

Abstract
Male mice were fed 40 ppm melatonin for 2 months prior to sacrifice at age 26 months, and compared with both 26 and 4 month-old untreated controls. The nuclear translocation of NF-κB increased with age in both brain and spleen and this was reversed by melatonin only in brain. Another transcription factor, AP-1 was increased with age in the spleen and not in brain and this could be blocked by melatonin treatment. The fraction of the active relative to the inactive form of several enabling kinases was compared. The proportion of activated ERK was elevated with age in brain and spleen but this change was unresponsive to melatonin. A similar age-related increase in glial fibrillary acidic protein (GFAP) was also refractory to melatonin treatment. The cerebral melatonin M1 receptor decreased with age in brain but increased in spleen. The potentially beneficial nature of melatonin for the preservation of brain function with aging was suggested by the finding that an age-related decline in cortical synaptophysin levels was prevented by dietary melatonin.
Keywords: Aging, Brain, Melatonin, Inflammation, Transcription factors, Synaptophysin, Kinases
Introduction
The aging brain is characterized by a shift in the homeostatic balance of inflammatory mediators to a proinflammatory state. Increased neuroinflammation is marked by higher steady-state levels of inflammatory cytokines and decreases in anti-inflammatory molecules [1]. Such elevated inflammatory responses in the brain, can lead to increased neuronal death [2–4]. Several innate immune molecules which can have a neuroprotective scavenging role in promoting clearance of pathogens, toxic cell debris and apoptic cells, can also express neurocytotoxic activity. An appropriate modulation of innate immune activity by exogenous agents may limit hyperinflammatory processes while sparing host defense and repair mechanisms [5]. We have previously shown that during senescence, there is a selective increase in mRNA levels of many genes associated with immune function. Such genes include those for several inflammatory cytokines. These age-related increases can be attenuated or even reversed by extended treatment with dietary melatonin [6–10]. The elevated expression of these genes provoked by a systemic injection of lipopolysaccharide (LPS) is also exacerbated in aged animals and the response to this exogenous inflammatory stimulus is also diminished by prior treatment of aged mice with melatonin [6, 11]. The current study sought to extend this work by examination of some proteins of the signaling pathways associated with the inflammatory cascade. The findings suggest that the effect of melatonin on transcription of a distinctive set of genes may be further reflected by parallel changes in translational events and in the regulation of gene expression by specific transcription factors. The spleen was also studied as it is an important non-neural immune regulator. It also showed both age related changes in transcription factor activity, that were attenuated by melatonin but the response profile differed significantly from that of the brain.
Methods
Animal Treatment
Male B6C3F1 mice, a hybrid between C57BL/6 and C3H from Harlan Labs (Indianapolis, IN), aged 5.5 months (young group) and 23.4 months (old group), were housed two to four per cage and were maintained on a 12 h light/dark cycle in a temperature controlled (22 ± 1°C) room. The B6C3F1 hybrid was used in order to take advantage of both the genetic and phenotypic uniformity and the vigor (increased disease resistance, better survival under stress and greater natural longevity) typical of hybrids, while maintaining genetic similarity to the published C57BL/6 mouse genome sequence [12]. Food and water were provided ad libitum. Young (YC) and old (OC) control animals were fed a pelleted minimal basal diet (AIN-93 M, Dyets #100900, Dyets Inc., Bethlehem, PA) consisting of 10% sucrose and 14% casein (w/w) as well as a minimal salt and vitamin mix. The basal diet of two similarly aged cohorts (YM and OM, respectively) was supplemented with 40-ppm (w/w) melatonin (Sigma, St. Louis, MO) for 9.3 weeks. All experiments were approved by the Institutional Animal Care and Use Committee at the University of California, Irvine, and conformed to the National Institute of Health guide for the care and use of laboratory animals.
Preparation of Samples
Cytoplasmic and nuclear fractions were prepared using the method of Lahiri and Ge [13]. The brain tissue from each animal was weighed and homogenized in an ice-cold buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF and 0.5% NP-40). The samples were incubated for 10 min and centrifuged (1,500×g) at 4°C for 1 min. The supernatant containing the cytoplasmic constituents was collected and protease inhibitor cocktail was added. The samples were aliquoted and stored at −80°C. The nuclear pellet was resuspended in a buffer composed of: 20 mM HEPES pH 7.9, 400 mM NaCl, 1 mM DTT, 1 mM EDTA, 1 mM EGTA and 1 mM PMSF. The samples were then centrifuged at 11,000×g for 5 min at 4°C. The supernatant that contained nuclear extract was aliquoted and stored at −80°C.
Electrophoretic Mobility Shift Assay
The extent of NF-κB and AP-1 activation was determined in the nuclear fraction of brain tissue using a protocol developed by Promega (Madison WI). The amount of protein in 2 μl of the nuclear extract was determined by the BCA protein assay kit (Pierce, Rockford, IL) and 50 μg of each sample, incubated with 32P-labeled oligonucleotides containing either the NF-κB or the AP-1 consensus sequence, was loaded onto a gel. A negative control containing no cell extract, as well as competitor reactions were included. The specific competitor contained unlabelled NF- κB or AP-1 consensus nucleotide while the nonspecific competitor contained unlabelled SP-1 consensus oligonucleotide. The competitor reactions also contained 50 μg of nuclear fraction derived from Al-treated mouse brains. This treatment has been shown not to alter the extent of NF-kB or AP-1 activation [14]. X-ray films were manually developed and the intensity of each band was measured and quantitated using the image analyzer, Eagle Eye, from Strategene (San Diego, CA).
Western Blots
The levels of ERK, pERK, JNK, pJNK, p38 and pp38 and the melatonin M1 receptor were determined using Western blotting. Protein content was determined using the BCA protein assay kit (Pierce, Rockford, IL). 25 μg of each sample was resolved on a SDS-10% PAGE and transferred onto a nitrocellulose membrane (Biorad, Hercules, CA). After blocking overnight in TBST (20 mM Tris–HCl, 150 mM NaCl and 0.1% Tween 20) containing 5% nonfat milk, membranes were washed 4× and incubated for 1 h with primary antibodies (mouse monoclonal antibody against pERK (1:500); rabbit polyclonal antibody against ERK (1:1500); goat polyclonal antibody against pJNK (1:500); rabbit polyclonal antibody against JNK-1 (1:500); rabbit polyclonal antibody against p38(1:500); and rabbit polyclonal antibody against p-p38 (1:500), rabbit polyclonal antibody against melatonin 1A/B receptors (1:4000) all purchased from Santa Cruz Biotechnology, Inc., Santa Cruz, California. After washing the membranes 4× with TBST, they were incubated with appropriate secondary HRP-conjugated antibodies (1:10,000). Bands were detected with ECL reagents (Amersham Pharmacia Biotech, Piscataway, NJ) following the manufacturer’s protocol. An antibody against actin (mouse monoclonal, 1:1500 dilution) purchased from Chemicon International (Temecula, CA) was used as an internal control to insure equal loading of protein for each sample. The intensity of specific bands was measured and quantitated using the image analyzer, Eagle Eye, from Strategene (San Diego, CA).
Immunohistochemistry
Immunohistochemistry was performed on paraffin embedded sections. Sections were immersed in antigen unmasking solution (Vector Laboratories, Burlingame, CA) for 20 min at 80°C in a water bath. Endogenous peroxidase in tissue was blocked by treating with 3% H2O2 in PBS for 20 min at room temperature. Nonspecific background staining was blocked by a 2 h incubation in 2% bovine serum albumin with 0.3% Triton X-100. Sections were then incubated with primary antibody (synaptophysin 1:1000, Santa Cruz, CA) overnight at 4°C, then rinsed in PBS with 0.1%Triton-X100 and incubated for 1 h at room temperature with biotinylated secondary antibody using an avidin DH and biotinylated enzyme complex kit (Vector Laboratories, Burlingame, CA). Finally, the sections were incubated for 4 min with diaminobenzidine (Vector Laboratories, Burlingame, CA). Sections were further processed by dehydration in a series of graded ethanol, cleared with xylene, and then coverslipped with 1,3-diethyl-8-phenylxanthine (DPX). Control sections were incubated in parallel without primary antibody and these sections failed to develop specific staining. Immunostaining was observed under a Nikon Eclipse 80i microscope mounted with a Nikon DS high-resolution digital color camera (1,280 × 1,024 pixel). Image acquisition and analysis was performed using NIS-Elements AR version 3.0 software. Digital images were analyzed using the Ver.3.00 analysis program (Nikon, Melville, NY). The percentage of immunostained area was quantitated by averaging several images per section. All experiments were repeated at least twice, with n = 6–8 animals per group. All quantitative comparisons were performed on sections processed at the same time.
Statistical Analysis
The differences among these groups were tested using one-way analysis of variance followed by the Tukey test. Values were accepted as significant if P < 0.05 level, using a two-tailed criterion.
Results
Treatment of mice with melatonin in the diet did not alter rates of food consumption or accretion of body weight in either young or old mice. Electrophoretic mobility shift analysis (EMSA) revealed that the level of active transcription factor NF-κB, which is associated with associated with inflammatory responses, was elevated in both brain and spleen with age. Treatment with melatonin reduced the activity of this factor in brains of both young and old animals (Fig. 1a). In the spleen there was a larger age-related increase in active NF-κB but this was not changed by dietary melatonin (Fig. 1b).
Fig. 1.
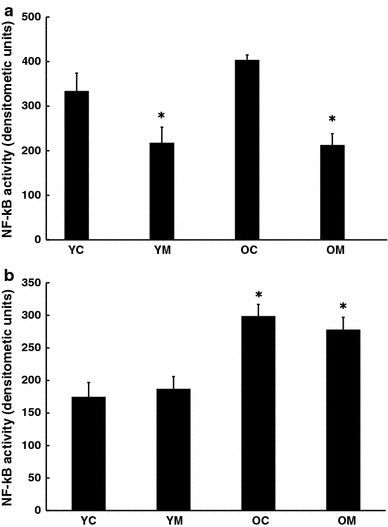
Effect of melatonin on levels of activation of NF-kB in a cortex and b spleen of 4.5 and 26.5 month-old mice as determined by EMSA. n = 8 for each group. * = Differs from value for untreated control (P < 0.05)
Active AP-1, which is related to immune activation, was also increased with age in the spleen Fig. 2a and this elevation was prevented by melatonin treatment. However, in the brain, no corresponding changes of AP-1 with age or melatonin treatment were found (Fig. 2b).
Fig. 2.
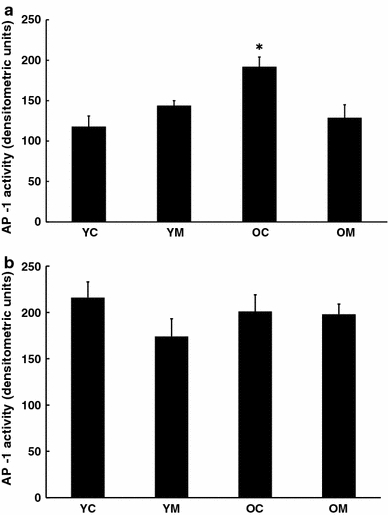
Effect of melatonin on levels of activation of AP-1 in a cortex and b spleen of 4.5 and 26.5 month-old mice. n = 8 for each group as determined by EMSA. * = Differs from value for untreated control (P < 0.05)
In order to gain insight into the regulation of these transcription factors, the fraction of several kinases present in the phosphorylated form was compared to the amount present as the inactive unphosphorylated form. In both cortical tissue (Fig. 3a) and spleen (Fig. 3b), the ratio of pERK/ERK was elevated with age, and this was unaltered by prior melatonin treatment. Neither pJNK/JNK nor pp38/p38 ratios were significantly altered with age in either brain or spleen (data not shown). However, in the spleen, dietary melatonin depressed pJNK/JNK values in both young and aged mice (Fig. 4).
Fig. 3.
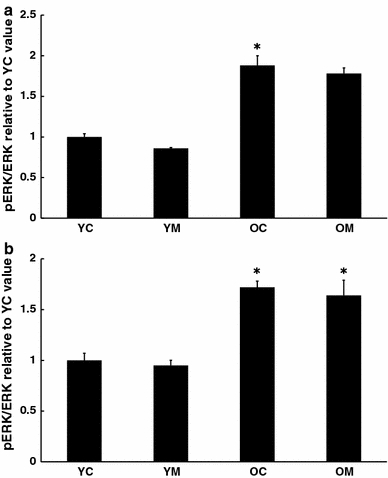
Effect of melatonin on levels of activation of ERK in a cortex and b spleen of 4.5 and 26.5 month-old mice as determined by Western blot. n = 7–8 for each group. * = Differs from value for untreated control (P < 0.05)
Fig. 4.
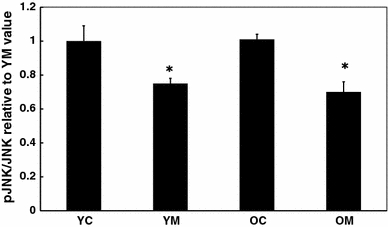
Effect of melatonin on levels of activation of JNK in spleen of 4.5 and 26.5 month-old mice. n = 7–8 for each group mice as determined by Western blot. * = Differs from value for untreated control (P < 0.05)
Melatonin M1 receptor density in brain was reduced with age (Fig. 5a), while in the spleen, the melatonin receptor content increased with age (Fig. 5b). Neither of these changes were altered by melatonin.
Fig. 5.
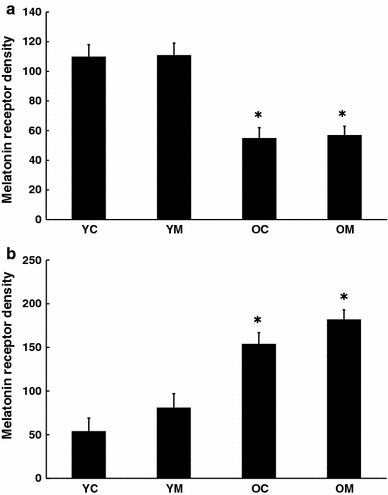
Effect of melatonin on melatonin receptor content in a cortex and b spleen of 4.5 and 26.5 month-old mice as determined by Western blot. n = 7–8 for each group. * = Differs from value for untreated control (P < 0.05)
The proportion of astrocytes expressing GFAP can be considered as a marker of brain immune activation. In accord with several prior reports, GFAP levels were increased with age. However, melatonin treatment did not alter this (Fig. 6). Synaptic nerve endings were visualized using immunohistochemical procedures for synaptophysin and staining intensity was quantitated. Synaptophysin density was reduced in cortex with aging and this was completely reversed by the melatonin treatment (Fig. 7a, b).
Fig. 6.
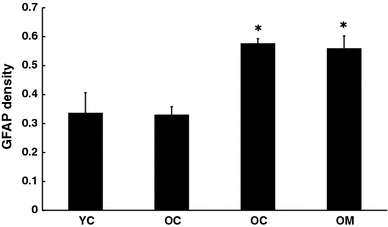
Effect of melatonin on GFAP levels in cortex of 4.5 and 26.5 month-old mice as determined by Western blot. n = 8 for each group. * = Differs from value for untreated control (P < 0.05)
Fig. 7.
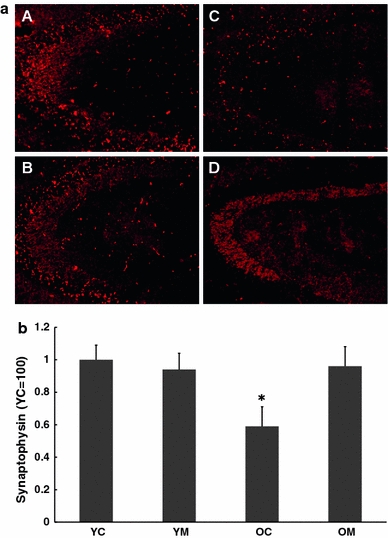
Immunohistochemical staining (a) together with quantitation (b) for synaptophysin in cortex of 4.5 and 26.5 month-old mice. Some animals of each group were treated with dietary melatonin. A = Young controls, B = Young + melatonin, C = Aged, D = Aged + melatonin. n = 7–10 for each group. * = Differs from value for untreated control (P < 0.05)
Discussion
Using gene expression analysis as a tool, we have previously reported a general age-related increase in unprovoked basal levels of inflammation for mouse brain [6]. With senescence, the levels of mRNAs of several inflammatory cytokines and other indices of inflammation are elevated and the responses to an exogenous stimulus (lipopolysaccharide, LPS) can be exaggerated and prolonged [8]. Many of these changes are partially or completely reversed following extended exposure to dietary melatonin [9]. While changes in gene expression have been demonstrated previously, transcriptional activity does not always correlate directly with activity of the protein product. The current study has investigated changes in activation levels of proteins involved in the inflammatory cascade. In parallel with findings from quantitative gene expression, evidence of elevated inflammation with age also derives from the current studies on transcription factors associated with inflammation. NF-κB was increasingly activated with age in cerebral cortex and spleen despite the absence of an exogenous stimulus. In nervous tissue, NF-κB is not merely a pro-inflammatory species but also has an important role in regulation of neurogenesis and short-term spatial memory [15] and in neuroprotection from stressors [16]. Neurogenesis, memory and intrinsic neuroprotective mechanisms are all increasingly impaired with age, in parallel with increased pro-inflammatory NF-κB. It therefore seems plausible that reversal of the age-related elevation of NF-κB could have a restorative effect on these cognitive and cellular processes. Indeed, our results suggest that melatonin can reduce NF-κB in the cortex of both young and aged animals.
However, since none of these events are improved with age, inhibition of an age-related elevation of NF-kB is likely to be beneficial rather than detrimental. In the case of brain, this increase was prevented by dietary melatonin.
The transcription factor AP-1 has recently been identified as an upstream regulator of several pro-inflammatory genes that may be involved in Alzheimer’s disease (AD), which is primarily an age-related memory disorder [17]. Nevertheless, in this study AP-1 activation in brain was unchanged with age and unaffected by melatonin treatment. In the spleen, levels of AP-1 rose with age and this increase was prevented by dietary melatonin. Thus, the responses of these two tissues to aging and to melatonin treatment, broadly resembled each other but differed in details.
When the degree of activation of several key kinases was quantitated, ERK was increased in aged brain but was reduced in aged spleen, Neither of these responses was altered by melatonin. The proportion of JNK and p38 that were activated by phosphorylation were unaltered with age. However, melatonin depressed activation of JNK in aged spleen but not brain. An age-related reduction in splenic ERK and JNK activation has been previously described and related to diminished lymphoproliferation in aged animals [18]. These results imply that melatonin treatment may modulate kinase activity in both a tissue-specific and pathway-specific manner. Further elucidation of these pathways may provide promising new therapeutic targets for age-related and inflammatory disorders.
GFAP levels were increased with age and this is in accord with our prior finding that GFAP mRNA expression is also elevated with age [9]. Not only is GFAP elevated with age but also its increase in response to brain injury is accentuated and prolonged. This may contribute to the worse cognitive outcome encountered following traumatic brain injury in the elderly [19]. We did not observe an effect on GFAP levels in melatonin treated animals, suggesting that age-related GFAP changes do not involve AP-1 or NF-κB signaling.
The overall effects of dietary melatonin upon proteinaceous indices of immune activation including transcription factors and their regulatory kinases, are not always concordant with levels of gene expression. In general the proteins are much less susceptible to alteration by melatonin that are the corresponding mRNAs. Such a disparity between mRNA and levels of the corresponding protein is found with the melatonin receptor. The receptor content is depressed in the aging brain, but its expression levels are elevated [9]. This may be due to a variety of regulatory steps between mRNA synthesis and the elaboration of protein, including mRNA stability, and siRNA. The lack of consonance between gene expression and levels of corresponding proteins may also reflect post-translational modification of proteins and consequent blunting of the changes occurring in mRNA production. Comparable to the current findings, an age-related reduction in melatonin receptor MT1 in the hypothalamic suprachiasmatic nucleus and a lack of response of levels of this receptor in brain to melatonin treatment has been described for both senescence-accelerated mice (SAMP8) and senescence-accelerated resistant mice (SAMR1) [20, 21].
The changes in profiles of immune-related transcription factors with age and their responses to melatonin vary between brain and spleen but are not always in accord. Since the spleen plays a major role in systemic immune regulation, it might be expected respond to aging with a profile differing from that of the brain, which has only intrinsic immune activity. What is remarkable is that the melatonin receptor density is depressed with age in the brain and elevated in the spleen. This difference cannot clearly be linked to responsivity to melatonin treatment. The age–related increase in spleen melatonin receptor that we found parallels a similar increase for the MT1 receptor of thymus, another immune-active tissue, while the levels of this receptor in all other tissues studied declined with age [22].
The actions of melatonin do not appear to depend entirely on melatonin receptor density. A more subtle dissection of melatonin receptor subtypes may be needed and this awaits the development of more selective antibodies. While dietary melatonin, is known to elevate plasma and brain levels of melatonin significantly [23], it did not alter melatonin receptor density. This may account for the lack of habituation to, and absence of, withdrawal effects of melatonin [24] and its analogs [25]. It has been reported the sensitivity to melatonin is reduced with age in the suprachiasmantic nucleus [26]. This may reflect reduction of melatonin receptor density and thus account for the ability of elevated levels of exogenous melatonin to reverse age-related changes in nervous tissue.
That the modulation of cortical levels of inflammation by melatonin may have physiological relevance for the aged organism, is suggested by the results for synaptophysin. Age-related losses of this protein have been reported by others [27] and these are exacerbated in double transgenic APP751SL/PS1M146L mice overexpressing both human mutant forms of amyloid precursor protein and presenilin [28]. These mice over-produce the AD-related amyloid-β (Aβ) peptide, which is known to stimulate inflammatory signaling [17]. Both the photomicrographs and the statistical analysis of quantitated immunostaining demonstrate that the age-related loss of synaptophysin can be prevented by melatonin treatment. Similarly, acute melatonin administration can protect against 1,3-bis(2- chloroethyl)-1-nitrosourea (BCNU)-induced or methamphetamine-induced reduction in synaptophysin [29, 30]. Since this protein may be an index of synaptic integrity, this implies that melatonin may be beneficial in the preservation of synaptic performance during senescence, and that melatonin may also have disease-specific application in the clinic.
The disparate effects of melatonin upon transcription factors and kinase activation in various tissues may give clues as to the mechanism of action of this agent. The original concept that melatonin primarily acts as an anti-oxidant has been questioned due to the very low levels of endogenous tissue melatonin which would be insufficient to provide marked scavenging of reactive oxygen radicals, and to the ability of melatonin to act as a pro-oxidant in isolated systems [10]. More likely is a more primary effect of melatonin upon the pattern of gene expression of a specific tissue. This could be enabled by very low levels of a triggering compound and could account for differing effects on the mRNA profile of each tissue.
Acknowledgments
This work was supported in part by grants from the National institutes of Health (AG 17694) to SCB and to DKL (AG18379 and AG18884).
With Respect
This issue is dedicated to honor Professor Abel Lajtha and a few reminiscences are appropriate. I have known Abel for 48 years. Even before this I knew of his work, and several of his papers are cited in my Ph. D. thesis from Birmingham University, England. This illustrates the lengthy period that Abel has had worldwide impact. He is unquestionably one of the fathers of modern neuroscience. His research accomplishments, while very extensive, are however only part of the overall contributions that he has made to our discipline. His leadership at first New York State’s Center for Neurochemistry at Ward’s Island, later at Orangeburg, his founding of “Neurochemical Research” and his editing a multi-volume series “Handbook of Neurochemistry” are all major milestones in the emergence of Neuroscience as an independent discipline. These contributions have been continuous and uninterrupted over a large span of time.
Others may elaborate on exceptional feats. I would like to mention Abel’s human qualities which are also outstanding and which have had a very positive effect on many people’s lives. I have always found Abel to be most sympathetic and to take on other people’s problems as if they were his. He has been unselfish with his time and courteous in responding to the needs of his fellows at whatever stage in their career they are at. Such a positive and generous personality, combined with his scientific insights goes far to account for his successful leadership in a wide range of areas. Although we have never collaborated directly together, several times in my career, Abel has rescued me from a difficult situation and I will always be grateful for his unstinting support.
I note that Abel has already 4 publications in 2010 (by May) and I wish him many more productive and happy years—SCB.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
Special issue article in Honor of Dr. Abel Lajtha.
References
- 1.Sparkman NL, Johnson RW. Neuroinflammation associated with aging sensitizes the brain to the effects of infection or stress. Neuroimmunomodulation. 2008;15:323–330. doi: 10.1159/000156474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finch CE, Morgan TE. Systemic inflammation, infection, ApoE alleles, and Alzheimer disease: a position paper. Curr Alzheimer Res. 2007;4:185–189. doi: 10.2174/156720507780362254. [DOI] [PubMed] [Google Scholar]
- 3.Marchalant Y, Brothers HM, Wenk GL. Inflammation and aging: can endocannabinoids help? Biomed Pharmacother. 2008;62:212–217. doi: 10.1016/j.biopha.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teeling JL, Perry VH. Systemic infection and inflammation in acute CNS injury and chronic neurodegeneration: underlying mechanisms. Neuroscience. 2009;158:1062–1073. doi: 10.1016/j.neuroscience.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 5.Griffiths M, Neal JW, Gasque P. Innate immunity and protective neuroinflammation: new emphasis on the role of neuroimmune regulatory proteins. Int Rev Neurobiol. 2007;82:29–55. doi: 10.1016/S0074-7742(07)82002-2. [DOI] [PubMed] [Google Scholar]
- 6.Sharman KG, Sharman E, Yang E, Campbell A, Bondy SC. Reduced basal levels and enhanced LPS response of IL-6 mRNA in aged mice. J Gerontol. 2001;56:B520–B523. doi: 10.1093/gerona/56.12.b520. [DOI] [PubMed] [Google Scholar]
- 7.Sharman KG, Sharman E, Bondy SC. Dietary melatonin selectively reverses age-related changes in cortical basal cytokine mRNA levels, and their responses to an inflammatory stimulus. Neurobiol Aging. 2002;23:633–638. doi: 10.1016/S0197-4580(01)00329-3. [DOI] [PubMed] [Google Scholar]
- 8.Sharman E, Sharman KG, Lahiri DK, Bondy SC. Age-related changes in murine CNS mRNA gene expression are modulated by dietary melatonin. J Pineal Res. 2004;36:165–170. doi: 10.1046/j.1600-079X.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- 9.Sharman EH, Sharman KZ, Bondy SC. Melatonin causes gene expression in aged animals to respond to inflammatory stimuli in a manner differing from that of young animals. Current Aging Sci. 2008;1:152–158. doi: 10.2174/1874609810801030152. [DOI] [PubMed] [Google Scholar]
- 10.Bondy SC, Sharman EH. Melatonin and the aging brain. Neurochem Int. 2007;50:558–571. doi: 10.1016/j.neuint.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Perreau VM, Bondy SC, Cotman CW, Sharman KZ, Sharman EH. Melatonin treatment in old mice enables a more youthful response to LPS in the brain. J Neuroimmunol. 2007;182:22–31. doi: 10.1016/j.jneuroim.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 13.Lahiri D, Ge Y. Electrophoretic mobility shift assay for the detection of specific DNA-protein complex in nuclear extracts from the cultured cells and frozen autopsy human brain tissue. Brain Res Protocols. 2000;5:257–265. doi: 10.1016/S1385-299X(00)00021-0. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Campbell A, Ali SF, Cong P, Bondy SC. Chronic exposure to low levels of aluminum alters cerebral cell signaling in response to acute MPTP administration. Toxicol Ind Health. 2007;23:515–524. doi: 10.1177/0748233708089027. [DOI] [PubMed] [Google Scholar]
- 15.Denis-Donini S, Dellarole A, Crociara P, Francese MT, Bortolotto V, Quadrato G, Canonico PL, Orsetti M, Ghi P, Memo M, Bonini SA, Ferrari-Toninelli G, Grilli M. Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. J Neurosci. 2008;28:3911–3919. doi: 10.1523/JNEUROSCI.0148-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L, Tao LY, Chen XP. Roles of NF-kappaB in central nervous system damage and repair. Neurosci Bull. 2007;23:307–313. doi: 10.1007/s12264-007-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vukic V, Callaghan D, Walker D, Lue LF, Liu QY, Couraud PO, Romero IA, Weksler B, Stanimirovic DB, Zhang W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol Dis. 2009;34:95–106. doi: 10.1016/j.nbd.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Torres C, Acuña-Castillo C, Walter R, Gardner EM, Murasko DM, Sierra F. Defect in ERK2 and p54(JNK) activation in aging mouse splenocytes. J Gerontol A Biol Sci Med Sci. 2002;57:B41–B47. doi: 10.1093/gerona/57.2.b41. [DOI] [PubMed] [Google Scholar]
- 19.Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol. 2008;213:372–380. doi: 10.1016/j.expneurol.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu YH, Zhou JN, Van Heerikhuize J, Jockers R, Swaab DF. Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer’s disease. Neurobiol Aging. 2007;28:1239–1247. doi: 10.1016/j.neurobiolaging.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Caballero B, Vega-Naredo I, Sierra V, Huidobro-Fernández C, Soria-Valles C, De Gonzalo-Calvo D, Tolivia D, Gutierrez-Cuesta J, Pallas M, Camins A, Rodríguez-Colunga MJ, Coto-Montes A. Favorable effects of a prolonged treatment with melatonin on the level of oxidative damage and neurodegeneration in senescence-accelerated mice. J Pineal Res. 2008;45:302–311. doi: 10.1111/j.1600-079X.2008.00591.x. [DOI] [PubMed] [Google Scholar]
- 22.Sánchez-Hidalgo M, Guerrero Montávez JM, Carrascosa-Salmoral Mdel P, Naranjo Gutierrez Mdel C, Lardone PJ, de la Romero CA. Decreased MT1 and MT2 melatonin receptor expression in extrapineal tissues of the rat during physiological aging. J Pineal Res. 2009;46:29–35. doi: 10.1111/j.1600-079X.2008.00604.x. [DOI] [PubMed] [Google Scholar]
- 23.Lahiri DK, Ge Y-W, Sharman EH, Bondy SC. Age-related changes in serum melatonin in mice, and the highest levels of combined melatonin and melatonin sulfate in the brain cortex than serum, heart, liver and kidney tissues. J Pineal Res. 2004;36:217–223. doi: 10.1111/j.1600-079X.2004.00120.x. [DOI] [PubMed] [Google Scholar]
- 24.Lemoine P, Nir T, Laudon M, Zisapel N. Prolonged-release melatonin improves sleep quality and morning alertness in insomnia patients aged 55 years and older and has no withdrawal effects. J Sleep Res. 2007;16:372–380. doi: 10.1111/j.1365-2869.2007.00613.x. [DOI] [PubMed] [Google Scholar]
- 25.Mayer G, Wang-Weigand S, Roth-Schechter B, Lehmann R, Staner C, Partinen M. Efficacy and safety of 6-month nightly ramelteon administration in adults with chronic primary insomnia. Sleep. 2009;32:351–360. doi: 10.1093/sleep/32.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jagota A, Kalyani D. Effect of melatonin on age induced changes in daily serotonin rhythms in suprachiasmatic nucleus of male Wistar rat. Biogerontology. 2010;11:299–308. doi: 10.1007/s10522-009-9248-9. [DOI] [PubMed] [Google Scholar]
- 27.Head E, Corrada MM, Kahle-Wrobleski K, Kim RC, Sarsoza F, Goodus M, Kawas CH. Synaptic proteins, neuropathology and cognitive status in the oldest-old. Neurobiol Aging. 2009;30:1125–1134. doi: 10.1016/j.neurobiolaging.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutten BP, Van der Kolk NM, Schafer S, van Zandvoort MA, Bayer TA, Steinbusch HW, Schmitz C. Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am J Pathol. 2005;167:161–173. doi: 10.1016/S0002-9440(10)62963-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uyanikgil Y, Baka M, Ateş U, Turgut M, Yavaşoğlu A, Ulker S, Sözmen EY, Sezer E, Elmas C, Yurtseven ME. Neuroprotective effects of melatonin upon the offspring cerebellar cortex in the rat model of BCNU-induced cortical dysplasia. Brain Res. 2007;1160:134–144. doi: 10.1016/j.brainres.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 30.Kaewsuk S, Sae-Ung K, Phansuwan-Pujito P, Govitrapong P. Melatonin attenuates methamphetamine-induced reduction of tyrosine hydroxylase, synaptophysin and growth-associated protein-43 levels in the neonatal rat brain. Neurochem Int. 2009;5:397–405. doi: 10.1016/j.neuint.2009.04.010. [DOI] [PubMed] [Google Scholar]