

Abstract
Compound effects on cloned human Ether-à-go-go related gene (hERG) potassium channels have been used to assess the potential cardiac safety liabilities of drug development candidate compounds. In addition to radioactive ligand displacement tests, two other common approaches are surrogate ion-based flux assays and electrophysiological recordings. The former has much higher throughput, whereas the latter measures directly the effects on ionic currents. Careful characterization in earlier reports has been performed to compare the relative effectiveness of these approaches for known hERG blockers, which often yielded good overall correlation. However, cases were reported showing significant and reproducible differences in potency and/or sensitivity by the two methods. This raises a question concerning the rationale and criteria on which an assay should be selected for evaluating unknown compounds. To provide a general basis for considering assays to profile large compound libraries for hERG activity, we have conducted parallel flux and electrophysiological analyses of 2,000 diverse compounds, representative of the 300,000 compound collection of NIH Molecular Library Small Molecular Repository (MLSMR). Our results indicate that at the conventional testing concentration 1.0 μM, the overlap between the two assays ranges from 32% to 50% depending on the hit selection criteria. There was a noticeable rate of false negatives by the thallium-based assay relative to electrophysiological recording, which may be greatly reduced under modified comparative conditions. As these statistical results identify a preferred method for cardiac safety profiling of unknown compounds, they suggest an efficient method combining flux and electrophysiological assays to rapidly profile hERG liabilities of large collection of naive compounds.
Introduction
Ion channels form a large class of integral membrane proteins. Their malfunction is causal to a variety of human diseases and they represent a class of attractive drug targets.1–3 As a group, they have not been exploited as efficiently as some other target classes, such as proteases and G-protein-coupled receptors.3–5 High-throughput screening (HTS) campaigns designed to identify active compounds for ion channel targets are of great interest, but have met considerable technical challenges.6,7 This limitation is primarily due to the inherent technical challenges associated with measuring electrical currents derived from ion channel activity in HTS formats. Currents passing through ion channels are often small and transient and require special conditions to activate, all of which can pose challenges for HTS implementation. In recent years, the rapid progress in developing functional assays and instrumentation has enabled HTS campaigns on an expanding variety of channel types.6,8
Ion channels are functionally diverse and exhibit a variety of properties critical for their physiological roles, most prominently their ability to allow selective permeation of ions. The major classes of channels can be defined based on ion permeation of potassium, sodium, calcium, chloride, or a mixture of these ions. Ionic selectivity is a key determinant of physiological function and an important factor in HTS assays' design and performance. Functional cell-based assays tend to yield a more physiological readout than some nonfunctional biochemical assays and are thus increasingly used in HTS ion channel assays.9 The two major types of nonelectrophysiological functional assays implemented using current technologies are based on detecting either the change of concentration of the permeated ions (or their surrogates) or the consequence of the concentration change. The former is usually carried out by taking advantage of fluorescent dyes that selectively bind to the targeted ions.8,10,11 The latter is often based on the membrane potential changes following ion fluxes.12,13 Using surrogate ionic fluxes, several large HTS campaigns have been recently described in published reports14,15 and in a public database (PubChem AIDs 1456, 1511, 1672, 1918, 2156, and 2239). Examples of HTS campaigns using direct measurement of ionic current have been less commonly reported.16
Voltage-gated potassium channels are an important class of drug targets.17 Modulators of these channels have been reported,17,18 some of which were identified via HTS compound screens using flux-based assays.19–21 The primary approach has involved the use of surrogate ions such as rubidium (Rb+) or thallium (Tl+), which may be detected by atomic absorption spectrometry22,23 or fluorescent dyes.8,10,11 With the advent of automated patch-clamps,6,24–27 it is now possible to consider whether the more direct measurements of channel electrophysiological activity obtained from the automated patch-clamp justifies its higher implementation cost in high-throughput screens. It is therefore important to obtain more specific and quantitative information to assess the cost and benefit comparatives between assays employing electrophysiological methods versus surrogate ion flux measurements.
Promiscuous block of the cardiac potassium channel encoded by human Ether-à-go-go related gene (hERG) is a major cause of cardiac toxicity by a variety of clinical drugs. Both flux assay and electrophysiological approaches have been applied to determine the potential cardiac toxicity risk of the developing compounds.16,28 We therefore used hERG as the testing channel and performed comparative analyses of Tl+-based flux assays and electrophysiological recording of a voltage-gated potassium channel. Our experiments employed a validation set (∼2,000 compounds) representative of the NIH Molecular Library Small Molecular Repository (MLSMR), which consists of more than 300,000 compounds and has been used in all screens of the molecular library initiative (http://mli.nih.gov/mli/). Therefore, the reported experiments favor a screening collection of diverse compounds instead of a drug-like collection. The hERG Tl+ assay was optimized to identify inhibitors (PubChem AID 1511). The electrophysiological assay used population-based recording methods and was designed to allow identification of both inhibitors and potentiators. The described assay conditions are comparable to what has been reported by others, as judged by IC50 values.11,29 The resultant hits from these screens under different selective criteria were analyzed and compared to assess these two assays. This approach differs from previously published analyses of hERG assays in which comparison of IC50 values of mostly known hERG blockers was performed to identify the preferred approach for safety evaluation and compound optimization. The present study employs a statistical approach to compare different HTS assays to quickly assess the effectiveness of assays to be employed for evaluation of hERG liabilities of large sets of compounds.
Materials and Methods
Cell Culture
A stable cell line expressing hERG was previously generated using Chinese hamster ovary cells. These cells were routinely cultured in complete Dulbecco's modified Eagle's medium/nutrient mixture F-12 medium (Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Gemini, Calabasas, CA), 2 mM l-glutamine (Invitrogen, Carlsbad, CA), and 500 μg/mL G418 (Invitrogen). They were cultured at 37°C under 5% CO2. Cells were maintained and passaged till 80% confluence was reached.
Compound Selection and Handling
The validation set of the MLSMR library, including 1,999 test compounds of various structures, was selected for the flux and electrophysiological assays. To determine whether the MLSMR validation set defines a more structurally representative subset of the larger MLSMR library than other possible small-molecule validation sets, we computed the number of structural neighbors in the entire MLSMR library for each compound in (1) the MLSMR validation set of 1,999 compounds (Biofocus DPI, San Francisco, CA) and (2) the Library of Pharmacologically Active Compounds (LOPAC) collection of 1,280 compounds (Sigma, St. Louis, MO), as well as the number of total unique structural neighbors in the MLSMR library for each set. We also performed these calculations for 100 random samples of MLSMR library subsets of size 1,000, 2,000, 3,000, 4,000, and 5,000 compounds. Finally, we performed a greedy optimization to estimate the subset with the maximal number of unique structural neighbors for a given compound subset size. This greedy approach used the standard algorithm for problems such as vertex cover: iterative selection of the compound with the greatest number of neighbors that are not neighbors of previously selected compounds. For these calculations, we computed ECFP_4 circular fingerprints hashed to binary strings of length 1,024 for each compound using Pipeline Pilot 6.1.5.0 Student Edition (Accelrys, San Diego, CA). Circular fingerprints measure structural patterns present in radial bond neighborhoods surrounding each nonhydrogen atom in a small molecule and have been previously demonstrated to contain high information content relative to other 2D structural descriptors.30–32 Because these descriptors sample an immense feature space of possible atom bond neighborhoods, they are hashed into binary strings of length suitable for computation. For each compound set, we removed fragments, standardized both coordinates and charges using Pipeline Pilot, and removed duplicate compound records (as determined by comparison of canonical SMILES strings computed in Pipeline Pilot). For each compound in the MLSMR validation set, LOPAC collection, and the whole library, we computed the Tanimoto coefficient with every compound in the MLSMR library:
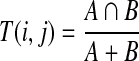 |
where A and B are the number of “on” bits in compounds i and j. Thus, each compound receives a score between 1 and 0 representing its similarity to a compound in the MLSMR library. Collections of compounds in the MLSMR library were judged similar to compounds in the validation set or LOPAC collection if their Tanimoto coefficient was greater than 0.3 or 0.7 compared with the target. The 0.3 threshold value has been previously observed as being the lowest score to correlate with the degree of structural similarity at which similar biological activity of small molecules may be expected.33,34 The 0.7 threshold value has been commonly used because it gives much higher confidence in correlating structural similarity. Theoretically, a Tanimoto coefficient of 1 should represent structurally identical compounds. However, because of the hashing step necessary to calculate binary fingerprints, we have observed in practice that sometimes nonidentical compounds may be assigned a Tanimoto coefficient of 1. Therefore, we set an upper limit on similarity between compounds in the validation set and LOPAC compared with the MLSMR library to be 1. To remove any comparisons of compounds to themselves, we subtracted 1 from the number of nearest neighbors for each compound in the validation set, and 1 from the number of nearest neighbors for those LOPAC compounds present in the MLSMR library (498/1,264 compounds). Random MLSMR subset selection and the greedy optimization algorithm were performed using in-house programs written in C++. Statistical analysis on the final datasets was performed with MATLAB (Mathworks, Natick, MA).
The validation compounds from BioFocus Discovery Partners International (Biofocus DPI) were dissolved in dimethyl sulfoxide (DMSO) with a concentration of 5 mM in 384-well polystyrene plates. All compounds are of >90% purity, as determined by liquid chromatography–mass spectrometry. They were diluted to the test concentration on the day of the experiments. The test compounds were examined at 1 and 10 μM in Tl+-based flux assays and at 1 μM for electrophysiological assays. The control compound, dofetilide (N-[4-[2-[2-[4-(methanesulfonamido)phenoxy]ethyl-methylamino]ethyl]phenyl] methane sulfonamide) (Fisher Scientific, Pittsburg, PA) was dissolved in DMSO at a concentration of 20 mM.
Tl+-Based Fluorescence Assay
Activity of potassium channels was monitored by the influx of a surrogate ion, Tl+, for potassium. Tl+ influx was detected through the use of a Tl+-sensitive fluorescent dye, FluxOR™ (Invitrogen). Cells were seeded at 6,500 cells per well into BD Biocoat poly-d-lysine–coated 384-well plates using a Multidrop (Thermo Scientific, Hudson, NH) and incubated overnight at 37°C under 5% CO2. The Tl+-based fluorescence assay protocol (see Appendix) was adapted from the manufacturer's recommended protocol. Briefly, medium was removed; cells were incubated with 1× FluxOR solution, 25 μL/well, for 90 min at room temperature in the dark; the 1× FluxOR solution was replaced by assay buffer (Hanks balanced salt solution containing 5.8 mM potassium; Catalog No. 14065; Invitrogen), 20 μL/well; 7.5× test compounds and controls in assay buffer were then added to cells, 4 μL/well; 20 min later, cell plates were loaded to Hamamatsu FDSS 6000 kinetic imaging plate reader (Hamamatsu Photonics, Hamamatsu, Japan); after establishing fluorescence baseline by 1 Hz scanning for 10 s, the hERG channels were activated by addition of 6 μL/well stimulus buffer containing 25 mM K2SO4 and 7 mM Tl2SO4 to the assay buffer, giving a final potassium concentration of 15.8 mM and final Tl+ concentration of 2.8 mM; and fluorescence measurement was continued at 1 Hz for another 110 s. To evaluate the robustness of the HTS Tl+-based fluorescence assays, dofetilide at 1 μM was applied as positive control, whereas assay buffer was employed as negative control. Both the positive and negative controls were prepared with either 0.02% (v/v) or 0.2% (v/v) DMSO, corresponding to 1 and 10 μM test concentrations, respectively. The fluorescence ratio, F(max–min)/F0 (ΔF/F0), was calculated for each well using the entire 120 s detection window and then normalized to the positive and negative control wells. Compared with the negative controls, if a compound caused signal decrease by more than three times the standard deviation (SD) in fluorescence ratio, it was classified an inhibitor.
Automated Patch-Clamp Assays with IonWorks
Automated patch-clamp experiments were performed using the population patch-clamp mode of automated voltage clamp recording with IonWorks Quattro™ (Molecular Devices Corporation, Sunnyvale, CA). Briefly, compound effects on the hERG channel were tested at 1 μM with a two-step voltage protocol. The Chinese hamster ovary cells stably expressing hERG channels were dislodged from flasks and dispensed into a 384-well population patch-clamp mode plate. After dispensing, seal resistance of cells was measured for each well and cells were perforated by incubation with 100 μg/mL amphotericin B (Sigma). Activity of hERG was then measured with the recording protocol as follows: cells were held at −70 mV, stepped down to −80 mV for 100 ms to estimate leak currents, and depolarized to −30 mV for 100 ms to estimate non-hERG currents. Then, hERG currents were evoked by two voltage pulses with a 20 mV difference at the depolarization steps. During the first pulse, cells were depolarized to +20 mV for 2 s and hyperpolarized to −30 mV for 2 s. Prior to the second pulse, cells were held at −70 mV for 3 s. Then cells were depolarized to +40 mV for 2 s followed by a hyperpolarization to −30 mV for 2 s. This series of pulses was applied to cells before and following a 3-min incubation of cells with 1 μM test compounds or assay buffer. Positive (1 μM dofetilide) and negative controls (external buffer with 0.02% [v/v] DMSO) were applied within each plate to evaluate the data quality. Only cells with a tail current amplitude bigger than 0.2 nA, a seal resistance >30 MOhms, and seal resistance drop rate lower than 25% were included for data analysis. The peak amplitudes of tail currents before and after compound treatment were measured. Compound effects were assessed by the percentage change in the hERG tail current. The percentage changes were calculated by dividing the difference between pre- and post-compound hERG tail currents by the respective pre-compound tail current in the same well and normalized to positive and negative controls in the same plate. Compared with the negative controls, if a compound decreased the hERG tail current by >3 SD, it was classified as an inhibitor.
Whole-cell currents were measured in the following recording buffers: 137 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4 adjusted with NaOH (extracellular solution); 40 mM KCl, 100 mM K-gluconate, 1 mM MgCl2, and 5 mM HEPES, pH 7.2 adjusted with KOH (intracellular solution). The voltages listed above were before correction for liquid junction potential. An online correction of −20 mV was applied to correct for junction potentials. The current signal was sampled at 0.625 kHz.
Data Analysis
The data from the Tl+-based flux assay were analyzed by the manufacturer's software package (Hamamatsu Photonics) and the automated patch-clamp experiments were analyzed in IonWorks 2.0.4.4 (Molecular Devices Corporation). Further calculations were performed in Excel (Microsoft, CA). Linear correlation and graphic plots were carried out in Origin 7.0 (OriginLab Corporation, Northampton, MA) and Spotfire (TIBCO, Somerville, MA).
Results
Selection of Test Compound Collection
To evaluate the extent of correlation between the Tl+-based flux assay and the population patch-based electrophysiological assay, we selected a validation set of 1,999 compounds. This group of compounds was assembled as a representation of the ∼300,000 MLSMR compound library (http://mlsmr.glpg.com/MLSMR_HomePage/). We use this collection to help gauge how different assays may perform if the entire MLSMR was to be tested.
To obtain a more quantitative assessment, we computed ECFP_4 circular fingerprints hashed to binary strings of length 1,024 for each compound (see Materials and Methods section). The number of neighbor compounds existing in the MLSMR library was higher for most compounds in the validation set compared with the LOPAC collection (Fig. 1A). The mean and standard error for the number of structural neighbors for the individual compounds in each set was, respectively, 829.5 ± 27.8 (validation set) and 306.7 ± 18.6 (LOPAC) if a 0.3 Tanimoto coefficient threshold is applied (Fig. 1A). When applying a 0.7 threshold, the value for LOPAC collection was close to zero (data not shown), consistent with the notion that more compounds in LOPAC are bioactive and unique relative to the validation set. We compared this to random samples of 100 of subsets of size 1,000, 2,000, 3,000, 4,000, and 5,000 of the MLSMR collection, which suggested that both the validation and LOPAC compound sets had lower numbers of individual neighbors per compound than random subsets of the library (Fig. 1A): the mean and standard error for the number of neighbors per compound for the entire library is 1,618 ± 31, suggesting that the values obtained for the random subsets are representative of the statistics of the library as a whole. Because compounds within a given collection may share the same neighbors, we also compared the number of unique neighbors in the MLSMR library for all compounds in the validation and LOPAC collections to both 100 random samples of subsets of size 1,000, 2,000, 3,000, 4,000, and 5,000 of the library, and a greedy optimization for maximum coverage (Fig. 1B). The results follow a similar trend to the calculations for number of neighbors per individual compounds. It should be noted that the validation set, although structurally more similar to the MLSMR library than the LOPAC collection using either a 0.3 or 0.7 threshold, actually appears to cover less of the collection with its structural neighbors than random MLSMR subsets (Fig. 1B). Because the MLSMR library contains products of combinatorial chemistry (Fig. 1D, Diversity compounds), in which a single scaffold is extensively augmented with diverse R groups, many compounds will have a large number of structural neighbors that are analogs in their respective combinatorial synthesis families. Conversely, subsets such as the validation compounds, which are more biased toward known bioactive compounds, may exclude such diversity compounds (Fig. 1D) and thus have an overall smaller number of unique neighbors. However, a recent study has suggested that the biologically active domain of chemical space is better covered by a relatively small set of natural-product-like compounds than a larger set of combinatorial compounds representing extensive structural diversity.6 Thus, although the validation set has an overall lesser number of unique neighbors in the MLSMR library than random subsets, it represents a mixture of diversity compounds and bioactives. Comparing only the LOPAC and validation sets, the number of structural neighbors per compound in the MLSMR library for the validation and LOPAC collections was significantly different as assessed by a two-sample t-test (P = 3.2603 e − 042) (Fig. 1C). Our analysis provides a quantitative basis that the validation set is, on a whole, more structurally representative of the chemical space spanned by the entire MLSMR library than the LOPAC collection. In addition, the results could allow us to examine whether bioactives and diversity compounds within the validation set display any collective difference.
Fig. 1.

Structural diversity of the MLSMR library captured by smaller curated compound sets. (A) Mean of number of neighbors per compound in the MLSMR library for compounds in the validation set (1,982 unique compounds as judged by canonical SMILES strings), LOPAC collection (1,264 unique compounds), and random samples of 100 of subsets of size 1,000, 2,000, 3,000, 4,000, and 5,000 of the MLSMR library, as judged by a Tanimoto coefficient of greater than 0.3 computed using 1,024-bit ECFP_4 fingerprints generated in Pipeline Pilot (Scitegic, San Diego, CA). For the validation and LOPAC collections, error bars are standard error for number of neighbors per compound; for random subsets, error bars represent a single SD of the mean number of neighbors per compound for each of the 100 samples of a given size. (B) Number of unique neighbors in MLSMR library for validation set, LOPAC, random subsets of the MLSMR library, and a greedy optimization for maximum coverage. Error bars for a single SD of random subsets are too small to be visualized. (C) The mean of the number of neighbors per compound in the validation and LOPAC sets are significantly different as assessed by a two-sample t-test (P = 3.2603 e −042). The triple asterisks indicate P < 0.001. Error bars represent the standard error of the two sets. (D) Composition of MLSMR and validation set compounds. LOPAC, Library of Pharmacologically Active Compounds; MLSMR, Molecular Library Small Molecular Repository; SD, standard deviation. Color images available online at www.liebertonline.com/adt.
Tl+-Based Assays
HTS campaigns including Tl+ and other flux-based assays are often performed using single concentrations of 1–20 μM. To gain additional information concerning the dynamic window, we performed our hERG assays at both 1 and 10 μM. An additional reason for including 1 μM drug concentration is that this concentration may be used to evaluate hERG-caused cardiotoxicity.35,36 The hERG Tl+-based assay was performed in duplicate at both concentrations to evaluate fidelity over a wider dynamic range and the results are plotted in Figure 2. Figures 2A and 2B show linear regression plots of two repeats for 1 and 10 μM concentrations as indicated. The R2 values were 0.974 and 0.972, respectively. This degree of convergence for data obtained at 1 and 10 μM suggests that this assay format may be used to identify hERG inhibitors with IC50 values less than or equal to the test concentrations. The control measurements made on each test plate were included in the data shown in Figure 2A and 2B. Analysis of variation in the normalized buffer control values was used to calculate criteria for hit identification as shown in Figure 2C, in which the dashed lines represent ±3 SD of the buffer control measurements and are plotted along with values obtained for test compounds. With this threshold, a total of 467 hits were identified at 1 μM test concentration and, as expected, more compounds (1,111) were identified as hits at 10 μM. Approximately 23% of compounds tested at 1 μM were active as inhibitors at the −3 SD level and 56% were active when tested at 10 μM (Table 1). This result is consistent with existing reports by others that the Tl+ assay is less sensitive than the electrophysiological assay.11,29 To assess the reliability of the hit selection process, we calculated the fraction of hits identified at 1 μM that were also identified as hits at 10 μM, which indicate that 94% (440/467) of the 1 μM hits confirmed at 10 μM. This suggests that this approach may be useful for large-scale classification of chemotypes. This hit rate is significantly higher than the expected rate of false positives expected for a normally distributed population. It is consistent with the existing data that a variety of chemotypes block hERG channels.36,37
Fig. 2.

Linear correlation of duplicates and signal distribution of compounds from the validation collection in flux assay. Compounds in the validation collection were tested in duplicate for hERG inhibition at 1 and 10 μM using a flux-based assay. Channels were activated by additional 10 mM K+ and 2.8 mM Tl+ in external buffer. Changes in fluorescence ratio (ΔF/F0) at the end of scanning (120 s) were employed to evaluate compound effects. Diagonal lines in panels A and B represent identical results. In both sets of experiments, duplicates exhibit high correlation. R2 values were 0.974 and 0.972, respectively. Hit selection was performed with the averaged fluorescence ratio change of duplicates. Four hundred sixty-seven and 1,111 compounds were identified as hits at 1 and 10 μM, respectively. Solid lines in panels C and D represent averages of buffer controls; dashed lines represent ±3 SD of buffer controls (SD = 4.94% and 4.64% for 1 and 10 μM testing compounds). For 1 and 10 μM, the averaged Z′ factor is 0.80 ± 0.04 and 0.82 ± 0.03, the signal-to-noise ratio is 27.66 ± 6.03 and 26.82 ± 5.13, and the signal window is 0.48 ± 0.06 and 0.61 ± 0.05, respectively. hERG, human Ether-à-go-go related gene.
Table 1.
Comparison of hERG Inhibitor Hit Rate Between Flux and Electrophysiological Assay Using Different Selection Criteria
| Hit Selection Criteria | Tl+ (%) | IonWorks (%) | Tl+ Positive, IonWorks Negative (%) | Tl+ Negative, IonWorks Positive (%) | Overlapped (%) |
|---|---|---|---|---|---|
| 1 μM Flux Assay vs. 1 μM Electrophysiology Assay | |||||
| 1 SD |
64.58 |
58.38 |
29.95 |
19.61 |
50.44 |
| 2 SD |
41.02 |
33.58 |
40.91 |
25.11 |
33.97 |
| 3 SD |
23.36 |
19.36 |
41.54 |
26.80 |
31.66 |
| 4 SD |
14.41 |
11.94 |
41.03 |
26.15 |
32.82 |
| 5 SD |
10.26 |
8.41 |
40.66 |
24.91 |
34.43 |
| 10 μM Flux Assay vs. 1 μM Electrophysiology Assay | |||||
| 1 SD |
86.59 |
58.38 |
38.76 |
5.77 |
55.47 |
| 2 SD |
71.89 |
33.58 |
58.42 |
7.65 |
33.93 |
| 3 SD |
55.58 |
19.36 |
68.68 |
6.72 |
24.60 |
| 4 SD |
41.17 |
11.94 |
73.71 |
5.94 |
20.34 |
| 5 SD | 31.82 | 8.41 | 75.64 | 4.36 | 20.00 |
SD, standard deviation; Tl+, thallium.
IonWorks-Based Electrophysiological Assay
Electrophysiological tests for hERG were carried out using the population patch recording mode on IonWorks Quattro (see Materials and Methods section). The promiscuous inhibition of hERG channels by a variety of chemical structures36,38 and the high hit rate for hERG inhibitors observed in the Tl+ assay favor the idea of focusing the analyses of effects in the IonWorks assay to data obtained at 1 μM compound concentration. The duplicate tests of 1,999 compounds yielded 1,373 pairs of data points from two repeats of separate plates, representing an average of 86% success rate of electrophysiological recording per plate. Failure of some compounds in electrophysiological tests may have resulted from specific chemical effects on cell health and seal quality. To explore this possibility, these compounds were analyzed for unique chemical properties that might be common to failed compounds, such as molecular weight, solubility, and hydrophobicity (ClogP). We did not detect any convincing pattern of shared properties for these failed compounds. Hence, the fluctuation of data quality was likely caused primarily by batch-to-batch cell variability, channel-dependent characteristics, and/or instrument stability.
Good agreement between replicates in the IonWorks assay was observed (R2 = 0.872) with a slope of 1 (Fig. 3A), suggesting the assay is sufficiently fit for reliable hit identification. Based on the high correlation coefficient between replicate tests, the hit selection process was applied to the set of 1,927 compounds for which one or two determinations were made. Application of a hit selection criterion of −3 SD for inhibitors, same as was used for Tl+ assay, has identified a total of 373 hits, yielding a 19% hit rate (Fig. 3B and Table 1).
Fig. 3.

Linear correlation between duplicates and signal distribution of compounds in the IonWorks electrophysiological assay. Compounds were tested in duplicate at 1 μM for hERG inhibition using an automated electrophysiological assay. (A) The peak tail currents of hERG were measured and replicate values from two trials are plotted; the diagonal line represents identical results. (B) The average of two repeats was used to evaluate compound effects. Three hundred eighty-seven compounds were identified as hits. Solid line in panel B represents the average of buffer controls; dashed lines represent ±3 SD of buffer controls. SD = 9.97%. The averaged Z′ factor is 0.70 ± 0.09, the signal-to-noise ratio is 14.08 ± 2.73, and the signal window is 93.64% ± 5.19%.
Correlation of Flux and Electrophysiological Data
The correlation between block of hERG tail currents in the electrophysiological assay and block of Tl+ influx in the fluorescent assay is plotted in Figure 4A for the 1,927 compounds for which both data types exist. A moderate positive correlation was observed (R2 = 0.609), with a significant number of compounds falling outside the predicted 95% confidence limits (dashed lines). Among the combined total of 638 hits identified by Tl+ and electrophysiological assays as hits (larger than −3 SD of buffer) at 1 μM test concentration, only 202 compounds were picked up by both assays, yielding ∼32% overlap (Table 1). Because the electrophysiological method directly measures ionic current, the false negatives identified by the Tl+ assay are of concern. We retested some of these compounds individually and confirmed their inhibitory effects. Figure 4B displays the structure of pyrilamine (CID 5284451) with potent inhibition of hERG currents (Fig. 4D). In contrast, it failed to significantly block Tl+ flux in the fluorescent assay over the entire 120-s recording period at 1 μM (Fig. 4C). The IC50 value of pyrilamine is 1.67 ± 0.04 μM by electrophysiological recording, but it is well above 10 μM in the Tl+ assay (data not shown). Pyrilamine is a previously known hERG blocker39 and has no detectable fluorescence that may cause interference to the Tl+-based measurement.
Fig. 4.

Correlation between the IonWorks electrophysiological- and flux- assays. The compound effects at 1 μM in the electrophysiological assay were compared with those in the flux assay (A). R2 is 0.609. Solid line in panel A plots a slope of 1; dashed lines represent 95% prediction lines. Some compounds exhibited little effect on hERG in the flux assay but significantly inhibited the hERG activity in the IonWorks electrophysiological assay. A representative compound, pyrilamine (solid dot in A), was shown in (B). Its fluorescence readout in the flux assay and recording trace were shown with the negative control (buffer) and the positive control (dofetilide) in (C) and (D) as indicated. Color images available online at www.liebertonline.com/adt.
Indeed, the degree of overlap between the assay results is a key parameter to gauge the effectiveness of the Tl+-based assay. To evaluate whether there are shared properties among the compounds positive in the Tl+ assay but not detectable in the electrophysiological assay, we analyzed their physical and chemical properties. There was no detectable trend (Supplementary Figs. S1 and S2; Supplementary Data are available online at www.liebertonline.com/adt). We further analyzed the data by comparing hit overlaps using different hit selection criteria for the two assays. Specifically, we examined how the threshold of hit selection might reduce the number of false negatives. We first plotted the percentage of hits by the electrophysiological assay that is selected by the Tl+-based assay. Using the data from 1 μM concentration for both assays, 58%–71% of IonWorks hits were identified using 1–5 SD selection criteria in the Tl+ assay (Fig. 5A). When similar analyses were performed using the data from the electrophysiological assay at 1 μM compared with data from the Tl+-based assay obtained at 10 μM, there is a noticeable improvement in the percentage of IonWorks hits identified, ranging from 79% to 90% coverage depending on hit selection criteria. However, the overlap of hits between the two assays was not improved by comparing the IonWorks data at 1 μM with the Tl+ data at 10 μM (Fig. 5B and Table 1).
Fig. 5.

Comparison of overlapped hits between the flux and electrophysiological assays. Hits in the flux- and electrophysiology-based assays were selected by comparing compound signals with the average of buffer controls. (A) The percentage of IonWorks hits identified by the Tl+-based flux assays is plotted in a histogram against different hit selection criteria for the Tl+ assay (1, 2, 3, 4, and 5 SDs) at 1 and 10 μM compound concentrations as indicated. (B) The hits distribution is plotted similarly as shown in Figure 4A except the data from 10 μM testing compounds in the flux assay were used with R2 = 0.562. Tl+, thallium.
Discussion
Using flux of surrogate ions to assess ion channel activity has dramatically improved the throughput of cell-based assays for ion channels. This approach is particularly useful when considering its general applicability to an entire class of ion channels, for instance, using Tl+ or Rb+ fluxes to assess potassium channel function. This method for detecting ion channel activity can be coupled with various approaches for triggering or modulating channel activity so that assays for channels with different modes of channel gating can be established in high-throughput formats. An ongoing concern when implementing high-throughput ion channel assays using surrogate ion flux to track channel activity is the ability of an assay to reliably monitor ion channel fluxes in a more physiological context. Electrophysiological measurements of ion channel activity are often performed under restrictive or nonphysiological conditions, but are still the most accessible current methods used to directly measure ion channel activity under conditions that may generate relevant pharmacological data.
The data in this report reveal 32% overlap between hits identified by a Tl+-based surrogate ion assay and those identified by automated patch-clamp recordings using criteria that defined inhibitory hits as signal values lower than −3 SD of the buffer control values on each plate. Although it is recognized that the SD values for different assays were different, the degree of hit overlap between the assays did not improve as the criteria for hit selection was increased to larger than −5 SD. Indeed, this modest level of overlap is surprising. A few factors may contribute to this lower than expected level. First, it is well known that the surrogate ions have considerably different permeabilities from that of physiologic ions. This has been proposed to contribute to a right shift of IC50 values for known compounds inhibiting hERG potassium channels.40 In this case, using a higher concentration of testing compounds may yield a better overlap of hits identified by two different assays. Analysis of the Tl+ assay hits identified at 10 μM testing compound concentration (Table 1 and Supplementary Fig. S3) revealed that, as expected, more hits were identified at each selection criteria compared with 1 μM testing, and fewer Tl+-negative/IonWorks-positive compounds were identified. However, this approach did not increase the overlap between the Tl+ assay hits and the IonWorks hits because of large increases in the numbers of Tl+-positive/IonWorks-negative compounds.
Second, based on studies using known inhibitors of hERG potassium channels, it is clear that different compounds display varying degrees of right shift of potency, as large as 100-fold, in surrogate ion-based flux assays.40 Therefore, the structural features of testing compounds may differentially affect the inhibitory potency in each assay. This factor may be especially more pronounced for hERG than for other channels because hERG is inhibited by a variety of structural classes. Indeed, the Tl-based assay may be further optimized to improve its correlation with electrophysiological recording. Under our assay conditions, IC50 values of dofetilide and terfenadine, two structurally distinct hERG inhibitors, have different degrees of affinity shift when tested in two assays. Based on the Tl+-based assay, their IC50 values are 35.3 ± 1.3 and 1,256.6 ± 77.1 nM. These values are similar to those reported in literature, for example, 26 and 1,278 nM by Bridal et al.29 Both ours and the earlier reports have shown different degrees of right-shift of dose–response curve depending on the given compound tested. It is conceivable that further optimization of the Tl+-based flux assay could yield a better correlation with electrophysiology, hence arguing for the need of channel-specific assay optimization.
Third, many ion channels display distinct gating characteristics in response to different physiological conditions. For hERG channels, the rapid and precise voltage protocol applied by the automated patch-clamp used to isolate tail currents has considerably higher resolution that is unattainable by a change of extracellular ion concentration introduced via liquid handling, which is commonly used for triggering channel opening in flux assays. As a result, the flux assay, although target specific, may not replicate gating-specific pharmacological effects measured in patch-clamp studies. Of course, the degree of correlation between different assay formats may depend on the target and the properties of active compounds sought by the primary screen.
The present study examines the correlations between compounds inhibiting hERG channels in a fluorescent Tl+ influx assay and an electrophysiological assay performed on an IonWorks 384-well automated patch-clamp instrument. A structurally diverse set of 1,999 compounds assembled from the NIH MLSMR library was used to examine these assays in an HTS format. The validation set contains both diverse compounds and bioactives. With the −3 SD criteria, the hit rate for non-“known bioactives” in the Tl+ assay is 23.3% at 1 μM and 54.9% at 10 μM. The hit rate for “known-bioactives” in the Tl+ assay is 23.4% at 1 μM and 56.9% at 10 μM. In the electrophysiology assay, the hit rate for naïve compounds is 20.4% and 17.2% for known bioactives. Although the validation set does not perfectly represent the MLSMR collection, its diversity compounds and known bioactives show similar performance in the two assays. One may speculate that the naive diversity compounds in MLSMR have uncharacterized bioactivity against hERG. Additionally, because there are considerable data obtained with the validation set available in PubChem, our results may be further analyzed in the context of other assays and targets.
The purpose of these experiments is not to determine the preferred or most reliable predictor of hERG liabilities for cardiac safety screening. That question has been addressed in a number of publications using IC50 values calculated from small sets of compounds typically with defined hERG-blocking properties (e.g., refs.11,29). The data presented here provide more relevant argument for the importance of electrophysiological assays using statistical data from a much larger collection of naïve compounds. The present experiments indicate that the hERG Tl+ flux assay used in the present format can be used to classify hERG-inhibitory properties of large sets of compounds. A significant fraction (1/4) of the combined hit set was negative in the flux assay and positive in the IonWorks assay when tested at 1 μM in both assays. This fraction decreased to 1/10 when comparing the flux assay results obtained at 10 μM with the IonWorks data at 1 μM. Therefore, a workflow utilizing a Tl+ flux high-density assay at a higher testing concentration followed by electrophysiological recording to validate the active compounds may remain an effective approach for rapid and efficient characterization of hERG inhibitory properties of large sets of compounds. Alternative approaches with lower numbers of false negatives and false positives may be preferable for cardiac safety profiling of limited sets of compounds.
Supplementary Material
Abbreviations:
- CHO
Chinese hamster ovary
- DMSO
dimethyl sulfoxide
- hERG
human Ether-à-go-go related gene
- HTS
high-throughput screening
- MLSMR
Molecular Library Small Molecular Repository
- SD
standard deviation
Appendix
Thallium Assay Protocol
| Step | Parameter | Value | Description | Notes |
|---|---|---|---|---|
| 1 |
Plate cells |
50 μL/well |
6,500 CHO-hERG cells |
BD Biocoat poly-d-lysine–coated 384-well plate, clear bottom for imaging |
| 2 |
Incubation time |
16–18 h |
Cell growth |
37°C and 5% CO2 |
| 3 |
FluxOR loading |
25 μL/well, 90 min |
Dye loading |
1:1,000 FluxOR at ambient temperature in the dark |
| 4 |
Assay buffer |
20 μL/well |
Extra dye removal and buffer replacement |
1 × HBSS buffer |
| 5 |
Compounds and controls |
4 μL/well |
1 or 10 μM |
Stock concentration is 7.5 or 75 μM; positive control (1 μM dofetilide) wells I1–P2 and A23–H24; negative control (assay buffer with 0.02% or 0.2% DMSO) wells I23–P24; blank controls (Tl+ free) wells A1–H2, library compounds wells A3–P22 |
| 6 |
Compound incubation |
20 min |
Compound equilibrium |
Ambient temperature in the dark |
| 7 |
Channel stimulation |
6 μL/well |
Channel activation |
Channels are activated by addition of 5× Cl−-free stimulus buffer containing 55.8 mM K+ and 14 mM Tl+ |
| 8 | Assay readout | 520 nm | Fluorescence detection | Hamamatsu FDSS 6000 kinetic imaging plate reader, excitation filter 480 nm, emission filter 520 nm, detect at 1 Hz for 120 s |
CHO, Chinese hamster ovary; DMSO, dimethyl sulfoxide; HBBS, Hanks balanced salt solution; hERG, human Ether-à-go-go related gene; Tl+, thallium.
IonWorks Assay Protocol
| Step | Parameter | Value | Description | Notes |
|---|---|---|---|---|
| 1 |
Add external buffer |
3.5 μL/well |
Prime plate |
External buffer: 137 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4 |
| 2 |
Hole test |
0 to −10 mV |
Test plate |
0 mV step to −10 mV |
| 3 |
Plate cells |
3.5 μL/well |
6,000 CHO-hERG cells |
Population patch-clamp mode plate, each well with 64 holes |
| 4 |
Seal test |
−70 to −80 mV |
Cell seal test |
−70 mV step to −80 mV for 160 ms |
| 5 |
Incubation with amphotericin B |
100 μg/mL |
Membrane perforation |
Amphotericin B in internal buffer (40 mM KCl, 100 mM K-gluconate, 1 mM MgCl2, 5 mM HEPES, pH 7.2) and perforate for 70 s |
| 6 |
Detect currents |
12 s |
Trace recording |
Cells held at −70 mV, stepped down to −80 mV for 100 ms, depolarized to −30 mV for 100 ms and to +20 mV for 2 s, hyperpolarized to −30 mV for 2 s, held at −70 mV for 3 s, depolarized to +40 mV for 2 s, and hyperpolarized to −30 mV for 2 s |
| 7 |
Compounds and controls |
3.5 μL/well, 3 min |
1 μM |
Stock concentration is 3 μM; positive control (1 μM dofetilide) columns 2 and 23; negative control (external buffer with 0.02% DMSO) columns 1 and 24, library compounds columns 3–22 |
| 8 | Detect currents | 12 s | Trace recording | Recording protocol is the same as step 6 |
Acknowledgments
The authors thank the members of both Li laboratory and JHICC for valuable discussions and comments on the manuscript. This work was supported by grants from the National Institutes of Health (GM078579, MH084691).
Disclosure Statement
No competing financial interests exist.
References
- 1.Camerino DC. Desaphy JF. Tricarico D. Pierno S. Liantonio A. Therapeutic approaches to ion channel diseases. Adv Genet. 2008;64:81–145. doi: 10.1016/S0065-2660(08)00804-3. [DOI] [PubMed] [Google Scholar]
- 2.Kaczorowski GJ. McManus OB. Priest BT. Garcia ML. Ion channels as drug targets: the next GPCRs. J Gen Physiol. 2008;131:399–405. doi: 10.1085/jgp.200709946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Overington JP. Al-Lazikani B. Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 4.Bridges TM. Lindsley CW. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3:530–541. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 5.Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5:785–799. doi: 10.1038/nrd2092. [DOI] [PubMed] [Google Scholar]
- 6.Dunlop J. Bowlby M. Peri R. Vasilyev D. Arias R. High-throughput electro-physiology: an emerging paradigm for ion-channel screening and physiology. Nat Rev Drug Discov. 2008;7:358–368. doi: 10.1038/nrd2552. [DOI] [PubMed] [Google Scholar]
- 7.Xu J. Chen Y. Li M. High-throughput technologies for studying potas-sium channels—progresses and challenges. Drug Discov Today Targets. 2004;3:32–38. [Google Scholar]
- 8.Beacham DW. Blackmer T. O’ Grady M. Hanson GT. Cell-based potassium ion channel screening using the FluxOR assay. J Biomol Screen. 2010;15:441–446. doi: 10.1177/1087057109359807. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, et al. Ion-channel assay technologies: quo vadis? Drug Discov Today. 2001;6:1278–1287. doi: 10.1016/s1359-6446(01)02095-5. [DOI] [PubMed] [Google Scholar]
- 10.Weaver CD. Harden D. Dworetzky SI. Robertson B. Knox RJ. A thallium-sensitive, fluorescence-based assay for detecting and characterizing potassium channel modulators in mammalian cells. J Biomol Screen. 2004;9:671–677. doi: 10.1177/1087057104268749. [DOI] [PubMed] [Google Scholar]
- 11.Titus SA, et al. A new homogeneous high-throughput screening assay for profiling compound activity on the human ether-a-go-go-related gene channel. Anal Biochem. 2009;394:30–38. doi: 10.1016/j.ab.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu CJ, et al. A high-capacity membrane potential FRET-based assay for NaV1.8 channels. Assay Drug Dev Technol. 2006;4:37–48. doi: 10.1089/adt.2006.4.37. [DOI] [PubMed] [Google Scholar]
- 13.Felix JP, et al. Functional assay of voltage-gated sodium channels using membrane potential-sensitive dyes. Assay Drug Dev Technol. 2004;2:260–268. doi: 10.1089/1540658041410696. [DOI] [PubMed] [Google Scholar]
- 14.Delpire E, et al. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc Natl Acad Sci USA. 2009;106:5383–5388. doi: 10.1073/pnas.0812756106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis LM, et al. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7.1. Mol Pharmacol. 2009;76:1094–1103. doi: 10.1124/mol.109.059840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown AM. High throughput functional screening of an ion channel library for drug safety and efficacy. Eur Biophys J. 2009;38:273–278. doi: 10.1007/s00249-008-0356-2. [DOI] [PubMed] [Google Scholar]
- 17.Wulff H. Castle NA. Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong Q. Gao Z. Wang W. Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci. 2008;29:99–107. doi: 10.1016/j.tips.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Zaks-Makhina E. Kim Y. Aizenman E. Levitan ES. Novel neuroprotective K+ channel inhibitor identified by high-throughput screening in yeast. Mol Pharmacol. 2004;65:214–219. doi: 10.1124/mol.65.1.214. [DOI] [PubMed] [Google Scholar]
- 20.Liu K, et al. High-throughput screening for Kv1.3 channel blockers using an improved FLIPR-based membrane-potential assay. J Biomol Screen. 2010;15:185–195. doi: 10.1177/1087057109356209. [DOI] [PubMed] [Google Scholar]
- 21.Xiong Q. Sun H. Li M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat Chem Biol. 2007;3:287–296. doi: 10.1038/nchembio874. [DOI] [PubMed] [Google Scholar]
- 22.Tang W, et al. Development and evaluation of high throughput functional assay methods for HERG potassium channel. J Biomol Screen. 2001;6:325–331. doi: 10.1177/108705710100600506. [DOI] [PubMed] [Google Scholar]
- 23.Gill S, et al. Flux assays in high throughput screening of ion channels in drug discovery. Assay Drug Dev Technol. 2003;1:709–717. doi: 10.1089/154065803770381066. [DOI] [PubMed] [Google Scholar]
- 24.Finkel A, et al. Population patch clamp improves data consistency and success rates in the measurement of ionic currents. J Biomol Screen. 2006;11:488–496. doi: 10.1177/1087057106288050. [DOI] [PubMed] [Google Scholar]
- 25.Mathes C. QPatch: the past, present and future of automated patch clamp. Expert Opin Ther Targets. 2006;10:319–327. doi: 10.1517/14728222.10.2.319. [DOI] [PubMed] [Google Scholar]
- 26.Schroeder K. Neagle B. Trezise DJ. Worley J. Ionworks HT: a new high-throughput electrophysiology measurement platform. J Biomol Screen. 2003;8:50–64. doi: 10.1177/1087057102239667. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, et al. A benchmark study with sealchip planar patch-clamp technology. Assay Drug Dev Technol. 2003;1:675–684. doi: 10.1089/154065803770381039. [DOI] [PubMed] [Google Scholar]
- 28.Sanguinetti MC. Mitcheson JS. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol Sci. 2005;26:119–124. doi: 10.1016/j.tips.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Bridal TR. Margulis M. Wang X. Donio M. Sorota S. Comparison of human ether-a-go-go-related gene screening assays based on IonWorks Quattro and thallium flux. Assay Drug Dev Technol. 2010;8:755–765. doi: 10.1089/adt.2010.0267. [DOI] [PubMed] [Google Scholar]
- 30.Bender A. Mussa HY. Glen RC. Reiling S. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J Chem Inf Comput Sci. 2004;44:1708–1718. doi: 10.1021/ci0498719. [DOI] [PubMed] [Google Scholar]
- 31.Bender A. Glen RC. Discussion of measures of enrichment in virtual screening: comparing the information content of descriptors with increasing levels of sophistication. J Chem Inf Model. 2005;45:1369–1375. doi: 10.1021/ci0500177. [DOI] [PubMed] [Google Scholar]
- 32.Glen RC, et al. Circular fingerprints: flexible molecular descriptors with applications from physical chemistry to ADME (vol 9, pg 199, 2006) Idrugs. 2006;9:311–311. [PubMed] [Google Scholar]
- 33.Young DW, et al. Integrating high-content screening and ligand-target prediction to identify mechanism of action. Nat Chem Biol. 2008;4:59–68. doi: 10.1038/nchembio.2007.53. [DOI] [PubMed] [Google Scholar]
- 34.Hert J, et al. New methods for ligand-based virtual screening: use of data fusion and machine learning to enhance the effectiveness of similarity searching. J Chem Inf Model. 2006;46:462–470. doi: 10.1021/ci050348j. [DOI] [PubMed] [Google Scholar]
- 35.Sanguinetti MC. Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 36.Vandenberg JI. Walker BD. Campbell TJ. HERG K+ channels: friend and foe. Trends Pharmacol Sci. 2001;22:240–246. doi: 10.1016/s0165-6147(00)01662-x. [DOI] [PubMed] [Google Scholar]
- 37.Aronov AM. Goldman BB. A model for identifying HERG K+ channel blockers. Bioorg Med Chem. 2004;12:2307–2315. doi: 10.1016/j.bmc.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 38.Recanatini M. Poluzzi E. Masetti M. Cavalli A. De Ponti F. QT prolongation through hERG K(+) channel blockade: current knowledge and strategies for the early prediction during drug development. Med Res Rev. 2005;25:133–166. doi: 10.1002/med.20019. [DOI] [PubMed] [Google Scholar]
- 39.Katchman AN. Koerner J. Tosaka T. Woosley RL. Ebert SN. Comparative evaluation of HERG currents and QT intervals following challenge with suspected torsadogenic and nontorsadogenic drugs. J Pharmacol Exp Ther. 2006;316:1098–1106. doi: 10.1124/jpet.105.093393. [DOI] [PubMed] [Google Scholar]
- 40.Rezazadeh S. Hesketh JC. Fedida D. Rb+ flux through hERG channels affects the potency of channel blocking drugs: correlation with data obtained using a high-throughput Rb+ efflux assay. J Biomol Screen. 2004;9:588–597. doi: 10.1177/1087057104264798. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.