2005;38:733–743. doi: 10.1021/ar0500012. [DOI] [PubMed] [Google Scholar]; b) Sergeeva NN, Shaker YM, Finnigan EM, McCabe T, Senge MO. Tetrahedron. 2007;63:12454–12464. [Google Scholar]; c) Senge MO, Richter J, Bischoff I, Ryan A. Tetrahedron. 2010;66:3508–3524. [Google Scholar]
- 17.Balazs YS, Saltsman I, Mahammed A, Tkachenko E, Golubkov G, Levine J, Gross Z. Magn Reson Chem. 2004;42:624–635. doi: 10.1002/mrc.1389. [DOI] [PubMed] [Google Scholar]
- 18.Du RB, Liu C, Shen DM, Chen QY. Synlett. 2009;16:2701–2705. [Google Scholar]
- 19.Paolesse R, Nardis S, Venanzi M, Mastroianni M, Russo M, Fronczek FR, Vicente MGH. Chem Eur J. 2003;9:1192–1197. doi: 10.1002/chem.200390136. [DOI] [PubMed] [Google Scholar]
- 20.Paolesse R. Synlett. 2008:2215–2230. [Google Scholar]
- 21.Nardis S, Mandoj F, Paolesse R, Fronczek FR, Smith KM, Prodi L, Montalti M, Battistini G. Eur J Inorg Chem. 2007;16:2345–2352. [Google Scholar]
- 22.Gross Z, Mahammed A. J Porphyrins Phthalocyanines. 2002;6:553–555. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.