

Abstract
We tested the hypothesis that primary afferent inputs play a role in astroglial hyperactivity after tissue injury. We first injected complete Freund’s adjuvant (CFA, 0.05 ml, 1:1 oil/saline) into the masseter muscle, which upregulated glial fibrillary acidic protein (GFAP), a marker of astrocytes, interleukin (IL)-1β an inflammatory cytokine, and phosphorylation of serine896 of the NR1 subunit (P-NR1) of the NMDA receptor in the subnuclei interpolaris/caudalis (Vi/Vc) transition zone, an important structure for processing trigeminal nociceptive input. Local anesthetic block with lidocaine (2%) of the masseter muscle at 10 min prior to injection of CFA into the same site significantly reduced the CFA-induced increase in GFAP, IL-1β and P-NR1 (p<0.05, n=4/group). We then tested the effect of peripheral electrical stimulation (ES). The ES protocol was burst stimulation consisting of trains of 4 square pulses (10–100 Hz, 0.1–3 mA, 0.5 ms pulse width). Under pentobarbital anesthesia, an ES was delivered every 0.2 s for a total of 30 min. The Vi/Vc tissues were processed for immunohistochemistry or western blot analysis at 10–120 min after ES. Compared to naive and SHAM-treated rats, there was increased immunoreactivity against GFAP, IL-1β and P-NR1 in the Vi/Vc in rats receiving ES. Double staining showed that IL-1β was selectively localized in GFAP-positive astroglia, and P-NR1-immunoreactivity was localized to neurons. These findings indicate that primary afferent inputs are necessary and sufficient to induce astroglial hyperactivity and upregulation of IL-1β, as well as neuronal NMDA receptor phosphorylation.
Keywords: Electrical stimulation, masseter nerve, astroglia, GFAP, IL-1β, neuron-glial interaction
INTRODUCTION
Recent studies have pointed out an important role of enhanced glial activity in persistent pain conditions. Evidence suggests that central nervous system (CNS) responses to injury involve neuron-to-glia signaling [1]. Functional neurotransmitter receptors are identified in glial cells [2–5] and there may exist signaling between axons and glial cells [6–8]. Interestingly, the increase in the expression of glial fibrillary acidic protein (GFAP), an astrocytic marker, after nerve injury may depend on N-methyl-D-aspartate (NMDA) receptor activity [9]. Zhuang et al. [10] show that ERK is sequentially activated in neurons, microglia, and astrocytes after spinal nerve ligation, suggesting a role of neuronal activity in triggering glial hyperactivity.
A few studies have tested the hypothesis that neuronal input generated at the site of injury contributes to CNS glial activation or hyperactivity. Local anesthetic block of peripheral nerves prevented injury-induced central glial hyperactivity [11–14]. However, a pitfall of this approach is that local anesthetics may act on Schwann cells [15], which may modify neuron-glial interactions. An alternative approach is to directly stimulate peripheral nerves. In fact, direct electrical stimulation (ES) of peripheral nerves induced glial hyperactivity in the spinal cord or related CNS structures [16–18].
Our previous studies show that masseter muscle inflammation in rats induced astroglial hyperactivity related to cytokine release and the activation of NMDA receptors in the trigeminal subnuclei interpolaris/caudalis (Vi/Vc) transition zone [13], a subregion of the spinal trigeminal nucleus involved in processing of orofacial nociceptive input particularly from muscle. To directly test the hypothesis that primary afferent input plays a role in injury-induced astroglial hyperactivity, cytokine release and NMDA receptor activation, the present study was undertaken to investigate the effect of electrical stimulation of the masseter nerve that provides afferent input to the spinal trigeminal nucleus including the transition zone [19]. We show that electrical stimulation of the masseter nerve afferent fibers induces time-dependent neuronal NMDA receptor phosphorylation, astroglial hyperactivity and upregulation of IL-1β associated with astroglia in the trigeminal transition zone, supporting a role of primary afferent input in triggering trigeminal glial activity after orofacial injury.
METHODS
Animals
Male Sprague-Dawley rats were used (200–300 g, Harlan, Indianapolis). To produce orofacial deep tissue inflammation, the rats received complete Freund adjuvant (CFA, 0.05 ml, 1:1 oil/saline suspension, Sigma) into the masseter muscle under isoflurane (2–3%) anesthesia [13]. Saline was used as a vehicle control. The CFA-injected rats show normal behavior and levels of activity and the effect of inflammation on normal behavior of the animal was minimal [20–22]. Local anesthetic block was produced by infiltration of lidocaine (lido, 2%, 0.05 ml) into the masseter at 10 min before CFA under isoflurane (2–3%). The effect of lidocaine typically lasts for about 1 h. Saline was injected as a control for the local anesthetic. The experiments were approved by the Institutional Animal Care and Use Committee of the University of Maryland Dental School.
Electrical Stimulation [23]
Under pentobarbital sodium anesthesia (40 mg/kg, i.p.), the masseteric nerve was exposed after retracting the temporalis muscle at the mandibular notch [24]. The nerve was suspended on a pair of silver hook electrodes. A segment of the nerve a few mm distal to the stimulating electrodes was either crushed or anesthetized with lidocaine to block efferent impulses. The electrical stimulation (ES) protocol was burst stimulation (BS) consisting of trains of 4 square pulses at 10–100 Hz (0.1–3 mA, 0.1–0.5 ms pulse width) and one train was delivered every 0.2 s for a total of 30 min by an A300 Pulsemaster (WPI) with the steady direct current output via a stimulus isolator (A360D). The rat was perfused with 4% paraformaldehyde and the Vi/Vc tissues processed for immunohistochemistry. Fresh tissues were collected for Western blotting.
Western Blot
The rats were anesthetized with isoflurane and decapitated. A block of caudal brainstem tissues was cut. The tissue block included a segment approximately 2 mm in length at the level of the obex. The full length of the Vi/Vc transition zone is enclosed in this block. The tissue block was then rotated and the ventral portions of the transition zone were harvested with punch biopsies. The tissues were homogenized in solubilization buffer (50 mM Tris. HCl, pH8.0; 150 mM NaCl, 1 mM EDTA, 1% NP40, 0.5% deoxycholic acid, 0.1% SDS, 1 mM Na3VO4, 1 U/ml aprotinin, 20 μg/ml leupetin, 20 μg/ml pepstatin A). The homogenate was centrifuged at 20,200 X g for 10 min at 4°C. The supernatant was removed. The protein concentration was determined using a detergent-compatible protein assay with a bovin serum albumin standard. Each sample contained proteins from one animal. The proteins (50 μg) were separated on a 7.5% SDS-PAGE gel and blotted to nitrocellulose membrane (Amersham). The blots were blocked with 5% milk in TBS buffer and then incubated with the respective antibody. The membrane was washed with TBS and incubated with anti-goat or mouse IgG (1:3000, Santa Cruz). The immunoreactivity was detected using Enhanced Chemiluminescence (ECL, Amersham). The loading and blotting of amount of proteins were verified by reprobing the membrane with anti-β-actin antiserum and with Coomassie blue staining.
Immunohistochemistry
Rats were deeply anesthetized with pentobarbital sodium (60–100 mg/kg, i.p.) and perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer at pH 7.4. The brain stem-upper cervical spinal cord was removed, postfixed, and transferred to 25% sucrose (w/v) for cryoprotection. Free-floating sections from the caudal brain stem including the trigeminal transition zone and caudal Vc-upper cervical spinal cord were incubated with relevant antibodies with 1–3% relevant normal sera. After washes in PBS, the sections were incubated with biotinylated secondary IgG (Vector, 1:400) and then with avidin and biotinylated horseradish peroxidase complex (Vector, 1:100). Tissue sections were finally reacted with 0.05% diaminobenzidine dihydrochloride (DAB, Sigma) in 0.1 M phosphate buffer containing 0.003% hydrogen peroxide. Control sections were processed by the same method except that the primary antisera were omitted or adsorbed by respective antigens. Double-labeling immunofluorescence was performed with Cy2 and Cy3 (1:500, Jackson ImmunoResearch, West Grove, PA) or Alexa Fluor 488 (1:500, Invitrogen Molecular Probes, Eugene, OR) and Cy3 after incubation with respective primary antibodies. Fos-like immunoreactivity (LI) was detected with polyclonal rabbit anti-Fos antibody (1:20,000) by the avidin-biotin method (Vectastain ABC Kit, Vector) followed by diaminobenzidine tetrahydrochloride (DAB, Sigma) with nickel intensification (DAB substrate Kit DAB-Ni, Vector).
Antibodies
All antibodies were purchased from commercial sources: Chemicon, mouse anti-GFAP, mouse anti-NeuN; EMD-Oncogene, rabbit anti-Fos (c-fos ab-5); Endogen, rat anti-IL-1β; Serotec, mouse anti-CD11b (OX-42); Sigma, mouse anti-β-actin; and Upstate, rabbit anti-NR1, rabbit anti-Phospho-ser896 NR1.
Data Analysis
Data are presented as mean ± S.E.M. For western blot analysis, the ECL-exposed films were digitized and densitometric quantification of immunoreactive bands was carried out using U-SCAN-IT gel (ver. 4.3, Silk Scientific Corp). The relative protein levels were obtained by comparing the respective specific band to the β-actin control from the same membrane. The deduced ratios were further normalized to that of the control rats on the same membrane and illustrated as percentage of controls. Raw data (e.g., ratio of the GFAP band over β-actin band) were used for statistical comparisons (ANOVA with Fisher’s PLSD test for post-hoc analysis). P < 0.05 was considered significant in all cases.
RESULTS
Effects of Local Anesthesia on CFA-Induced Increases in GFAP, IL-1β and NMDA Receptor Phosphorylation
Consistent with our previous report [13], injection of CFA into the masseter muscle induced significant upregulation of GFAP, IL-1β and phosphorylation of the NMDA receptor NR1 subunit at ser896 (P-NR1), as shown by immunofluorescence staining (Fig. 1A, B, D, E, G, H, CFA vs. NAÏVE) and Western blot analysis (Fig. 1J-L, saline vs. naïve, p < 0.05, n = 3–4/group). Our previous studies have shown that these biochemical changes correlated with inflammation of the masseter muscle and the time course of orofacial hyperalgesia [13, 19, 25].
Fig. 1.

Effects of local anesthesia on CFA-induced increases in GFAP, IL-1β and NMDA receptor phosphorylation. Lidocaine (lido, 0.05 ml, 2%) was infiltrated into the masseter muscle at 10 min prior to the injection of CFA into the same site and the Vi/Vc tissues were removed at 30 min after CFA. Saline was injected as a control. A–I: Immunostaining. J–L: Western blot (n=4/group). β-actin was used as a loading control. *, p<0.05, vs. naïve. Compared to saline-injected rats, the increase in GFAP, a marker of astrocyte, IL-1β, a prototypic inflammatory cytokine, and P-NR1 (phosphorylated NR1 at ser896) was blocked by lidocaine treatment, suggesting that the upregulation was dependent on the nerve input from the injured site.
To verify the involvement of primary afferent input in neurochemical changes in the trigeminal transition zone after masseter inflammation, we produced local anesthetic block in rats receiving CFA injection into the masseter muscle. A local anesthetic, lidocaine (2%, 0.05 ml), was infiltrated into the masseter muscle at 10 min prior to the injection of CFA into the same site. The Vi/Vc tissues were punched out at 30 min after CFA. Compared to rats pretreated with saline (0.9%, 0.05 ml), the CFA-induced increase in GFAP was blocked in lidocaine-treated rats, (p < 0.05, n=4) (Fig. 1C, J). The inflammation-induced increases in IL-1β and P-NR1 were also blocked (p < 0.05, n = 3–4) (Fig. 1F, K and I, L). The effect of lidocaine was unlikely mediated through a systemic effect since the same amount of local anesthetic applied to the site neighboring the site of injury was ineffective against nociception [26]. These results are consistent with our previous report that local anesthetic block of the masseter nerve remote from the site of inflammation blocked inflammation-induced upregulation of GFAP and IL-1β [13] and suggest that primary afferent inputs associated with peripheral nociceptor activation after inflammation are necessary for the hyperactivity of astroglia, upregulation of IL-1β and increased NMDA receptor phosphorylation.
Peripheral Electrical Stimulation Induces an Increase in GFAP Expression
Although the results of local anesthetic block experiments support the contribution of primary afferent input to central neuronal and glial hyperactivity, local anesthetics may produce temporary injury to Schwann cells and their neurotoxic properties may account in some part for the effect of local anesthetics [15]. Thus, we directly tested the dependence of glial hyperactivity on primary afferent input by ES of the masseter nerve. A masseteric nerve was exposed and the BS protocol was used to deliver ES. Efferent impulses to the muscle were blocked by crushing or anesthetizing a distal segment of the nerve.
We first verified that our ES protocol activated trigeminal neurons as shown by Fos protein expression, a marker of neuronal activation. Naive and SHAM rats were used as controls. Burst stimulation consisting of trains of 4 square pulses at 100 Hz (0.1–3 mA, 0.5 ms pulse width) and one train was delivered every 0.2 s for a total of 30 min. The animals were perfused at 1 h after the cessation of ES. In the caudal Vc-upper cervical dorsal horn (medullary dorsal horn), intense Fos protein expression was induced in the medial portion of the superficial dorsal horn ipsilateral to ES (Fig. 2). ES at the 3-mA intensity was most effective. The ES-induced Fos expression in the medullary dorsal horn was predominantly ipsilateral, although a few Fos-positive cells were seen on the contralateral side (Fig. 2B). In the Vi/Vc transition zone, Fos expression was observed, particularly in the ventral portion (Fig. 3). Fos-labeled cells were also found in bilateral nucleus tractus solitarius (NTS), lateral reticular nucleus (LRN) and inferior olivary nucleus (IO). The pattern of trigeminal Fos expression following ES of the masseter nerve is consistent with previous similar studies [20], (also see [27]).
Fig. 2.
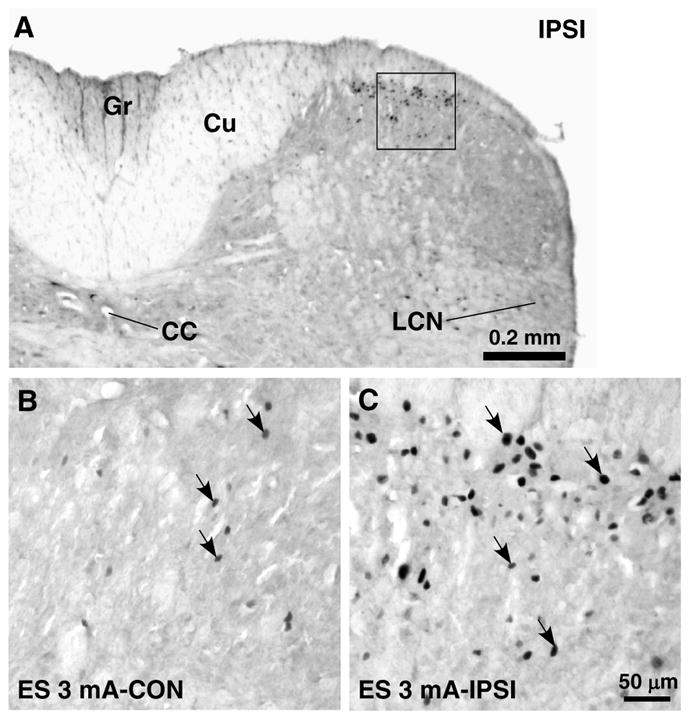
Effect of ES on Fos protein expression in the upper cervical spinal cord. Burst stimulation consisting of trains of 4 square pulses at 100 Hz (3 mA, 0.5 ms pulse width) and one train was delivered every 0.2 s for a total of 30 min. The same stimulation parameters were used in Figs. 3–6. A. Low power spinal cord section at the C1 level. Fos protein expression was induced in the medial portion of the superficial dorsal horn ipsilateral (IPSI) to ES of the masseter nerve. CC, central canal; Cu, cuneate fasciculus; Gr, gracile fasciculus; LCN, lateral cervical nucleus. B–C. Fos protein expression in the areas equivalent to that shown in the square in A under different conditions as indicated in each panel. Panel C was directly enlarged from the square in A. Arrows indicate examples of Fos-labeled nuclei.
Fig. 3.

Effect of ES on Fos protein expression in the Vi/Vc transition zone. A. Low power brainstem section ipsilateral to ES at the level approximately 14.0 mm posterior to Bregma. Fos protein expression was induced in the dorsal and ventral portions of the Vi/Vc transition zone. Nucleus tractus solitarius (NTS), lateral reticular nucleus (LRN) and inferior olivary nucleus (IO) also exhibited ES-induced Fos protein expression. Py, pyramidal tract; Sp5, spinal trigeminal tract. B–C. Fos protein expression in the areas equivalent to that shown in the square in A. Panel C was directly enlarged from the square in A. Arrows indicate examples of Fos-labeled nuclei.
We then examined the effect of ES on astroglial markers in the trigeminal transition zone (Fig. 4A). Compared to naive rats (Fig. 4B) and SHAM-operated rats (Fig. 4C) without ES, ES (3 mA, 0.5 ms, 100 Hz) of the masseter nerve afferents produced an apparent increase in GFAP-LI in the Vi/Vc transition zone at 30 min after ES (Fig. (4D)). The effect of ES on GFAP expression was quantified with western blot (Fig. 4E). Normalized with the β-actin loading control and compared to naive rats (n=4), there were significant increases in the relative levels of GFAP at 30 and 60 min after ES (p<0.05, n=4). Stimulation with lower pulse intensity and frequency (0.1 mA, 0.1 ms, 10 Hz) did not produce an effect on GFAP expression.
Fig. 4.

Increased GFAP immunoreactivity in the ventral Vi/Vc after ES of the masseter nerve. A. Low power brain stem section at the level approximately 14.0 mm posterior to Bregma. Dashed circle indicates the area analyzed, which lies in the ventral portion of the Vi/Vc transition zone. Compared to naïve (B) and SHAM-operated (C) rats, ES (3 mA for 30 min) induced an increased GFAP staining (D). Green immunostaining shows Fos-labeled nuclei, a marker of neuronal activation. N=4/group. Abbreviations: cc, central canal; IPSI, ipsilateral; NTS, nucleus tractus solitarius; Py, pyramidal tract; Vc, subnucleus caudalis; Vi, subnucleus interpolaris. E. Western blot illustrating the effect of ES of the masseter nerve on GFAP expression in the ventral Vi/Vc transition zone. β-actin was used as a loading control. A representative blot is shown on top. The relative protein levels are shown in the bottom histogram. *, p<0.05 vs. NAIVE. N=3–4 for each time point. Error bars represent S.E.M.
Increased IL-1β Immunoreactivity After Electrical Stimulation
We have shown previously that masseter muscle inflammation induced an increase in the level of inflammatory cytokine IL-1β, associated with increased astroglial activity in the trigeminal transition zone [13]. We examined the effect of ES of the masseter nerve on the IL-1β in the Vi/Vc. Western blot showed ES-induced increase in IL-1β (Fig. 5). There were significant increases in IL-1β protein levels at 30 and 60 min after ES (3 mA, 0.5 ms, 100 Hz) of the masseter nerve (p<0.05, n=4/group).
Fig. 5.
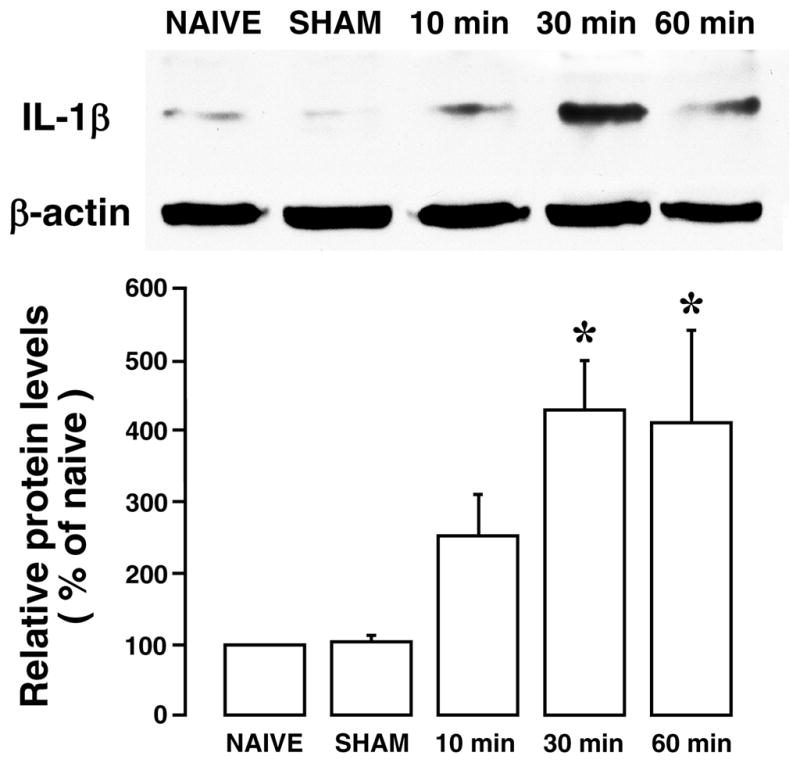
Western blot illustrating the effect of ES of the masseter nerve on IL-1β expression in the ventral Vi/Vc transition zone. β-actin was used as a loading control. A representative blot is shown on top. The relative protein levels are shown in the bottom histogram. *, p<0.05 vs. NAIVE. N=3–4 for each time point. Error bars represent S.E.M.
Brainstem tissue sections were processed for double immunofluorescence labeling after ES (Fig. 6). IL-1β immunoreactivity was found to co-localize with GFAP (Fig. 6A–C), but not with CD11b (Fig. 6D), a marker of microglia, or NeuN (Fig. 6E), a neuronal marker. These results are consistent with findings that after masseter inflammation [13] and suggest that astroglia are a source of IL-1β release in response to peripheral stimulation.
Fig. 6.

Brainstem tissue sections were processed for immuno-fluorescence labeling after ES of the masseter nerve. Images are from the ventral Vi/Vc. A–C: IL-1β immunoreactivity (green) co-localizes with GFAP (red), but not with CD11b (D), a marker of microglia, and NeuN (E), a neuronal marker, after ES of the masseter nerve. A, B: Single labeling of IL-1β (A) and GFAP (B) on the same section. C shows overlay of A and B. F: P-NR1 (green) immunoreactivity was localized to neurons (NeuN, red).
Peripheral Electrical Stimulation Enhances NMDA Receptor NR1 ser896 Phosphorylation
ES of the masseter nerve also induced an increased NMDA receptor phosphorylation in the Vi/Vc transition zone, as assessed with anti-phospho ser896 NR1 antibodies. As shown in Fig. (7), P-NR1-LI increased at 30 min after ES (3 mA) (Fig. 7C), compared to naive (Fig. 7A) and SHAM (Fig. 7B) rats. Western blot showed that significant increases in the P-NR1 levels occurred as early as 10 min after ES and maintained throughout the observation period (p<0.05, n=3/group) (Fig. 7D). Double immunofluorescence with NeuN staining indicated that P-NR1-LI was localized to neurons (Fig. 6F).
Fig. 7.

Increased NMDA receptor NR1 ser896 phosphorylation in the Vi/Vc after ES of the masseter nerve. A–C: Immunostaining. Compared to NAIVE (A) and SHAM-operated (B) rats, ES at 3 mA induced an increased P-NR1 staining (C). D: Western blot illustrating the effect of ES of the masseter nerve on the NMDA receptor subunit NR1 phosphorylation in the ventral Vi/Vc transition zone. β-actin was used as a loading control. A representative blot is shown on top. The relative protein levels are shown in the bottom histogram. *, p<0.05 vs. NAIVE. n=3–4 for each time point. Error bars represent S.E.M.
DISCUSSION
The present experiments were designed to provide direct evidence that injury-related trigeminal glial hyperactivity and cytokine induction after inflammation were dependent on primary afferent input. The results show that 1) masseter muscle inflammation-induced upregulation of GFAP, IL-1β and enhanced NMDA receptor phosphorylation in the Vi/Vc was blocked by local anesthesia of the peripheral injured site, 2) ES of the masseter nerve afferents induced increases in GFAP, IL-1β, and NMDA receptor phosphorylation similar to that after masseter inflammation, and 3) IL-1β selectively colocalizes with GFAP, confirming an increased expression in astroglia. These results indicate that primary afferent inputs associated with peripheral nociceptor activation after inflammation are necessary and sufficient to induce hyperactivity of astroglia and upregulation of IL-1β, as well as neuronal NMDA receptor phosphorylation.
Removing primary afferent input by local anesthesia of either nerve terminals at the injured site (present study) or afferent nerve remote from the injury [13] prevented central glial hyperactivity and cytokine induction. Pretreatment with a long-lasting local anesthetic, bupivacaine-loaded microspheres, above the nerve injury site prevented activation of p38 mitogen-activated protein kinase in the spinal cord after nerve injury [12]. Thus, blocking action potential conduction appears to prevent central glial hyperactivity, which suggests a role of nerve input in triggering an increased glial activity. However, nerve input may play a less important role in maintaining the established glial hyperactivity [12]. One should be cautious that local anesthetic may also act on glial cells. Local anesthetics bupivacaine and lidocaine can enhance calcium-independent nitric oxide synthase catalytic activity and nitrite production in rat glioma cell lines [28]. Nav1.6 sodium channels are found on Schwann cells [29] and local anesthetics may damage Schwann cells directly [15]. These reports with local anesthetics are therefore inconclusive.
In the present study, we employed an alternative approach of ES of peripheral nerve to address the role of nerve input in glial hyperactivity. Electrical stimulation has been widely used to produce afferent input that triggers activation of synaptic activity in dorsal horn neurons [30] and release of mediators of nociception [23, 31]. Stimulation of the hypoglossal nerve in the rat induces Fos protein expression in the spinal trigeminal nucleus [27] and upregulates GFAP immunoreactivity in the hypoglossal nucleus [16]. It should be cautioned that electrical stimulation could damage axons in peripheral nerves [32]. Molander et al. [17] showed that electrical stimulation of the sciatic nerve produced degeneration of nerve fibers associated with increased glial marker staining at 7–14 days after stimulation. However, the nerve damage occurs at least 48 hours after stimulation, requires continuous stimulation and can be prevented by applying stimulation with intermittent cycles [17, 32].
To avoid nerve damage, we used a BS stimulation protocol to generate a primary afferent barrage and the effect of ES was examined within two hours after stimulation. At C-fiber intensity, BS of dorsal roots induces co-release of substance P, glutamate and brain-derived neurotrophic factor (BDNF) into the dorsal horn [23, 33]. The effects of ES on primary afferent neurotransmitter/modulator release are stimulus pattern-dependent. Simple tetanic stimulation (up to 100 Hz) of dorsal root C-fibers does not induce BDNF release [33]. We have used the BS protocol to induce BDNF release in the supraspinal pain modulatory circuitry [34]. In the present study, BS of the masseter nerve afferents was able to induce trigeminal Fos protein expression, a marker of neuronal activation, in a manner that is consistent with neuronal responses to orofacial noxious stimulation [20]. Stimulation of the masseter nerve further produced similar increases in GFAP- and IL-1β-LI in the Vi/Vc transition zone, which were consistent with the results of CFA-induced inflammation [13]. BS-induced IL-1β also selectively colocalized with GFAP as seen in the inflammation model [13], suggesting its upregulation in reactive astrocytes after afferent stimulation. The combination of local anesthesia and afferent stimulation experiments provide convergent evidence that injury-related trigeminal glial hyperactivity is dependent on primary afferent input.
It is still unclear, however, how primary afferent input would convey its signal to glial cells. We observed in the present study that NMDA receptor phosphorylation was also enhanced after ES of the masseter nerve, which is consistent with our finding after masseter inflammation [13]. Our results also showed that the increased NMDA receptor phosphorylation occurred in neurons and appeared within 10 minutes after ES. In contrast, significant upregulation of astrocytic proteins and IL-1β did not occur until at least 30 min after ES. This suggests that the initiation of NMDA receptor phosphorylation may not be due to IL-1β activation in astrocytes and that a relay of primary afferent input via secondary neurons may be necessary for the increased astroglial activity. Sequential activation of ERK in neurons and glia following spinal nerve ligation is consistent with this hypothesis [10]. Although initiation of NMDA receptor phosphorylation does not appear to depend upon glial activity, positive feedback from glial cells and associated mediators may play a role in the maintenance of enhanced NMDA receptor and neuronal excitability after injury. We have shown that IL-1 receptor antagonists blocked NMDA receptor phosphorylation after orofacial injury [13, 35], suggesting a role of IL-1β and associated astroglial activity in the maintenance of enhanced NMDA receptor phosphorylation. Alternatively, neurotransmitters released from primary afferent central terminals may directly activate glia. Substance P and calcitonin gene related peptide have been shown to induce upregulation of GFAP and IL-1β in the Vi/Vc region from medullary slices [13], which may account for the maintenance of NMDA receptor phosphorylation.
Our findings further support that neuronal input related to peripheral injury not only activates neural circuitry in the sensory pathways but also affects glial cells and associated cytokines. Although cytokines can be released at the site of injury [22, 36–38], central cytokine induction is a result of sequential or reciprocal neuron-glia interactions and may not be attributed to cytokines released at the periphery. This may explain the findings that the IL-1β level increases in both the inflamed paw and cerebrospinal fluid, but not in the circulation [39] and that the levels of IL-1β are increased after masseter inflammation at the site of injury but not in the circulation [22]. Apparently, IL-1β is released from glial cells that are activated by nerve input and act back on neurons to further enhance the maintenance of hyperexcitability [13, 40]. Thus, injury-related nerve input initiates a cascade of events in the CNS involving both neuronal and non-neuronal components, ultimately leading to hyperalgesia.
Acknowledgments
This work was supported by National Institutes of Health, DE11964, DE018573, NS060735, NS059028. H.W. is supported by T32 DE007309.
Footnotes
This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/-licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
References
- 1.Ren K, Dubner R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr Opin Anaesthesiol. 2008;21:570–9. doi: 10.1097/ACO.0b013e32830edbdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shelton MK, McCarthy KD. Mature hippocampal astrocytes exhibit functional metabotropic and ionotropic glutamate receptors in situ. Glia. 1999;26:1–11. doi: 10.1002/(sici)1098-1136(199903)26:1<1::aid-glia1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 3.Gallo V, Ghiani CA. Glutamate receptors in glia: new cells, new inputs and new functions. Trends Pharmacol Sci. 2000;21:252–8. doi: 10.1016/s0165-6147(00)01494-2. [DOI] [PubMed] [Google Scholar]
- 4.Schipke CG, Ohlemeyer C, Matyash M, Nolte C, Kettenmann H, Kirchhoff F. Astrocytes of the mouse neocortex express functional N-methyl-D-aspartate receptors. FASEB J. 2001;15:1270–2. doi: 10.1096/fj.00-0439fje. [DOI] [PubMed] [Google Scholar]
- 5.Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci. 2006;26:2673–83. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu SY, Kriegler S. Neurotransmitter-mediated signaling between axons and glial cells. Glia. 1994;11:191–200. doi: 10.1002/glia.440110213. [DOI] [PubMed] [Google Scholar]
- 7.Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–81. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steward O, Torre ER, Tomasulo R, Lothman E. Neuronal activity up-regulates astroglial gene expression. Proc Natl Acad Sci USA. 1991;88:6819–23. doi: 10.1073/pnas.88.15.6819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrison CJ, Dougherty PM, Carlton SM. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp Neurol. 1994;129:237–43. doi: 10.1006/exnr.1994.1165. [DOI] [PubMed] [Google Scholar]
- 10.Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–59. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 11.Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79:163–75. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- 12.Wen YR, Suter MR, Kawasaki Y, et al. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–21. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- 13.Guo W, Wang H, Watanabe M, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–18. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie W, Strong JA, Zhang JM. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience. 2009;160:847–57. doi: 10.1016/j.neuroscience.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powell HC, Kalichman MW, Garrett RS, Myers RR. Selective vulnerability of unmyelinated fiber Schwann cells in nerves exposed to local anesthetics. Lab Invest. 1988;59:271–80. [PubMed] [Google Scholar]
- 16.Hall LL, Borke RC, Anders JJ. Transection or electrical stimulation of the hypoglossal nerve increases glial fibrillary acidic protein immunoreactivity in the hypoglossal nucleus. Brain Res. 1989;490:157–61. doi: 10.1016/0006-8993(89)90443-5. [DOI] [PubMed] [Google Scholar]
- 17.Molander C, Hongpaisan J, Svensson M, Aldskogius H. Glial cell reactions in the spinal cord after sensory nerve stimulation are associated with axonal injury. Brain Res. 1997;747:122–9. doi: 10.1016/s0006-8993(96)01230-9. [DOI] [PubMed] [Google Scholar]
- 18.Hathway GJ, Vega-Avelaira D, Moss A, Ingram R, Fitzgerald M. Brief, low frequency stimulation of rat peripheral C-fibres evokes prolonged microglial-induced central sensitization in adults but not in neonates. Pain. 2009;144:110–8. doi: 10.1016/j.pain.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Wei F, Dubner R, Ren K. Selective distribution and function of primary afferent nociceptive inputs from deep muscle tissue to the brainstem trigeminal transition zone. J Comp Neurol. 2006;498:390–402. doi: 10.1002/cne.21062. [DOI] [PubMed] [Google Scholar]
- 20.Imbe H, Dubner R, Ren K. Masseter inflammation-induced Fos protein expression in brainstem neurons depends on somatosensory-vagal-adrenal integration. Brain Res. 1999;845:165–75. doi: 10.1016/s0006-8993(99)01913-7. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda T, Terayama R, Jue S-S, Sugiyo S, Dubner R, Ren K. Differential rostral projections of caudal brainstem neurons receiving trigeminal input after masseter inflammation. J Comp Neurol. 2003;465:220–33. doi: 10.1002/cne.10836. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe M, Guo W, Zou S-P, Sugiyo S, Dubner R, Ren K. Antibody array analysis of peripheral and blood cytokine levels in rats after masseter inflammation. Neurosci Lett. 2005;382:128–33. doi: 10.1016/j.neulet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Lever IJ, Bradbury EJ, Cunningham JR, et al. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21:4469–77. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greene EC. The anatomy of the rat. Philadelphia: The American Philosophical Society; 1935. [Google Scholar]
- 25.Sugiyo S, Takemura M, Dubner R, Ren K. Trigeminal transition zone/rostral ventromedial medulla connections and facilitation of orofacial hyperalgesia after masseter inflammation in rats. J Comp Neurol. 2005;493:510–23. doi: 10.1002/cne.20797. [DOI] [PubMed] [Google Scholar]
- 26.Guo W, Wang H, Zou S, Wei F, Dubner R, Ren K. Long lasting pain hypersensitivity following ligation of the tendon of the masseter muscle in rats: a model of myogenic orofacial pain. Mol Pain. 2010;6:40. doi: 10.1186/1744-8069-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bereiter DA, Bereiter DF, Hirata H, Hu JW. c-fos expression in trigeminal spinal nucleus after electrical stimulation of the hypoglossal nerve in the rat. Somatosens Mot Res. 2000;17:229–37. doi: 10.1080/08990220050117583. [DOI] [PubMed] [Google Scholar]
- 28.Feinstein DL, Murphy P, Sharp A, Galea E, Gavrilyuk V, Weinberg G. Local anesthetics potentiate nitric oxide synthase type 2 expression in rat glial cells. J Neurosurg Anesthesiol. 2001;13:99–105. doi: 10.1097/00008506-200104000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Musarella M, Alcaraz G, Caillol G, Boudier JL, Couraud F, Autillo-Touati A. Expression of Nav1.6 sodium channels by Schwann cells at neuromuscular junctions: role in the motor endplate disease phenotype. Glia. 2006;53:13–23. doi: 10.1002/glia.20252. [DOI] [PubMed] [Google Scholar]
- 30.Liu XG, Sandkuhler J. Activation of spinal N-methyl-D-aspartate or neurokinin receptors induces long-term potentiation of spinal C-fibre-evoked potentials. Neuroscience. 1998;86:1209–16. doi: 10.1016/s0306-4522(98)00107-9. [DOI] [PubMed] [Google Scholar]
- 31.Klein CM, Coggeshall RE, Carlton SM, Sorkin LS. The effects of A- and C-fiber stimulation on patterns of neuropeptide immunostaining in the rat superficial dorsal horn. Brain Res. 1992;580:121–8. doi: 10.1016/0006-8993(92)90935-3. [DOI] [PubMed] [Google Scholar]
- 32.Agnew WF, McCreery DB, Yuen TG, Bullara LA. Histologic and physiologic evaluation of electrically stimulated peripheral nerve: considerations for the selection of parameters. Ann Biomed Eng. 1989;17:39–60. doi: 10.1007/BF02364272. [DOI] [PubMed] [Google Scholar]
- 33.Pezet S, Malcangio M, McMahon SB. BDNF: a neuromodulator in nociceptive pathways? Brain Res Brain Res Rev. 2002;40:240–9. doi: 10.1016/s0165-0173(02)00206-0. [DOI] [PubMed] [Google Scholar]
- 34.Guo W, Robbins MT, Wei F, Zou S, Dubner R, Ren K. Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation. J Neurosci. 2006;26:126–37. doi: 10.1523/JNEUROSCI.3686-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008;28:10482–95. doi: 10.1523/JNEUROSCI.3593-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–5. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 37.Griffin BD, Moynagh PN. Persistent interleukin-1beta signaling causes long term activation of NFkappaB in a promoter-specific manner in human glial cells. J Biol Chem. 2006;281:10316–26. doi: 10.1074/jbc.M509973200. [DOI] [PubMed] [Google Scholar]
- 38.Lin HW, Basu A, Druckman C, Cicchese M, Krady JK, Levison SW. Astrogliosis is delayed in type 1 interleukin-1 receptor-null mice following a penetrating brain injury. J Neuroinflamm. 2006;3:15. doi: 10.1186/1742-2094-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–5. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- 40.Kawasaki Y, Xu ZZ, Wang X, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14:331–6. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]