

Abstract
The implementation of multiconfigurational quantum chemistry methods into a quantum-mechanics/molecular-mechanics protocol has allowed the construction of a realistic computer model for the sensory rhodopsin of the cyanobacterium Anabaena PCC 7120. The model, which reproduces the absorption spectra of both the all-trans and 13-cis forms of the protein and their associated K and L intermediates, is employed to investigate the light-driven steps of the photochromic cycle exhibited by the protein. It is found that the photoisomerizations of the all-trans and 13-cis retinal chromophores occur through unidirectional, counterclockwise 180° rotations of the ═C14─C15═ moiety with respect to the Lys210-linked end of the chromophore axis. Thus, the sequential interconversions of the all-trans and 13-cis forms during a single photochromic cycle yield a complete (360°) unidirectional rotation of the ═C14─C15═ moiety. This finding implies that Anabaena sensory rhodopsin is a biological realization of a light-driven molecular rotor.
Keywords: molecular motors, molecular modeling, photoreceptors, stereochemistry, chirality
The building of molecular-level motors is essential for implementing a bottom-up approach to nanoscale devices (1). One major challenge in this regard is the production of unidirectional (rotary) motion. In 1999, Feringa and co-workers demonstrated that repetitive, unidirectional rotations about a carbon–carbon double bond could be achieved in chiral biarylidenes (2). In these molecules each clockwise (CW) or counterclockwise (CCW) 360° rotation involves four discrete steps, which are activated by UV light or heat. As shown in Fig. 1A for the R, R biphenanthrylidene I, a light-induced trans-cis isomerization of the carbon–carbon double bond brings the system to station II, where further isomerization continuing in the same direction is prevented by steric hindrance. However, by supplying heat, a change from M to P helicity occurs (II → III), which enables a second light-induced cis-trans isomerization (III → IV) to proceed in the original direction. Similarly, in station IV a thermal step is needed to obtain the initial stereoisomer and for starting a new cycle.
Fig. 1.

(A) The biphenanthrylidene molecular rotor fabricated by Feringa and co-workers (2). Full curly arrows indicate CCW (stations I and III) double bond isomerization. The right-handed (M) and left-handed (P) helicities (see the dashed curly arrows in stations I and II) result from steric clashing of the double bond substituents and the 3R and 3′R stereogenic centers. The II → III and IV → I thermal steps are driven by 11 and 9 kcal mol-1 stabilization of the III and I structures, respectively. (B) The photochromic cycle of ASR. Full curly arrows indicate alternating CCW and CW isomerizations about the ─C13═C14─ and ─C15═N─ retinal double bonds, respectively. The helicities of the ─C15═NH─Cε─ and ─C12─C13═C14─ moieties (dashed curly arrows) are assigned by determining whether the helicity of the corresponding ─H(C14)─C15═NH─Cε─ and ─C12─C13═C14─(C15)H─ fragments is M or P.
Although biological systems that accomplish rotary motion using chemical energy are known at the level of complex protein assemblies [e.g., ATPase (3) and flagella motors (4)], such behavior is yet to be detected at the single-molecule level. However, it has been suggested that rhodopsins, a class of ubiquitous photoreceptors that include the rod visual pigment (Rh) of superior animals, could be biological analogues to the biarylidene rotors (5). Rhodopsins (6) are transmembrane proteins featuring seven α-helices that form a barrel-like structure with a cavity accommodating the chromophore: a protonated Schiff base of retinal that forms a covalent linkage to a lysine residue of the protein. The photocycles of rhodopsins are initiated by the selective trans-cis photoisomerization of either the ─C11═C12─ (in visual pigments) or the ─C13═C14─ (in microbial rhodopsins) retinal double bond. Although retinal itself does not contain stereogenic centers, the chromophore is chiral as a result of the chirality of the protein cavity. Indeed, for Rh, crystallographic (7, 8), NMR (9, 10), and computed (11) structural data point to a chromophore that displays a negatively twisted 11-cis double bond and M helicity in its backbone. Upon light absorption, the chromophore isomerizes to bathorhodopsin, the first isolable photocycle intermediate that exhibits a ca. -140° twisted ─C11═C12─ bond and P helicity (12). This M → P change in helicity is consistent with a CW isomerization motion, which has also been confirmed by excited state reaction path and trajectory computations (13, 14).
Besides olefin chirality, a second requirement for achieving a complete (360°) light-driven unidirectional rotation in a molecular system is photochromism, whereby two thermostable forms of the system can be reversibly converted into one another through the absorption of light. Recently, Spudich and co-workers have reported that the sensory rhodopsin of the cyanobacterium Anabaena PCC 7120 (ASR) fulfills this requirement (15, 16). As shown in Fig. 2, ASR exists in two thermostable forms (ASRAT and ASR13C in Fig. 2A) that harbor different stereoisomers of the retinal chromophore (AT: all-trans, 15-anti; 13C: 13-cis, 15-syn) and are part of a photochromic cycle that involves a number of intermediate states with different absorption maxima (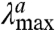 ). Daylight adaptation gives an equilibrium dominated by ASR13C (e.g., 78% at > 560 nm in phosphatidylcholine liposomes) (17). The photostationary ASRAT/ASR13C ratio depends on the wavelength of illumination and is believed to provide a mechanism for single-pigment color sensing (15, 16). The efficiency of the photoinduced ASRAT → ASR13C and ASR13C → ASRAT conversions has been quantitatively investigated by Kandori and co-workers (17). These authors showed that both processes occur with unity quantum yield, without any significant influence from competitive thermal processes.
). Daylight adaptation gives an equilibrium dominated by ASR13C (e.g., 78% at > 560 nm in phosphatidylcholine liposomes) (17). The photostationary ASRAT/ASR13C ratio depends on the wavelength of illumination and is believed to provide a mechanism for single-pigment color sensing (15, 16). The efficiency of the photoinduced ASRAT → ASR13C and ASR13C → ASRAT conversions has been quantitatively investigated by Kandori and co-workers (17). These authors showed that both processes occur with unity quantum yield, without any significant influence from competitive thermal processes.
Fig. 2.

(A) Structures of the retinal chromophores of ASRAT and ASR13C. (B) The intertwined photocycles of ASRAT and ASR13C as represented by the ─C12─C13═C14─C15═N─Cε─ chromophore moiety in the different states. The wavelength values refer to observed (17) and computed (in bold) absorption maxima of the full protein. In each state the isomerizing bond is highlighted.
The photochromism of ASR coupled to the chirality of the embedded retinal chromophore constitutes a unique feature among rhodopsins. Therefore, it is worthwhile to investigate whether ASR can accomplish the type of rotary motion that Feringa and co-workers, using NMR and circular dichroism spectroscopy, demonstrated in photochromic chiral biarylidenes (2). To this end, and because relevant experimental techniques are cumbersome to apply to a full protein, we have built a computer model of ASR and performed quantum chemical calculations in the framework of a hybrid quantum-mechanics/molecular-mechanics (QM/MM) scheme to investigate the chromophore structural changes during the first steps of its photochromic cycle. The calculations are based on the ab initio complete-active-space self-consistent-field (CASSCF) method (18), which is a multiconfigurational quantum chemical method that offers maximum flexibility for an unbiased description of the electronic structure and geometries of molecules in their ground and excited states. CASSCF wave functions can also (19) be used in the context of second-order perturbation theory (CASPT2), which recovers the dynamic correlation energy and allows for a quantitative prediction of excitation energies. Indeed, in previous work we have shown that CASPT2//CASSCF-based (i.e., CASSCF geometry optimization and CASPT2 energy evaluation) QM/MM calculations reproduce both absorption and emission spectra of proteins incorporating chemically different chromophores (20–22). For example, such calculations reproduce the 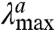 value of Rh with an error of 3 kcal mol-1 only (13). CASPT2//CASSCF is also a viable, and arguably the only, quantum chemical approach for computing excited state reaction paths, trajectories, and conical intersections at a common level of theory (23). Hence, such calculations are the method of choice for simulating the photochemical steps and primary photoproducts of the photocycle of ASR. In the present work, we show that CASPT2//CASSCF-based QM/MM calculations not only reproduce available structural and spectroscopic data for ASR, but also provide valuable previously undescribed insight into the chromophore structural changes during the initial stages of the photocycle of ASR.
value of Rh with an error of 3 kcal mol-1 only (13). CASPT2//CASSCF is also a viable, and arguably the only, quantum chemical approach for computing excited state reaction paths, trajectories, and conical intersections at a common level of theory (23). Hence, such calculations are the method of choice for simulating the photochemical steps and primary photoproducts of the photocycle of ASR. In the present work, we show that CASPT2//CASSCF-based QM/MM calculations not only reproduce available structural and spectroscopic data for ASR, but also provide valuable previously undescribed insight into the chromophore structural changes during the initial stages of the photocycle of ASR.
Results and Discussion
To assess the accuracy of the electrostatic embedding QM/MM scheme employed in the calculations, the computed models of the parent (ASRAT and ASR13C), photoproduct (ASRAT-K and ASR13C-K), and L (ASRAT-L and ASR13C-L) states that are part of the photocycle of ASR were validated by comparison with available spectral data (17, 24). From inspection of Fig. 2B, it is clear that the observed changes in absorption between the different states are well reproduced. Furthermore, the experimental data are throughout matched with a less than 4 kcal mol-1 error. We also find (Table S1) that the computed decrease in oscillator strength between ASRAT and ASRAT-K (1.29 → 1.18) and the increase between ASR13C and ASR13C-K (1.05 → 1.10) are consistent with the changes in absorbance reported by Kandori and co-workers (24). These authors also investigated the chromophore conformational changes occurring during the early stages of the ASR photocycle using FTIR spectroscopy at 77 K (24). It was shown that the Schiff base N-D stretching frequency increases when the protein evolves from the ASRAT and ASR13C states to the corresponding K intermediates. This effect was assigned to the weakening/breaking of the N-H---O hydrogen bond involving the water molecule (W402) located between the Schiff base and its Asp75 counterion (15, 16). In particular, the ca. 350 cm-1 increase in N-D frequency observed upon the ASRAT → ASRAT-K transformation would reflect a complete breaking of the hydrogen bond, whereas the smaller increase (ca. 200 cm-1) during the ASR13C → ASR13C-K transformation would reflect a weakening—not a complete breaking—of the bond.
Using the experimental FTIR data as reference (24), we have evaluated the changes in N-D frequency in the K states relative to the parent states (Table S2). Consistently with the FTIR data, we find that the N-D frequency increases in the K states and that the increase in ASRAT-K (ca. 100 cm-1) is more pronounced than that in ASR13C-K (ca. 50 cm-1). Indeed, the distances and angles between the N-H moiety and W402 reported in Fig. 3 indicate that our ASRAT-K and ASR13C-K models are in agreement with the interpretation of the FTIR signal changes. In particular, whereas the N-H---O distance is 3.13 Å in ASR13C-K, it is 3.36 Å in ASRAT-K, thus reflecting a more substantial weakening in hydrogen bond strength in the latter state.
Fig. 3.

(A and B) Structural evolution of the retinal chromophore during the photochromic cycle of ASR. The crystallographic water molecule (W402) in the vicinity of the Schiff base nitrogen is also shown, together with the corresponding N─H---O distances and N─H---O angles. The ASRAT → ASRAT-K and ASR13C → ASR13C-K transitions occur with the same mechanism corresponding to a ca. 180° rotation of the plane containing the ═C14─C15═ fragment (plane β). As indicated by the dashed rotation axis and “eye” symbol in the ASRAT and ASR13C structures, this rotation occurs CCW with respect to the Lys210 side chain. The N-H bond undergoes a large and reversible reorientation due to the change in ─C15═N─Cε─Cδ─ dihedral angle that accompanies the isomerization of the ─C13═C14─ bond. During the subsequent K → L transition, the N-H bond rotates in the reverse direction, bringing about the isomerization of the ─C15═N─ bond. This backward rotation is driven by the reconstitution of the N─H---O hydrogen bond. The Newman projections display the pretwisting of the ─C15═N─ bond, which clearly favor CW isomerization of this bond during the K → L transition. The evolution of the ═C14─C15═NH─Cε─ and ═C12─C13═C14─C15═ dihedrals describing the two isomerizations is reported below each structure in degrees. (C) Top (i.e., cytoplasm side) view of the orientation of the “rotating” ═C14─C15═ moiety in ASRAT and ASR13C. The retinal backbone axis is represented by a dashed line.
Through an analysis of the structural changes along the photoisomerization coordinates (Fig. 4) and the structures of the corresponding K and L intermediates (Fig. 3), we are now in the position to characterize the molecular motion driving the photochromism of ASR. The ASRAT and ASR13C chromophores feature a negatively (-167°) and a positively (+13°) pretwisted ─C13═C14─ double bond, respectively. This pretwisting, which is also found in the corresponding crystallographic structures, induces local helicity in the molecular backbone (Fig. 1B). Thus, the chromophore displays P helicity in both ASRAT and ASR13C. The computed reaction paths (Fig. 4) show that, upon photoexcitation, both ASRAT and ASR13C undergo CCW isomerization about the ─C13═C14─ bond, leading to a conical intersection and, following decay to the ground state, to ASRAT-K and ASR13C-K. Indeed, the K intermediates show highly twisted ─C13═C14─ bonds of -26° (AT) and +150° (13C). Through the CCW isomerization to reach the K intermediates, the P helicities of the parent states are changed into M helicities. A parallel behavior is found for the ─C15═N─ double bond, whose initial P helicity in the parent states is turned, via a limited CW conformational change, into M helicity in ASRAT-K and ASR13C-K. These stereochemical relationships reveal an analogy between the photochromic cycle of ASR with the photoisomerization steps of the light-driven biarylidene rotor fabricated by Feringa and co-workers (2). Namely, as apparent from Fig. 1, the ASRAT → ASRAT-K and ASR13C → ASR13C-K steps are analogous to the I → II and III → IV steps of the biarylidene system, with the only difference being that in ASR the stereochemical signatures pertain to two double bonds rather than to one double bond. This is consistent with the fact that there are two double bonds (─C13═C14─ and ─C15═N─) that isomerize in the ASR system, yielding a rotation of the ═C14─C15═ chromophore moiety. This twofold isomerization motion allows for the rotation of the ═C14─C15═ moiety in the limited space offered by the protein cavity.
Fig. 4.

(A) CASPT2//CASSCF/AMBER energy profiles along the S1 photoisomerization path of ASRAT. S0 and S2 energy profiles along the S1 path are also given. The S1 path is computed in terms of a relaxed scan along the reactive ═C12─C13═C14─C15═ dihedral angle. The location of the conical intersection ASRAT-CI is revealed by the change in the character of the S1 electronic state. Computed molecular structures and QM/MM energies are given in Table S1. (Top) Changes of key dihedral angles with the leftmost value referring to ASRAT and the Δ value referring to the difference between ASRAT and ASRAT-CI. The preferential CCW twisting of the ─C13═C14─ bond is confirmed via additional calculations showing that CW twisting is energetically less favorable (see further Fig. S1). (B) The corresponding path and structural details for ASR13C.
As shown in Fig. 1A, the photochemical rotary motion in the biarylidene system is governed by two thermal steps corresponding to the formation of thermodynamically favored stereoisomers (i.e., steps II → III and IV → I). These steps act as molecular ratchets in the sense that they prevent back-rotations by establishing the pretwisting of the central double bond, thereby controlling the directionality of the subsequent photochemical steps. Comparing with ASR, the K → L transformations involve CW isomerization about the ─C15═N─ bond and a limited CCW conformational change about the ─C13═C14─ bond. Accordingly, for both forms of the protein, the M helicity displayed by the K intermediate is turned into P helicity in the L intermediate (Fig. 1B), in full analogy with the thermal steps of the biarylidene system. Furthermore, just as the thermal steps of the biarylidene system are driven by the greater stabilities of stereoisomers III and I over II and IV (Fig. 1A), the K → L transformations are driven by the greater stabilities of the L intermediates over the K intermediates. This stabilization is due to the reconstitution of planar double bonds and the reformation of the N-H---O hydrogen bond between the Schiff base and W402 in the L intermediates. The stereochemical and thermodynamic analogies between the K → L transformations of ASR and steps II → III and IV → I of the biarylidene system indicate that the K → L transformation (and therefore ─C15═N─ isomerization) provides an effective ratchet-like mechanism for maintaining the rotary motion of the ═C14─C15═ chromophore fragment. Indeed, as shown in Fig. S2, the excited state evolution of the ─C13═C14─ bond is controlled by the stereochemistry of the ─C15═N─ bond.
In conclusion, the chromophore stereochemical changes during the photocycle of ASR indicate that this protein undergoes unidirectional cycling between its photochromic forms. The cycle is closed when the L intermediates are converted into the parent states. Our calculations, as well as the crystal structure data (15, 16), further indicate that these conversions involve conformational changes of the side chain of Lys210. As documented by the structures of Fig. 3, the unidirectional cycling is achieved through alternating and sequential CCW and CW twisting of the ─C13═C14─ and ─C15═N─ bonds, respectively. Such motion is readily interpreted in terms of the CCW (right-hand rule) rotation of the ═C14─C15═ moiety of the chromophore with respect to its backbone axis. Indeed, the plane containing this unit (plane β in Fig. 3) rotates by 360° in a CCW fashion throughout the photochromic cycle through alternating photochemical and thermal double bond isomerization steps, overall following a bicycle-pedal (or crankshaft) mechanism. Thus, our calculations extend the concept of space-saving bicycle-pedal isomerization established for the excited state evolution of the visual pigment chromophore almost thirty-five years ago (25) to an entire photochromic cycle.
Computational Methods
Details of the calculations can be found in the SI Appendix. Briefly, the models of ASRAT and ASR13C were constructed starting from the crystallographic structure of ASR (15, 16) available in the Protein Data Bank (PDB ID code 1XIO). This structure provides an average representation of the protein and, with the exception of the Lys210 residue bound to the retinal chromophore, was kept fixed in all calculations, whereas the chromophore and internal water molecules were fully flexible. The models of the primary photoproducts (ASRAT-K and ASR13C-K) were derived from the parent ASRAT and ASR13C models via excited state (S1) isomerization path computations. These provide minimum energy paths connecting the Franck–Condon points on S1 to the points where decay to the ground state (S0) occurs. As shown in Fig. 4 and consistently with what was found for Rh (13), both paths end at a S1/S0 conical intersection (ASRAT-CI and ASR13C-CI, respectively) where the S0 and S1 potential energy surfaces cross and the decay probability is unity (26). Starting from the highly twisted ASRAT-CI and ASR13C-CI structures (with ─C13═C14─ angles of -108° and +84°, respectively), the ASRAT-K and ASR13C-K structures were obtained by performing S0 geometry optimizations. Because both K structures feature a twisted ─C15═N─ bond, the subsequent K → L transitions are likely to involve 15-anti → 15-syn (AT) and 15-syn → 15-anti (13C) rotations, which is also supported by the data of Kandori and co-workers (17, 24). Therefore, the ASRAT-L and ASR13C-L models were derived from the corresponding K models by further twisting the ─C15═N─ bond in the same CW direction, followed by S0 geometry optimization yielding equilibrium structures with nearly planar ─C15═N─ bonds.
Supplementary Material
Acknowledgments.
Prof. H. Kandori is acknowledged for useful discussions. B.D. was supported by Linköping University, the Carl Trygger Foundation, and a Marie Curie Intra-European Fellowship (MEIF-CT-2006 023430) of the 6th European Community Framework Programme. We are grateful to the Ohio Supercomputer Center for granted computer time. M.O. is grateful to the Center for Photochemical Sciences and School of Arts and Sciences of the Bowling Green State University for a startup grant.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1015085107/-/DCSupplemental.
References
- 1.Browne WR, Feringa BL. Making molecular machines work. Nat Nanotechnol. 2006;1:25–35. doi: 10.1038/nnano.2006.45. [DOI] [PubMed] [Google Scholar]
- 2.Koumura N, Zijlstra RWJ, van Delden RA, Harada N, Feringa BL. Light-driven monodirectional molecular rotor. Nature. 1999;401:152–155. doi: 10.1038/43646. [DOI] [PubMed] [Google Scholar]
- 3.Kinbara K, Aida T. Toward intelligent molecular machines: Directed motions of biological and artificial molecules and assemblies. Chem Rev. 2005;105:1377–1400. doi: 10.1021/cr030071r. [DOI] [PubMed] [Google Scholar]
- 4.Berg HC. The rotary motor of bacterial flagella. Annu Rev Biochem. 2003;72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- 5.Feringa BL. The art of building small: From molecular switches to molecular motors. J Org Chem. 2007;72:6635–6652. doi: 10.1021/jo070394d. [DOI] [PubMed] [Google Scholar]
- 6.Spudich JL, Yang CS, Jung KH, Spudich EN. Retinylidene proteins: Structures and functions from archaea to humans. Annu Rev Cell Dev Biol. 2000;16:365–392. doi: 10.1146/annurev.cellbio.16.1.365. [DOI] [PubMed] [Google Scholar]
- 7.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 8.Okada T, et al. The retinal conformation and its environment in rhodopsin in light of a new 2.2 Å crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 9.Salgado GFJ, et al. Deuterium NMR structure of retinal in the ground state of rhodopsin. Biochemistry. 2004;43:12819–12828. doi: 10.1021/bi0491191. [DOI] [PubMed] [Google Scholar]
- 10.Struts AV, et al. Structural analysis and dynamics of retinal chromophore in dark and meta I states of rhodopsin from 2H-NMR of aligned membranes. J Mol Biol. 2007;372:50–66. doi: 10.1016/j.jmb.2007.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pescitelli G, et al. Inherent chirality dominates the visible/near-ultraviolet CD spectrum of rhodopsin. J Am Chem Soc. 2008;130:6170–6181. doi: 10.1021/ja711009y. [DOI] [PubMed] [Google Scholar]
- 12.Yan EC, et al. Resonance Raman analysis of the mechanism of energy storage and chromophore distortion in the primary visual photoproduct. Biochemistry. 2004;43:10867–10876. doi: 10.1021/bi0400148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andruniòw T, Ferré N, Olivucci M. Structure, initial excited-state relaxation, and energy storage of rhodopsin resolved at the multiconfigurational perturbation theory level. Proc Natl Acad Sci USA. 2004;101:17908–17913. doi: 10.1073/pnas.0407997101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frutos LM, Andruniów T, Santoro F, Ferré N, Olivucci M. Tracking the excited-state time evolution of the visual pigment with multiconfigurational quantum chemistry. Proc Natl Acad Sci USA. 2007;104:7764–7769. doi: 10.1073/pnas.0701732104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogeley L, et al. Anabaena sensory rhodopsin: A photochromic color sensor at 2.0 Å. Science. 2004;306:1390–1393. doi: 10.1126/science.1103943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sineshchekov OA, Trivedi VD, Sasaki J, Spudich JL. Photochromicity of Anabaena sensory rhodopsin, an atypical microbial receptor with a cis-retinal light-adapted form. J Biol Chem. 2005;280:14663–14668. doi: 10.1074/jbc.M501416200. [DOI] [PubMed] [Google Scholar]
- 17.Kawanabe A, Furutani Y, Jung KH, Kandori H. Photochromism of Anabaena sensory rhodopsin. J Am Chem Soc. 2007;129:8644–8649. doi: 10.1021/ja072085a. [DOI] [PubMed] [Google Scholar]
- 18.Roos BO. In: Advanced Chemistry and Physics: Ab Initio Methods in Quantum Chemistry II. Lawley KP, editor. Chicester, UK: Wiley; 1987. pp. 399–445. [Google Scholar]
- 19.Andersson K, Malmqvist P-A, Roos BO, Sadlej AJ, Wolinski K. Second-order perturbation theory with a CASSCF reference function. J Phys Chem. 1990;94:5483–5488. [Google Scholar]
- 20.Coto PB, Strambi A, Ferrè N, Olivucci M. The color of rhodopsins at the ab initio multiconfigurational perturbation theory resolution. Proc Natl Acad Sci USA. 2006;103:17154–17159. doi: 10.1073/pnas.0604048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coto PB, et al. Origin of the absorption maxima of the photoactive yellow protein resolved via ab initio multiconfigurational methods. J Phys Chem B. 2008;112:7153–7156. doi: 10.1021/jp711396b. [DOI] [PubMed] [Google Scholar]
- 22.Sinicropi A, Andruniow T, Ferré N, Basosi R, Olivucci M. Properties of the emitting state of the green fluorescent protein resolved at the CASPT2//CASSCF/CHARMM level. J Am Chem Soc. 2005;127:11534–11535. doi: 10.1021/ja045269n. [DOI] [PubMed] [Google Scholar]
- 23.Migani A, Olivucci M. In: Conical Intersections: Electronic Structure, Dynamics and Spectroscopy. Domcke W, Yarkony DR, Köppel H, editors. Singapore: World Scientific; 2004. pp. 271–320. [Google Scholar]
- 24.Kawanabe A, Furutani Y, Jung KH, Kandori H. FTIR study of the photoisomerization processes in the 13-cis and all-trans forms of Anabaena sensory rhodopsin at 77 K. Biochemistry. 2006;45:4362–4370. doi: 10.1021/bi052324b. [DOI] [PubMed] [Google Scholar]
- 25.Warshel A. Bicycle-pedal model for the first step in the vision process. Nature. 1976;260:679–683. doi: 10.1038/260679a0. [DOI] [PubMed] [Google Scholar]
- 26.Bernardi F, Olivucci M, Robb MA. Potential energy surface crossings in organic photochemistry. Chem Soc Rev. 1996;25:321–328. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.