

Abstract
Aims
Acute myocardial ischaemia induces a decrease in resting membrane potential [which leads to reduction of action potential (AP) Vmax] and intracellular acidification (which closes gap junctions). Both contribute to conduction slowing. We hypothesized that ventricular expression of the skeletal muscle Na+ channel, Nav1.4 (which activates fully at low membrane potentials), or connexin32 (Cx32, which is less pH-sensitive than connexin43) would support conduction and be antiarrhythmic. We tested this hypothesis in a murine model of ischaemia and reperfusion arrhythmias.
Methods and results
Empty adenovirus (Sham) or adenoviral constructs expressing either SkM1 (gene encoding Nav1.4) or Cx32 genes were injected into the left ventricular wall. Four days later, ventricular tachycardia (VT) occurred during reperfusion following a 5 min coronary occlusion. In Nav1.4- and Cx32-expressing mice, VT incidence and duration were lower than in Sham (P < 0.05). In vitro multisite microelectrode mapping was performed in the superfused right ventricular wall. To simulate ischaemic conditions, [K+] in solution was increased to 10 mmol/L and/or pH was decreased to 6.0. Western blots revealed Cx32 and Nav1.4 expression in both ventricles. Nav1.4 APs showed higher Vmax and conduction velocity (CV) than Shams at normal and elevated [K+]. Exposure of tissue to acid solution reduced intracellular pH to 6.4. There was no difference in CV between Sham and Cx32 groups in control solution. Acid solution slowed CV in Sham (P < 0.05) but not in Cx32.
Conclusion
Nav1.4 or Cx32 expression preserved normal conduction in murine hearts and decreased the incidence of reperfusion VT.
Keywords: Reperfusion arrhythmias, Gene transfer, Na+ channel, Connexins, Acidosis
1. Introduction
Experimental studies have shown that reperfusing an ischaemic region after a brief period of coronary artery ligation frequently leads to ventricular tachyarrhythmias.1,2 A clinical example in which a sudden decrease in coronary flow persists for several minutes, after which flow is re-established, is Prinzmetal's or variant angina,3 which may represent 20–30% of patients admitted to coronary care units because of unstable angina.4 Electrocardiogram (ECG) monitoring of patients with variant angina reveals cardiac arrhythmias in ∼50% of cases.5,6 Moreover, patients with variant angina and severe ventricular arrhythmias have a mortality significantly higher than those without arrhythmias and are more likely to die suddenly.6–8 In many such patients, ventricular fibrillation during reperfusion is responsible for sudden death.9 A major issue regarding prevention/treatment is that there is no time to administer a drug or device when severe ventricular arrhythmias result from acute infarction and reperfusion. In this study, we used a murine model to test a novel gene therapy that speeds conduction as a means for preventing reperfusion arrhythmias.
Reperfusion arrhythmias accompany recovery from acute myocardial ischaemia which is associated with a gradual and progressive decrease in resting membrane potential10 and intracellular acidification11 occurring over minutes. Loss of resting membrane potential leads to reduction of action potential (AP) upstroke velocity (Vmax)10 and low pH decreases gap junctional coupling:12 both contribute to marked slowing of conduction.1,10,13 The alterations in electrophysiological properties and the heterogeneous distribution of recovery on reperfusion1,10,14 can predispose tissue to re-entrant arrhythmias.2,15 We hypothesized that introduction into ventricular tissue of the skeletal muscle Na+ channel, Nav1.4 (which activates at lower membrane potentials than the cardiac Na+ channel, Nav1.516,17), or introduction of the liver gap junctional protein, Cx32 (which is less pH-sensitive than the myocardial connexin, Cx4312,18), would normalize AP Vmax or gap junctional conductance, respectively, and, thereby, improve conduction velocity (CV) during ischaemia and reperfusion to decrease the incidence of reperfusion arrhythmias.
2. Methods
2.1. SkM1 and Cx32 adenovirus preparation
Preparation of the adenoviral construct of rat skeletal Na+ channel gene, SkM1 (kindly provided by Dr Gail Mandel) that encodes Nav1.4, has been described previously.19,20 Murine Cx32 cDNA was transferred from its original vector into a pDC516 shuttle vector. The new pDC516-Cx32 plasmid was co-transfected with a viral genomic plasmid pBHGfrt▵E1,3FLP (Microbix Biosystem) into E1-complementing HEK293 cells. After successful recombination, resulting in production of a replication-deficient adenovirus of Cx32 (AdCx32), the virus was plaque-purified and sequenced prior to further amplification. Large scale viral stock was concentrated via CsCl binding, consequently titrated with mouse anti-adenovirus hexon antibodies (Advanced ImmunoChemical, Long Beach, CA,USA), and the titre was defined as fluorescence forming units per millilitre (ffu/mL).
2.2. Gene delivery
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Heath (NIH Publication No. 85-23, revised 1996). All protocols were approved by the Columbia University Institutional Animal Care and Use Committee. Male C57B mice weighing 23–32 g were anaesthetized with sodium pentobarbital (50–70 mg/kg ip) and fixed in the supine position on a heating pad. Intubation was attained by direct laryngoscopy using the outer sheath of a 24 G intravenous catheter. The catheter was then connected to a rodent ventilator for continuous mechanical ventilation (respiratory rate 120/min with a tidal volume of 0.5 mL, Harvard Apparatus Respirator, model 707). Rectal temperature was maintained within 37.5–38.0°C. A left thoracotomy was performed via the fourth intercostal space, the heart was exposed, and 25 μL of saline containing 1.6 × 109 ffu of either empty adenovirus (Sham) or adenoviral constructs with SkM1 or Cx32 was injected into the anterior left ventricular (LV) wall using an insulin syringe with a 31 G needle. The chest and skin were then closed with 6-0 silk suture, the endotracheal tube was withdrawn, and the animal was allowed to recover.
2.3. In situ experiments
Four days after surgery, the animals were re-anaesthetized. Electrodes were placed to record the standard limb lead ECG. The hearts were exposed and 8-0 nylon threads passed around the left coronary artery using a tapered needle ∼1 mm from the tip of the left atrium. The ends of the nylon were threaded through a PE-10 tubing to make a snare,21 and the artery was occluded by tightening the snare. Myocardial ischaemia was confirmed by regional cyanosis and ST-segment elevation. Five minutes later, the artery was reperfused by releasing the ligation. We used 5min occlusion because the relationship between ischaemia duration and incidence of ventricular tachycardia (VT) upon subsequent reperfusion in the murine heart is ‘bell-shaped’ with a maximum at 5 min of ischaemia.21 Reperfusion was confirmed by a rapid colour change in the surface of the myocardium. When VT (five or more consecutive ventricular beats) was observed, its incidence, rate, and duration were analysed.
2.4. Microelectrode methods
The right ventricular (RV) wall was isolated, pinned to the bottom of a 4 mL tissue bath epicardial surface up, and superfused with Tyrode's solution (T = 35°C) at 12 mL/min. The bath was connected to ground via a 3 M KCl/Ag/AgCl junction. The preparation was continuously paced at a cycle length of 250 ms via bipolar Teflon-coated silver electrodes located at an antero-basal site. To confirm the stability of propagation, a bipolar electrogram was constantly recorded in the apical region. Multisite microelectrode mapping was performed using conventional microelectrode techniques: APs were sequentially recorded at 25–35 sites evenly covering the entire ventricular surface. Conduction time to each site was determined as the time interval between the stimulus artefact and AP upstroke. Measurement was facilitated via photography using a grid and a video camera. Distribution of conduction time over the ventricular surface was approximated by a polynomial function with precision of 0.5 ms. This function was used to plot isochrones and calculate CV at any site.
2.4.1. SkM1-treated mice
Bicarbonate-buffered Tyrode's solution was equilibrated with 95% O2/5% CO2 and contained (mmol/L): NaCl 131, NaHCO3 18, KCl 4, CaCl2 1.8, MgCl2 0.5, NaH2PO4 1.8, and dextrose 5.5 (pH 7.35 ± 0.05). After 1 h of equilibration, a control map was obtained. Then, the Tyrode's KCl concentration was increased to 10 mmol/L (to simulate ischaemia-induced depolarization). After 15 min of equilibration, multisite microelectrode mapping was repeated.
2.4.2. Cx32-treated mice
To simulate ischaemia-induced intracellular acidification, sodium acetate-containing solutions were used. Acetate, a weak acid that shuttles H+ by diffusing through the plasma membrane in its neutral H+-associated form, significantly reduces intracellular pH (pHi).22 Control HEPES-buffered Tyrode's solution contained (mmol/L): NaCl 130, KCl 4, CaCl2 1.8, MgCl2 1.0, dextrose 5.5, sodium acetate 20, and HEPES 10 (pH 7.4). For the low pH solution (pH 6.0), HEPES was replaced by MES buffer (10 mmol/L). After 1 h of equilibration in control solution and obtaining a control map, the tissue was superfused with low pH solution for 15 min and mapping was repeated. To test the effects of both factors (depolarization and acidosis), KCl concentration in low pH solution was increased to 10 mmol/L.
2.5. Measurement of pHi
The drop in pHi induced by low pH solution was measured with pH-sensitive microelectrodes in a separate group of experiments (see Supplementary materials online, Methods). Values averaged for eight RV preparations showed that switching the pH in bathing solution from 7.4 to 6.0 resulted in a drop of pHi from 7.01 ± 0.03 to 6.41 ± 0.04 (P < 0.05).
2.6. Assessment of area at risk
Surgical preparation was performed as described above, and the mouse was stabilized for 10 min. The coronary artery was then ligated as above, the aorta was clamped, and 1 mL of 1% Evans Blue dye was injected into the LV lumen to negatively mark the ischaemic zone. The heart was excised, washed in ice-cold 0.9% saline, frozen in liquid nitrogen, and cut transversely into 1 mm thick slices. The area at risk was determined via planimetry, using NIH Image software.
2.7. Western blot analysis
Western blotting was performed as described previously23 (for details, see Supplementary materials online, Methods).
2.8. Immunohistochemistry
2.9. Statistical analysis
Data are expressed as mean ± SEM. Differences in the incidence of reperfusion arrhythmias between sham and treated animals were analysed by Fisher's exact test. Continuous parameters were analysed with Student's t-test for repeated or non-repeated measures or with one- or two-way ANOVA where appropriate. P < 0.05 was considered significant.
3. Results
3.1. Assessment of area at risk and expression of Cx32 and Nav1.4
In six shams, area at risk expressed as a percentage of LV volume was 42 ± 5%. The size of area at risk in Nav1.4- and Cx32-expressing mice did not differ from Sham: 47 ± 7 and 41 ± 5%, respectively (n = 5 for each).
Following adenoviral injection into the LV free wall, the expression of Cx32 and of Nav1.4 was confirmed by western blot analysis (Figure 1A). Note that Cx32 and Nav1.4 were only observed in hearts treated with adenovirus expressing the corresponding protein, whereas those treated with empty adenoviral vector showed no expression. The overexpressed protein was detected in both ventricles, with more in the LV than in the RV. Figure 1B explores the distribution patterns of Cx32 and Nav1.4. The results show that each protein was expressed throughout the both ventricles.
Figure 1.

(A) Representative western blots and loading controls showing Cx32 and Nav1.4 (32 and 260 kDa bands, respectively) expression in adenovirus-treated hearts 4 days after adenoviral gene transfection. In each panel, bands from left to right: positive control (liver for Cx32 and skeletal muscle for Nav1.4); LV and RV of mouse injected with Cx32- or SkM1-expressing adenovirus; negative control (LV of mouse injected with empty adenovirus). (B) Confocal images showing (in red) expression of Cx32 or Nav1.4 in LV and RV tissues from a mouse injected with either Ad-Cx32 (left) or Ad-SkM1 (right). Cryosections (10 μm) were cut perpendicular to the apico-basal axis with 1.5 mm steps. LV sections from a mouse injected with empty adenovirus were used as a negative control. The liver and skeletal muscle were used as positive controls for Cx32 and Nav1.4, respectively.
3.2. Intact animal studies
3.2.1. ECG changes
Figure 2A shows three representative ECG complexes and Table 1 summarizes the baseline ECG parameters in the three groups. No significant differences in RR, PR, QT, and QTc intervals and in P-wave durations were observed among the three groups. However, QRS duration was significantly shorter in the Nav1.4 group.
Figure 2.

(A) Representative ECG complexes (lead II) obtained in Sham, Nav1.4-, and Cx32-expressing mice before occlusion of the coronary artery. Each trace begins and ends with a P-wave. (B) Examples of lead II ECG during reperfusion in one Sham, one Nav1.4-, and one Cx32-expressing mouse. Arrows indicate the release of ligation. Middle and right panels show ECG 10 and 15 s after beginning reperfusion, respectively. Two fragments of Sham ECG (before and during VT) are shown at a high sweep speed.
Table 1.
Baseline lead II ECG parameters in Sham (n = 20), Nav1.4-expressing (n = 20) and Cx32-expressing (n = 20) mice
| Group | RR (ms) | P-wave (ms) | PR (ms) | QRS (ms) | QT (ms) | QTc |
|---|---|---|---|---|---|---|
| Sham | 127.4 ± 2.8 | 13.2 ± 0.6 | 37.9 ± 0.7 | 11.2 ± 0.3 | 69.4 ± 2.5 | 61.4 ± 1.9 |
| Nav1.4 | 127.0 ± 5.0 | 12.0 ± 0.5 | 36.0 ± 0.8 | 9.2 ± 0.3* | 69.8 ± 2.0 | 62.4 ± 1.9 |
| Cx32 | 124.0 ± 4.3 | 13.5 ± 0.7 | 38.1 ± 0.7 | 12.1 ± 0.4 | 70.2 ± 2.8 | 53.0 ± 2.0 |
RR, RR interval duration; P-wave, P-wave duration; PR, PR interval duration; QRS, QRS interval duration; QT, QT interval duration; QTC, rate-corrected QT interval. In each mouse, five consecutive PQRST complexes were measured and averaged. QT intervals were corrected for heart rate using the formula, QTc = QT/(RR/100)1/2 with QT and RR measured in milliseconds.36
*P < 0.05 vs. Sham.
3.2.2. Reperfusion arrhythmia
Figure 2B (upper panel) is an example of VT induced upon reperfusion in a sham-operated mouse. Insets (shown at a high sweep speed) clearly demonstrate the atrial origin of excitation before VT and the ventricular origin during VT. VT appeared within 3–15 s after starting reperfusion. Figure 2B (middle and lower panels) shows—for representative SkM1- and Cx32-injected animals—that no VT occurred. Table 2 summarizes the data for reperfusion VT in the three groups. There was no difference in the rate of VT, but VT incidence was lower and the duration was shorter in Nav1.4- and Cx32-expressing mice than in Sham.
Table 2.
Summary of reperfusion-induced VT in Sham, Nav1.4, and Cx32 groups
| Group | n | Weight (g) | VT incidence (%) | VT rate (b.p.m.) | VT duration (s) |
|---|---|---|---|---|---|
| Sham | 20 | 26.3 ± 0.5 | 60 | 973 ± 21 | 11.7 ± 3.1 |
| Nav1.4 | 20 | 26.8 ± 0.6 | 15* | 962 ± 12 | 3.3 ± 1.7* |
| Cx32 | 20 | 27.1 ± 0.7 | 15* | 971 ± 18 | 4.2 ± 1.6* |
VT, ventricular tachycardia; VT incidence, percentage of mice with VT in each study group.
*P < 0.05 vs. Sham.
3.3. Isolated tissue studies
To gain further insight into the mechanisms by which Nav1.4 or Cx32 prevent VT induction, multisite microelectrode epicardial mapping of isolated RV was performed. We studied RV rather than LV because of the better survival in the tissue bath of the relatively thin RV preparations and the demonstrable presence of Nav 1.4 and Cx32 protein in RV tissues (Figure 1).
3.3.1. Nav1.4-expressing mice
There were two obvious AP differences between Nav1.4 and sham mice (Figure 3A and Table 3): that is, SkM1-injected mice had higher Vmax and amplitude. There were no differences in maximum diastolic potential (MDP) and AP duration between the two groups. Superfusion of preparations with Tyrode's solution containing normal (4 mmol/L) and elevated potassium (10 mmol/L) concentrations provided a wide MDP range and allowed recording of APs at membrane potentials as low as –50 mV (Figure 3B). At all membrane potentials, Vmax in Nav1.4-expressing mice was significantly greater than in Shams. Distributions of conduction times over the RV surface in one Sham and one SkM1-injected mouse at two potassium concentrations are shown in Figure 4A–D. At both potassium concentrations, CV was greater in the Nav1.4-expressing mouse. There was no difference in MDP between Sham and Nav1.4 mice at 4 mmol/L KCl, and elevation of KCl to 10 mmol/L led to significant and similar depolarization in both groups (Figure 4E). At both KCl concentrations, Vmax was higher in the Nav1.4 group (Figure 4F). At a normal potassium concentration, CV was significantly greater in preparations from Nav1.4-expressing mice than in Shams (Figure 4G). Elevation of potassium slowed CV in both groups; however, in the Nav1.4 group, it remained higher than in Shams and was similar to that in Shams at 4 mmol/L KCl.
Figure 3.

(A) Representative AP recorded from the RV free wall of one Sham and one Nav1.4-expressing mouse in Tyrode’ solution containing 4 mmol/L KCl. Note that AP in Nav1.4 mouse has higher amplitude and Vmax. (B) Dependence of Vmax on MDP. Values were collected from multisite microelectrode mapping at 4 and 10 mmol/L KCl. In each preparation, Vmax values were grouped and averaged for each 5 mV step along the abscissa. Each individual experiment was allowed to contribute one point in each step. *P < 0.05 (n = 9 for each group).
Table 3.
AP parameters in the RV epicardium from Sham, Nav1.4-, and Cx32-expressing mice superfused with solutions containing 4 mmol/L KCl at pH 7.40
| Group | n | MDP (mV) | APA (mV) | Vmax (V/s) | APD30 (ms) | APD50 (ms) | APD90 (ms) |
|---|---|---|---|---|---|---|---|
| Bicarbonate buffer | |||||||
| Sham | 9 | −74 ± 1 | 79 ± 3 | 156 ± 12 | 2.1 ± 0.2 | 3.9 ± 0.3 | 27.7 ± 2.1 |
| Nav1.4 | 9 | −75 ± 1 | 97 ± 2* | 239 ± 10* | 2.4 ± 0.1 | 4.4 ± 0.1 | 31.8 ± 2.3 |
| HEPES buffer | |||||||
| Sham | 8 | −70 ± 1 | 78 ± 2 | 148 ± 14 | 1.8 ± 0.2 | 3.0 ± 0.2 | 20.5 ± 3.0 |
| Cx32 | 8 | −71 ± 2 | 80 ± 1 | 156 ± 11 | 2.0 ± 0.1 | 3.3 ± 0.2 | 20.1 ± 2.9 |
MDP, maximum diastolic potential; APA, action potential amplitude; Vmax, maximum upstroke velocity; APD30, APD50, and APD90, action potential duration to 30, 50, and 90% of full repolarization, respectively. In each preparation, values were averaged over all recording sites. Each preparation contributed one point to the average value.
*P < 0.05 vs. Sham in the same buffer.
Figure 4.

Representative maps of conduction times in the RV epicardium of one Sham and one Nav1.4-expressing mouse at 4 mmol/L (A and B) and 10 mmol/L (C and D) KCl. The pacing site is marked with a cross and isochrones are drawn at 3 ms intervals. CV averaged over all recording sites in each preparation is shown at the bottom of each panels. (E) MDP, (F) Vmax, and (G) CV show respective mean values for Sham and Nav1.4 groups at 4 and 10 mmol/L KCl. Each preparation contributed one point to the overall average value, and for each preparation, values were averaged over all recording sites. *P < 0.05 vs. 4 mM KCl in the same group and +P < 0.05 vs. Sham at the same potassium concentration (n = 9 for both groups).
3.3.2. Cx32-expressing mice
Representative maps of conduction times in preparations from one sham and one Cx32-expressing mouse are shown in Figure 5A–D. To test whether acidification slows conduction and whether Cx32 expression supports conduction in this environment, the tissues were exposed to low pH (6.0) solution. At normal pH, CV was similar in both preparations (Figure 5A and B). Low pH led to a marked decrease in CV in the sham (Figure 5C) but not the Cx32-expressing preparation (Figure 5D). There were no differences in AP parameters between Sham and Cx32 groups at pH 7.4 (Table 3).
Figure 5.

Representative maps of conduction times in the RV epicardium of one Sham and one Cx32-expressing mouse at pH in bathing solution = 7.4 (A and B) and 6.0 (C and D). The pacing site is marked with a cross and isochrones are drawn at 3 ms intervals. CV averaged over all recording sites in each preparation is shown at the bottom of each panel. (E) MDP, (F) Vmax, and (G) CV show respective mean values for Sham and Cx32 groups at normal and low pH. Each preparation contributed one point to the overall average value, and for each preparation, values were averaged over all recording sites. *P < 0.05 vs. pH 7.4 in the same group, +P < 0.05 vs. Sham at the same pH (n = 8 for both groups).
Summary data for the effects of low pH are shown in the lower panels of Figure 5. Low pH had no effects on MDP (Figure 5E) and Vmax (Figure 5F) in both groups. Acidosis induced a significant decrease in CV in Shams and had no effect in Cx32 (Figure 5G). As a result, at low pH, CV was higher in Cx32-expressing mice in comparison to Shams.
3.3.3. Combined effects of elevated potassium and low pH
Because the ischaemia–reperfusion setting is not associated with changes in 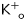 or pH alone, experiments were performed to examine the combined effects of both factors, depolarization and acidosis, in Sham and in Nav1.4- and Cx32-expressing mice. Each preparation was equilibrated with HEPES-buffered solution having normal pH and potassium, and a microelectrode map was obtained. Then, the tissue was superfused with MES-buffered low pH solution with increased potassium concentration (10 mmol/L) (low pH–high K+ solution) and mapping was repeated. The results are summarized in Figure 6. Low pH–high K+ solution produced similar depolarization in all three groups of preparations (Figure 6A). Vmax was significantly reduced in all groups but in Nav1.4, it remained significantly higher than in Sham and Cx32 (Figure 6B). In Sham, low pH–high K+ solution slowed CV by 50% (Figure 6C). CV was slowed in Nav1.4 and Cx32 mice as well, but in these groups it was significantly faster than in Sham.
or pH alone, experiments were performed to examine the combined effects of both factors, depolarization and acidosis, in Sham and in Nav1.4- and Cx32-expressing mice. Each preparation was equilibrated with HEPES-buffered solution having normal pH and potassium, and a microelectrode map was obtained. Then, the tissue was superfused with MES-buffered low pH solution with increased potassium concentration (10 mmol/L) (low pH–high K+ solution) and mapping was repeated. The results are summarized in Figure 6. Low pH–high K+ solution produced similar depolarization in all three groups of preparations (Figure 6A). Vmax was significantly reduced in all groups but in Nav1.4, it remained significantly higher than in Sham and Cx32 (Figure 6B). In Sham, low pH–high K+ solution slowed CV by 50% (Figure 6C). CV was slowed in Nav1.4 and Cx32 mice as well, but in these groups it was significantly faster than in Sham.
Figure 6.

Effects of high potassium and low pH on MDP (A), Vmax (B), and CV (C) in Sham, Nav1.4, and Cx32 groups. In each preparation, values were averaged over all recording sites. Each preparation contributed one point to average value. *P < 0.05 vs. pH 7.4 and 4 mmol/L KCl in the same group and +P < 0.05 vs. Sham at the same potassium concentration and pH (n = 6 for Sham and n = 5 for Nav1.4 and Cx32 groups).
4. Discussion
Slowing of conduction is one of the major electrophysiological alterations induced by acute myocardial ischaemia.1,10,13 A decrease in CV shortens the wavelength of excitation and can predispose the myocardium to re-entry. The theory of impulse propagation in a continuous electrical medium states that changes in ionic depolarizing current flow and changes in extracellular and intracellular resistances have independent effects on CV.24 Formally, CV is proportional to the square root of Vmax and independently, inversely proportional to the square root of (extracellular + intracellular resistances).25 Values for intracellular resistance are higher than those for extracellular resistance26 and a major contributor to intracellular resistance is that of intercellular connections (gap junctions) that form resistive obstacles during propagation.24 Therefore, we tried to decrease the extent of conduction slowing within the ischaemia/reperfusion zone by either increasing Vmax (Nav1.4 expression) or decreasing intracellular resistance (Cx32 expression) and tested whether these interventions could suppress arrhythmia.
The voltage-gated sodium channel is a key determinant of cardiac excitability. The magnitude of the sodium current determines the upstroke velocity and, in conjunction with electrical coupling among cells, the CV of the electrical impulse. We selected the skeletal muscle sodium channel gene, SkM1, as a therapeutic intervention. The steady-state inactivation curve for the SkM1-encoded channel (Nav1.4) is at voltages about 20 mV positive to that of the cardiac sodium channel Nav1.5.16,17 Thus, overexpressing the Nav1.4 channel in the myocardial tissue can not only increase the number of Na+ channels in the membrane, but also make more of them available for opening in a depolarized tissue. This can improve conduction through ischaemia-induced depolarized regions and might be antiarrhythmic.
We have recently demonstrated in experiments using rat ventricular myocyte cultures that expression of Nav1.4 but not Nav1.5 Na+ channels preserves AP Vmax and CV in a depolarized syncytium.19 The results presented in the present study demonstrate that expressing Nav1.4 in the mouse heart exerts antiarrhythmic effects on reperfusion arrhythmias following brief ischaemic episodes. Our electrophysiological data are consistent with the suggested mechanism of this protective action.19 We found that even in the RV wall of mice injected with SkM1 (where Nav1.4 was expressed less than in LV), AP Vmax was significantly greater than in Shams. The high Vmax resulted in greater CV in the preparations from Nav1.4-expressing mice. Increased CV was reflected on the surface ECG as an abbreviated QRS duration in SkM1-injected mice.
Intracellular potassium loss and extracellular potassium accumulation (up to 10 mmol/L within 5 min after the onset of coronary artery occlusion27) are early consequences of myocardial ischaemia. Our in vitro experiments simulated this condition via the increase in external potassium concentration from 4 to 10 mmol/L. Despite a similar degree of elevated potassium-induced depolarization, CV in Nav1.4-expressing tissue was significantly greater than in Shams and was similar to that in sham animals at a normal potassium concentration. These results suggest the property of Nav1.4 channels to support conduction in a depolarized myocardium as a mechanism for prevention of reperfusion-induced VT.
The resistivity of Cx43 gap junctions is a major determinant of intracellular resistance in the ventricular myocardium. Cx43 is highly pH-sensitive and closes at low pH.18 The liver gap junctional protein, Cx32, is less sensitive to acidification-induced uncoupling than Cx4318,28 and therefore can support conduction in the ischaemic myocardium and might be antiarrhythmic. Our results confirm this suggestion: similar to Nav1.4, expression of Cx32 in mouse heart protects against reperfusion arrhythmias.
Acute ischaemia induces a rapid and prominent decrease in pHi (to about 6.4 in 5 min after the onset of ischaemia11). Our isolated tissue experiments simulated these conditions by superfusion with a low pH solution. Measurements with pH-sensitive electrodes showed that superfusion with Na-acetate containing pH 6.0 solution led to a drop of pHi to about 6.4. At these pH values, most Cx43 gap junctions should be closed, whereas significant numbers of Cx32 junctions should stay open.18,28
Simulations and experimental studies have demonstrated a complex non-linear relationship between cell–cell coupling and CV: a 95% decrease in coupling induces about a 50% decrease in CV.29 In accordance with these data, we found that CV decreased by about 40% at low pH in sham preparations, whereas no changes in CV were seen in Cx32-expressing tissue. Because low pH had no effects on MDP or Vmax, the results suggest the effectiveness of Cx32 gap junctions to support conduction in the acidic myocardium as a mechanism for protection against reperfusion-induced VT. In sum, under conditions simulating two major ischaemia-induced deteriorations (acidosis and high potassium) in both treated groups (Nav1.4 and Cx32), CV was faster than in shams. Our logic in using a simulated ischaemic superfusate was that prevention of conduction slowing during ischaemia would decrease heterogeneity of conduction during reperfusion and forestall arrhythmia.
The mechanism of antiarrhythmic action of Nav1.4 and Cx32 overexpression presented above is based on the assumption that circus movement re-entry is a key factor in the genesis of reperfusion arrhythmias. Indeed, it has been demonstrated that even normal mouse ventricles (without regional ischaemia) can maintain sustained re-entrant activity.30 However, the precise electrophysiological mechanisms underlying the genesis of reperfusion arrhythmias (especially in the mouse heart) remain unclear. Coronel et al.31 studied the pig and found a marked AP duration shortening in the reperfused tissue that could contribute to reperfusion arrhythmias caused by re-entry. However, in the murine model, the VT rate approached 1000 b.p.m., resulting in a cycle length of ∼60 ms which is much longer than the AP duration measured in the RV under conditions of simulated ischaemia. Moreover, in the Coronel et al. study in pigs, AP shortening was seen only after 10 min (but not 5 min) of coronary occlusion and the shortest duration occurred 1 min after the onset of reperfusion. In our experiments, the duration of ischaemia was 5 min and VT developed within 15 s after releasing the ligation. In sum, this information indicates that AP shortening did not contribute substantially to reperfusion VT in our study.
Although re-entry is a major arrhythmogenic mechanism, enhanced automaticity and delayed afterdepolarization-induced triggered activity are alternative mechanisms that can induce re-entry or induce sustained arrhythmias in their own right.2 The development of delayed afterdepolarizations implies increased calcium uptake/release during reperfusion. However, there is little evidence for reperfusion-induced calcium overload after short periods of ischaemia when the tissue is only moderately injured and capable of full recovery.32 In any case, an increase in Na+ current would not be expected to inhibit automaticity or triggered activity. Also, expression of Cx32 is not expected to affect automaticity or triggered activity. At the same time, our data demonstrate that Nav1.4 and Cx32 both sped conduction as they reduced the incidence of arrhythmias. These results are consistent with re-entry as a mechanism for reperfusion VT in our experiments.
The model used in the present study is relevant to situations in which reperfusion after seconds or minutes of ischaemia is associated with the release of coronary artery spasm. Our results demonstrate that expressing either Nav1.4 or Cx32 proteins in the ventricular myocardium can protect against reperfusion arrhythmias after a short-lasting coronary artery spasm. Importantly, we have shown recently that expression of Nav1.4 reduces the incidence of inducible ventricular tachyarrhythmia/fibrillation in 1-week-old canine infarcts.20 Thus, Nav1.4 expression can be beneficial over a range of intervals of coronary occlusion. In contrast, expression of Cx32 might be detrimental following long-lasting occlusion because the reduction of intercellular coupling by decreasing Cx43 junction conductance is a response of the myocardium that can limit the damage caused by coronary occlusion. Indeed, myocardial expression of Cx32 (in the presence of Cx43) does not influence infarct size during short-lasting ischaemia33 but increases infarct size 24 h after coronary ligation in mice34 and promotes monomorphic VT 1 week after coronary ligation in dogs.35 Therefore, although our study demonstrates that myocardial overexpression of non-native channels having biophysical properties different from those of the normal heart may be beneficial, any benefit will vary with the nature of the channel and the nature and duration of the injury. Hence, in considering the gene therapy of arrhythmias, the selectivity and specificity of any intervention in any setting become key factors in assessing risk/benefit ratio, not unlike the situation we encounter in evaluating drugs.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by USPHS-NHLBI grant HL-094410.
Supplementary Material
Acknowledgements
The authors express their gratitude to Eugene Bobkov for his excellent technical assistance.
Conflict of interest: none declared.
References
- 1.Murdock DK, Loeb JM, Euler DE, Randall WC. Electrophysiology of coronary reperfusion: a mechanism for reperfusion arrhythmias. Circulation. 1980;61:175–182. doi: 10.1161/01.cir.61.1.175. [DOI] [PubMed] [Google Scholar]
- 2.Manning AS, Hearse DJ. Reperfusion-induced arrhythmias: mechanisms and prevention. J Mol Cell Cardiol. 1984;16:497–518. doi: 10.1016/s0022-2828(84)80638-0. [DOI] [PubMed] [Google Scholar]
- 3.Prinzmetal M, Kennamer R, Merliss R, Wada T, Naci B. Angina pectoris. I. A variant form of angina pectoris. Am J Med. 1959;27:375–388. doi: 10.1016/0002-9343(59)90003-8. [DOI] [PubMed] [Google Scholar]
- 4.Unstable angina pectoris: national co-operative study group to compare surgical and medical therapy. Results in patients with S-T segment elevation during pain. Am J Cardiol. 1980;45:819–823. doi: 10.1016/0002-9149(80)90127-7. [DOI] [PubMed] [Google Scholar]
- 5.Keller KB, Lemberg L. Prinzmetal's angina. Am J Crit Care. 2004;13:350–354. [PubMed] [Google Scholar]
- 6.Miller DD, Waters DD, Szlachcic J, Theroux P. Clinical characteristics associated with sudden death in patients with variant angina. Circulation. 1982;66:588–592. doi: 10.1161/01.cir.66.3.588. [DOI] [PubMed] [Google Scholar]
- 7.Myerburg RJ, Kessler KM, Mallon SM, Cox MM, de Marchena E, Interian A, Jr, et al. Life-threatening ventricular arrhythmias in patients with silent myocardial ischemia due to coronary-artery spasm. N Engl J Med. 1992;326:1451–1455. doi: 10.1056/NEJM199205283262202. [DOI] [PubMed] [Google Scholar]
- 8.MacAlpin RN. Cardiac arrest and sudden unexpected death in variant angina: complications of coronary spasm that can occur in the absence of severe organic coronary stenosis. Am Heart J. 1993;125:1011–1017. doi: 10.1016/0002-8703(93)90108-l. [DOI] [PubMed] [Google Scholar]
- 9.Tzivoni D, Keren A, Granot H, Gottlieb S, Benhorin J, Stern S. Ventricular fibrillation caused by myocardial reperfusion in Prinzmetal's angina. Am Heart J. 1983;105:323–325. doi: 10.1016/0002-8703(83)90534-3. [DOI] [PubMed] [Google Scholar]
- 10.Downar E, Janse MJ, Durrer D. The effect of acute coronary artery occlusion on subepicardial transmembrane potentials in the intact porcine heart. Circulation. 1977;56:217–224. doi: 10.1161/01.cir.56.2.217. [DOI] [PubMed] [Google Scholar]
- 11.Garlick PB, Radda GK, Seeley PJ. Studies of acidosis in the ischaemic heart by phosphorus nuclear magnetic resonance. Biochem J. 1979;184:547–554. doi: 10.1042/bj1840547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stergiopoulos K, Alvardo JL, Mastroianni M, Ek-Vitorin JF, Taffet SM, Delmar M. Hetero-domain interactions as a mechanism for the regulation of connexin channels. Circ Res. 1999;84:1144–1155. doi: 10.1161/01.res.84.10.1144. [DOI] [PubMed] [Google Scholar]
- 13.Penkoske PA, Sobel BE, Corr PB. Disparate electrophysiological alterations accompanying dysrhythmia due to coronary occlusion and reperfusion in the cat. Circulation. 1978;58:1023–1035. doi: 10.1161/01.cir.58.6.1023. [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto T, Peter T, Hamamoto H, Mandel WJ. Electrophysiologic observations on ventricular tachyarrhythmias following reperfusion. Am Heart J. 1983;105:201–209. doi: 10.1016/0002-8703(83)90514-8. [DOI] [PubMed] [Google Scholar]
- 15.Corr PB, Witkowski FX. Potential electrophysiologic mechanisms responsible for dysrhythmias associated with reperfusion of ischemic myocardium. Circulation. 1983;68(Suppl. I):I-16–I-24. [PubMed] [Google Scholar]
- 16.Chahine M, Deschene L, Chen LQ, Kallen RG. Electrophysiological characteristics of cloned skeletal and cardiac muscle sodium channels. Am J Physiol. 1996;271:H498–H506. doi: 10.1152/ajpheart.1996.271.2.H498. [DOI] [PubMed] [Google Scholar]
- 17.Wang DW, George AL, Bennett PB. Comparison of heterologously expressed human cardiac and skeletal muscle sodium channels. Biophys J. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu S, Taffet S, Stoner L, Delmar M, Vallano ML, Jalife J. A structural basis for the unequal sensitivity of major cardiac and liver gap junctions to intracellular acidification: the carboxyl tail length. Biophys J. 1993;64:1422–1433. doi: 10.1016/S0006-3495(93)81508-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Protas L, Dun W, Jia Z, Lu J, Bucchi A, Kumari S, et al. Expression of skeletal but not cardiac Na+ channel isoform preserves normal conduction in a depolarized cardiac syncytium. Cardiovasc Res. 2009;81:528–535. doi: 10.1093/cvr/cvn290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lau DH, Clausen C, Sosunov EA, Shlapakova IN, Anyukhovsky EP, Danilo P, Jr, et al. Epicardial border zone overexpression of skeletal muscle sodium channel SkM1 normalizes activation, preserves conduction, and suppresses ventricular arrhythmia. An in silico, in vivo, in vitro study. Circulation. 2009;119:19–27. doi: 10.1161/CIRCULATIONAHA.108.809301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakamoto J, Miura T, Tsushida A, Fukuma T, Hasegawa T, Shimamoto K. Reperfusion arrhythmias in the murine heart: their characteristics and alteration after ischemic preconditioning. Basic Res Cardiol. 1999;94:489–495. doi: 10.1007/s003950050165. [DOI] [PubMed] [Google Scholar]
- 22.Lewandowski R, Procida K, Vaidyanathan R, Coombs W, Jalife J, Nielsen MS, et al. RXP-E—a connexin43-binding peptide that prevents action potential propagation block. Circ Res. 2008;103:519–526. doi: 10.1161/CIRCRESAHA.108.179069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozgen N, Guo J, Gertsberg Z, Danilo P, Jr, Rosen MR, Steinberg SF. Reactive oxygen species decrease cAMP response element binding protein expression in cardiomyocytes via a protein kinase D1-dependent mechanism that does not require Ser133 phosphorylation. Mol Pharmacol. 2009;76:896–902. doi: 10.1124/mol.109.056473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleber AG, Janse MJ, Fast VG. Normal and abnormal conduction in the heart. In: Page E, Fozzard HA, Solaro RJ, editors. Handbook of Physiology. Section 2 the Cardiovascular System. Vol. 1. New York, NY; Oxford University Press; 2001. pp. 455–530. The Heart. [Google Scholar]
- 25.Walton MK, Fozzard HA. The conducted action potential: models and comparison to experiments. Biophys J. 1983;44:9–26. doi: 10.1016/S0006-3495(83)84273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleischhauer J, Lehmann L, Kleber AG. Electrical resistances of interstitial and microvascular space as determinants of the extracellular electrical field and velocity of propagation in ventricular myocardium. Circulation. 1995;92:587–594. doi: 10.1161/01.cir.92.3.587. [DOI] [PubMed] [Google Scholar]
- 27.Hill JL, Gettes LS. Effect of acute coronary artery occlusion on local myocardial extracellular K+ activity in swine. Circulation. 1980;61:768–778. doi: 10.1161/01.cir.61.4.768. [DOI] [PubMed] [Google Scholar]
- 28.Morley GE, Taffet SM, Delmar M. Intramolecular interactions mediate pH regulation of connexin43 channels. Biophys J. 1996;70:1294–1302. doi: 10.1016/S0006-3495(96)79686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas SP, Kucera JP, Bircher-Lehman L, Rudy Y, Saffitz JE, Kleber AG. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res. 2003;92:1209–1216. doi: 10.1161/01.RES.0000074916.41221.EA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaidya D, Morley GE, Samie FH, Jalife J. Reentry and fibrillation in the mouse heart: a challenge to the critical mass hypothesis. Circ Res. 1999;85:174–181. doi: 10.1161/01.res.85.2.174. [DOI] [PubMed] [Google Scholar]
- 31.Coronel R, Wilms-Schopman FJG, Opthof T, Cinca J, Fiolet JWT, Janse MJ. Reperfusion arrhythmias in isolated pefused pig hearts. Inhomogeneities in extracellular potassium, ST and QT potentials, and transmembrane action potentials . Circ Res. 1992;71:1131–1142. doi: 10.1161/01.res.71.5.1131. [DOI] [PubMed] [Google Scholar]
- 32.Shen AC, Jennings RB. Kinetics of calcium accumulation in acute myocardial injury. Am J Pathol. 1972;67:441–452. [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez-Sinovas A, Sanchez JA, Gonzalez-Loyola A, Barba I, Morente M, Augilar R, et al. Effects of substitution of Cx43 by Cx32 on myocardial energy metabolism, tolerance to ischemia and preconditioning protection. J Physiol. 2010;588:1139–1151. doi: 10.1113/jphysiol.2009.186577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prestia K, Kelly CW, Sosunov EA, Anyukhovsky EP, Brink PR, Rosen MR, et al. Connexin 32 influences infarct size following left anterior descending artery ligation in mice. Heart Rhythm. 2009;6(5S):S417. Abstract. [Google Scholar]
- 35.Lau DH, Shlapakova I, Sosunov E, Anyukhovsky E, Danilo P, Rosen T, et al. Overexpression of Cx32, a pH insensitive gap junction subunit, in the canine epicardial border zone promotes monomorphic ventricular tachycardia. Heart Rhythm. 2009;6(5S):S458. Abstract. [Google Scholar]
- 36.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mice. Am J Physiol. 1998;274:H747–H751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.