

Abstract
Genomic rearrangements are common, occur by largely unknown mechanisms, and can lead to human diseases. We previously demonstrated that some genome rearrangements occur in budding yeast through the fusion of two DNA sequences that contain limited sequence homology, lie in inverted orientation, and are within 5 kb of one another. This inverted repeat fusion reaction forms dicentric chromosomes, which are well-known intermediates to additional rearrangements. We have previously provided evidence indicating that an error of stalled or disrupted DNA replication forks can cause inverted repeat fusion. Here we analyze how checkpoint protein regulatory pathways known to stabilize stalled forks affect this form of instability. We find that two checkpoint pathways suppress inverted repeat fusion, and that their activities are distinguishable by their interactions with exonuclease 1 (Exo1). The checkpoint kinase Rad53 (Chk2) and recombination protein complex MRX(MRN) inhibit Exo1 in one pathway, whereas in a second pathway the ATR-like kinases Mec1 and Tel1, adaptor protein Rad9, and effector kinases Chk1 and Dun1 act independently of Exo1 to prevent inverted repeat fusion. We provide a model that indicates how in Rad53 or MRX mutants, an inappropriately active Exo1 may facilitate faulty template switching between nearby inverted repeats to form dicentric chromosomes. We further investigate the role of Rad53, using hypomorphic alleles of Rad53 and null mutations in Rad9 and Mrc1, and provide evidence that only local, as opposed to global, activity of Rad53 is sufficient to prevent inverted repeat fusion.
Keywords: Genome instability, Global versus local checkpoint, Replication error, Mec1, Rad53
Large-scale chromosomal changes have a profound impact on all genomes, from microbial to human. Chromosomal changes contribute to speciation by forming reproductive barriers, and are associated with genetic disorders and cancers as well (1). Chromosomal changes leading to duplication, deletion, or translocation of genomic information have been associated with diseases (2, 3). Malignant tumors have complex karyotypes, and a significant fraction harbor large-scale changes, including dicentric chromosomes (4), a focus of the present study.
How large-scale chromosomal changes occur remains a matter of speculation. Recent studies suggest that defects in DNA replication are major contributors to genome instability (5–7). Replication errors may occur when replication forks encounter and attempt to replicate through “lesions” on the template strand. Lesions can consist of protein complexes bound to DNA or lesions in the DNA itself. Along with lesions, several other factors contribute to replication-associated rearrangements, including repeat sequences prone to homology-driven recombination. Repeat sequences are present in all genomes; for example, the human genome contains >1,000,000 Alu sequences that encompass ∼10% of the total DNA (8). Repeats can lead to instability; studies in bacteria (8), yeast (9, 10), and mouse cells (11) indicate that repeats lying in inverted orientation are unstable and mediate large-scale chromosomal changes. A third contributor to replication-associated rearrangements is regulatory proteins, which play a plethora of roles that are still being defined (12). Many regulators stabilize replication forks, preventing them from undergoing error. Examining the roles of inverted repeats and regulators of replication fork biology is the focus of the present study.
We previously described a system in Saccharomyces cerevisiae in which nearby inverted repeats fuse to form dicentric chromosomes, which then cause chromosome-wide instability (10). Dicentric chromosomes are inherently unstable (13). We showed that fusion of inverted repeats is general in the yeast genome (10). The inverted repeat sequences that fuse share as little as 20 bp of incomplete homology and are separated by up to 5 kb of DNA. Fusion appears to occur as a consequence of an error in DNA replication (9, 10). In our previous study, we also found several major DNA repair and recombination pathways prevent inverted repeat fusion (10). Here we report a study of how proteins in DNA checkpoint pathways that stabilize replication forks and recruit repair proteins also regulate inverted repeat fusion. Using a genetic approach, along with a key insight from a previous study (14), we identify proteins that prevent fusion of inverted repeats and place them into distinct pathways. We also provide evidence that local, as opposed to global, Rad53 activity appears to be sufficient to prevent inverted repeat fusion, presumably by stabilizing single replication forks.
Results
Chr VII Disome System.
We previously developed a genetic system that allows us to evaluate the role of regulators in the fusion of inverted repeats to form dicentric chromosomes (9). We can also follow the fate of the dicentrics (Fig. 1). In brief, we created a haploid yeast strain that contains an extra copy of chromosome VII (ChrVII). The CAN1 gene is present near the left telomere of this extra chromosome. Cells that retain CAN1 die when grown in the presence of the drug canavanine; thus, selection for loss of CAN1 allows us to identify cells (CanR cells) that have undergone any chromosomal change that includes loss of CAN1. Using appropriate genetic markers (Fig. 1), we identified three types of CanR colonies. We identified colonies that had suffered chromosome loss and others that experienced allelic recombination. These colonies are round on selective plates, and most of the cells in a single colony have the same genotype, as might be expected (Fig. 1A). The third type of CanR colony is unusual in two respects: The colonies have a strikingly “sectored” appearance, and the cells in a single colony have multiple genotypes. We refer to these as “mixed colonies,” because they contain cells of different, or mixed, genotypes (SI Materials and Methods). We previously demonstrated that these mixed colonies arise from cells with unstable dicentric chromosomes (10). We suggested that any of several inverted repeats might fuse to form dicentrics, although we have identified only one dicentric thus far, the product of fusion between two repeats, termed S2 and S3 (Fig. 1B). Once formed, in each cell division dicentrics either duplicate faithfully or are resolved by chromosome loss, by allelic recombination, or by a nonreciprocal translocation (forming the D7–D11 translocation shown in Fig. 1B).
Fig. 1.

Chromosome system to detect instability. (A) Two homologs of ChrVII and mutant alleles on each allow for genetic detection of chromosome changes. The CAN1 gene has been removed from ChrV and inserted into one copy of ChrVII. Selection for the loss of the CAN1 gene allows the growth of cells with any of three types of chromosome changes, including simple loss, allelic recombinants, and mixed colonies. Mixed colonies contain cells of multiple genotypes, including a specific translocation. See SI Materials and Methods for details. (B) Configuration of elements in the ChrVII403 site and the geometry and order of how fusion might occur. Two tRNA genes (pentagons) transcribe toward the oncoming fork and slow replication. Fusion between the two LTR σ repeats (S2 and S3), shown diagrammatically, forms a dicentric, followed by recombination between the two LTR δ sequences (D7 and D11) to form the specific translocation. (Reprinted from reference 10. Copyright © 2009 Cold Spring Harbor Laboratory Press.)
To determine the mechanisms that affect the formation and fates of dicentric chromosomes, we introduced mutations of DNA damage regulatory genes into the ChrVII disome strain. We tested two separate mutant isolates for each gene and found concordant results for these pairs of mutant strains in all cases. For each mutant strain, we determined genetically the frequency of chromosome loss, allelic recombination, and mixed colonies. We also determined molecularly, using PCR assays (see below), the frequency of formation of one specific dicentric chromosome and of the one specific translocation that forms from that specific dicentric. For the molecular assay of dicentrics, we perform qualitative or quantitative assay, asking how frequently dicentrics arise in cultures of unselected cells (see SI Materials and Methods for details). We report the average of six independent cultures of mutant cells. For the molecular assay of the translocation, we isolated genomic DNA from cells in six mixed colonies and performed a qualitative PCR assay to determine whether the translocation was present in cells in each of the six mixed colonies. All of the mutants reported herein are proficient for formation of the specific translocation, so we focus here on mechanisms involved in inverted repeat fusion to form dicentrics. Taken together, these genetic and molecular analyses allow us to identify the roles of genes in either preventing or assisting in the fusion of inverted repeats.
Mec1 and Tel1 Prevent Nearby Inverted Repeat Fusion.
We begin this analysis with the phosphotidyl inositol kinase-like kinases Mec1(ATR) and Tel1(ATM). Mec1 and Tel1 are master regulators of multiple checkpoint pathways that respond to stalled replication forks or DNA damage and act to stabilize forks and facilitate repair (12). We previously reported a high frequency of mixed colonies in mec1 mutants compared with WT cells (9). All strains carrying mec1 or rad53 mutations (discussed below) also contain an sml1 mutation that is necessary for the survival of these mutants. Deletion of sml1 caused no detectable phenotype in our instability assay (9) (Table 1). We analyzed the role of Mec1 in the fusion of inverted repeats and instability in more detail, and found that the formation of the dicentric chromosome was much more frequent (up to 500-fold) in mec1 sml1 cells compared with WT or sml1 cells, and that the resultant translocation was readily formed in a mec1 mutant (Table 1). Based on our quantitative PCR results, we estimate that dicentrics are formed in 1 in 100,000 cells in WT and in nearly 1 in 200 cells in mec1 mutants.
Table 1.
Role of checkpoint regulators in chromosome rearrangements
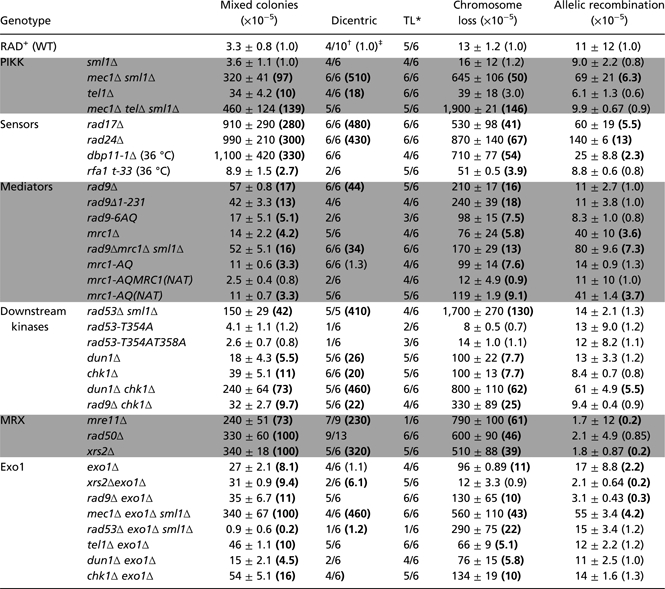
Frequencies of mixed colony, allelic, and chromosome loss were determined from 12 independent colonies. SD and fold-change compared to WT are shown. Statistically significant in bold P < 0.01, Kruskal-Wallis analysis.
*TL indicates D7-D11 translocation.
†Result of qualitative PCR analysis. Shown is the ratio of dicentric positive to total number of colonies tested.
‡Indicates fold changes in the frequency of dicentric compared to WT. Dicentric level of 1 corresponds to frequency of 1 in 100,000. (n = 6)
We also found that the Tel1 PIKK has a more minor, yet readily detectable, role in preventing mixed-colony formation; a tel1 mutant had a 10-fold higher frequency of mixed colonies compared with WT cells. The frequencies of allelic recombination and chromosome loss were unchanged compared with WT (Table 1). We also created a mec1 tel1 double mutant, which demonstrated no synergistic affect; it had a similarly high level of instability as a mec1 mutant (not synergistic; Table 1). We suggest that both Mec1 and Tel1 are acting on replication forks to suppress instability, although we cannot rule out the formal possibility that instability in these mutants might involve telomere defects (Discussion).
Checkpoint Sensors Prevent Inverted Repeat Fusion.
We next analyzed checkpoint sensors needed to activate Mec1 and Tel1 kinases. The activity of Mec1 is dependent on RPA-coated single-stranded DNA, the checkpoint clamp loader (Rad24/RFC complex), the checkpoint sliding clamp (Rad17-Mec3-Ddc1), and Dpb11 (TopBP1) (15, 16). We evaluated the roles of these Mec1-activating regulators in preventing nearby inverted repeat fusion. We found that rad17 and rad24 single mutants have similar phenotypes, both with extremely high levels of nearby inverted repeat fusion (∼300-fold higher than in WT; Table 1). To evaluate the role of Dpb11, we used a thermosensitive Dpb11 allele, dpb11-1, which fails to interact with the Rad17-Mec3-Ddc1 sliding clamp (17). We found that when grown at a maximum permissive temperature of 36 °C, dbp11-1 mutants were as unstable as rad17 and rad24 mutants and readily formed dicentric chromosomes (Table 1). The instability of these sensor mutants even exceeded that of mec1. Finally, we tested the roles of replication- or recombination-defective RPA alleles (18) and found, surprisingly, that these alleles had no affect on inverted repeat fusion (Table 1 and Table S1). A simple possible explanation for the characteristics of this phenotype is that the level of RPA activity in these alleles might be sufficient to support the activity of other regulators (e.g., Mec1) that suppress inverted repeat fusion.
Taken together, these results are consistent with the general view that Mec1, Tel1, the Rad17-Mec3-Ddc1 sliding clamp complex and its Rad24-dependent clamp loader, and Dpb11 (but not RPA) play major roles in preventing the fusion of nearby inverted repeats and the formation of dicentrics. These proteins likely have central functions in replication fork stability, thus explaining why inverted fusion readily occurs in their absence.
Rad53 Prevents Inverted Repeat Fusion and Does So Independently of Rad9 and Mrc1.
One of the key downstream components of the replication stress signal transduction pathway is the effector kinase Rad53 (19–21). Activation of Rad53 depends mainly on the Mec1/Ddc2 complex and on Rad9 and Mrc1 mediators (12). We first evaluated the role of the Rad53 kinase in suppressing inverted repeat fusion and found that rad53 mutants had extremely high levels of dicentric products and mixed colonies (Table 1).
We next evaluated the roles of Rad9 and Mrc1, which are required for genome-wide activation of Rad53 and assist in its autoactivation (22–25). As we reported previously (9), inactivation of Rad9 or Mrc1 alone leads to a modest level of instability that is much lower than that of rad53 nulls (Table 1). The lesser roles of Rad9 and Mrc1 compared with Rad53 has one of two possible explanations: Either Rad53 acts independently of Rad9 and Mrc1 to prevent inverted repeat fusion, or Rad9 and Mrc1 are redundant for Rad53 activation. To test for functional redundancy, we constructed a rad9mrc1sml1 triple mutant and, surprisingly, found a similar level of instability as in a rad9 single mutant or a rad9sml1 double mutant (Table 1 and Table S1). The frequency of the dicentric chromosome was the same in the rad9 mrc1 sml1 mutant and the rad9sml1 double mutant. This suggests that Rad53’s roles in stabilizing single stalled replication forks is largely independent of Rad9 and Mrc1 (Discussion).
To further test the role of Rad53 in suppressing inverted repeat fusion, we used two alleles of Rad53 that are defective for autophosphorylation and global checkpoint response (26). Normally, both Rad53 and Rad9 are phosphorylated by Mec1, and the phosphorylated forms of Rad53 and Rad9 interact, resulting in autophosphorylation of Rad53 and induction of a global checkpoint response. Two previously characterized alleles of rad53—contain mutations in threonines whose autophosphorylation is required for checkpoint signal amplification (26). We introduced both of these rad53 hypomorphic alleles into our ChrVII disome and found that both alleles conferred the expected damage sensitivity to the disome. We then measured instability and found that both rad53 hypomorphic mutations suppressed inverted repeat fusion and instability as effectively as did RAD53 (WT) cells (Table 1). In addition, we also tested an allele of Rad9, rad9-6AQ, that is defective for Rad53 binding (27). We found that the rad9-6AQ allele had a low level of instability, ∼3-fold more stable than a rad9 null mutant (Table 1). The low instability of rad9-6AQ and rad53 nulls indicates that Rad9 might account for at most 10% of the activity of Rad53 in preventing inverted repeat fusion. These data suggest that Rad9 and Mrc1 do not contribute much to the activity of Rad53 in preventing inverted repeat fusion, and that only a low level of Rad53 activity is needed to prevent inverted repeat fusion (Discussion).
Roles of Rad9 and Mrc1 in Instability.
As stated above, it appears that Rad9 and Mrc1 do not regulate Rad53 in preventing inverted repeat fusion and instability in our system. What are the roles of Rad9 and Mrc1, then? We address the role of Rad9 below in the discussion of the Chk1 effector kinase. In addition to regulating Rad53 and checkpoint signaling, Mrc1 is known to play a role in the normal progression of the replisome. To investigate whether the modest level of instability in a mrc1 single mutant is due to defects in replisome progression, we used the mrc1-AQ allele, a separation-of-function allele of Mrc1 constructed by Osborn and Elledge (28) that is replication-proficient but checkpoint-defective. We found that the checkpoint-defective mrc1-AQ allele mutant was as unstable as a mrc1 mutant (Table 1), suggesting that Mrc1’s role in the suppression of inverted repeat fusion and instability is not linked to its role in DNA replication fork progression. Consistent with this view, we found that a mutation in another fork progression mediator, Tof1, also had no detectable change in the frequency of mixed colonies compared with WT cells (Table S1). We do not know why mrc1 mutants are unstable. We argue below that Rad9 suppresses inverted repeat fusion mainly through Chk1 regulation.
Chk1 and Dun1 Kinases.
Mec1 and Rad53 kinases activate two downstream effector kinases, Chk1 and Dun1. We found that cells harboring a chk1 mutation have elevated instability, comparable to that of rad9 single mutants (Table 1). Interestingly, the level of instability was similar in a rad9 chk1 double mutant and a rad9 single mutant, suggesting that Rad9 acts in the same pathway as Chk1 to suppress inverted repeat fusion. Previously, Blankley and Lydall reported that a specific domain of Rad9 is required for activation of the Chk1 kinase (29). To directly test the hypothesis that instability in rad9 mutants is due to its failure to regulate Chk1, we tested the instability of a rad9Δ1-231 allele that is defective for Chk1 but proficient for Rad53 activation, and found that it had comparable instability to that of rad9 null mutants. Together with the results for Rad53 presented above, this suggests that Rad9 acts on Chk1, not on Rad53, to suppress inverted repeat fusion through fork stability.
A second protein kinase, Dun1, is regulated by Rad53 and functions in a separate pathway than Chk1 during the G2/M arrest after DNA damage (30). We introduced a dun1 mutation into our system and found that it led to a moderate increase in mixed colonies and dicentrics compared with WT cells (Table 1). We also found that a dun1 chk1 double mutant exhibited a synergistic increase in the frequency of mixed colonies compared with dun1 and chk1 single mutants. Thus, we conclude that Dun1 and Chk1 act in two separate pathways to regulate replication fork behavior and inverted repeat fusion. The actions of Dun1 and Chk1 in parallel to maintain fork stability remain unknown.
Checkpoint Genes Act in Two Pathways Distinguishable by the Function of Exo1.
Thus far, we have established that all checkpoint genes that we tested play some role in preventing nearby inverted repeat fusion. Our evaluation of Rad53, Rad9, Mrc1, Dun1, and Chk1 revealed to be least two apparent pathways. Dun1 and Chk1 are in separate pathways, as are Rad53 and Rad9. We cannot yet place Mrc1 in a specific pathway. In an attempt to obtain further evidence on pathways of regulation, we made use of recent insights from Segurado and Diffley (14). Those authors found that under certain conditions, replication fork instability in rad53 mutants was completely suppressed by an exo1 mutation. Exo1 is a 5′-3′ single-strand nuclease. Remarkably, they also found that deletion of exo1 was not sufficient to prevent replication fork breakdown in mec1 cells. Finally, they found that Chk1 acts in the Exo1-independent pathway (Fig. 2) and suggested that in absence of Rad53, Chk1 plays an important role in stabilizing stalled replication forks.
Fig. 2.

Pathways that prevent inverted repeat fusion. (A) Summary of different checkpoint pathways that prevent fusion of nearby inverted repeats and cell death. A stalled fork (purple cloud) leads to Mec1 activation and fork stabilization in Rad53-dependent (3) or -independent (1 and 2) manner. Mec1 acts via Rad9 and Chk1 to preserve stability (1). Rad53 and MRX prevent instability, and thus repeat fusion, by regulating the Exo1 nuclease (3 and 4). Rad53- and Exo1-mediated regulation of stability and prevention of inverted repeat fusion does not require Rad9 or Dun1 kinase. Rad53 signal amplification by Rad9 and Mrc1 is also dispensable for suppression of inverted repeat fusion. (B and C) Possible roles of Exo1. Faithful template switch to the newly replicated sister strand (B), and faulty template switch to a template that is made accessible by Exo1-dependent DNA degradation (C).
Consequently, we decided to test whether Exo1 might have a role in inverted repeat fusion and might further distinguish the Rad53 and Chk1 pathways that regulate inverted repeat fusion. We measured the instability occurring after the introduction of an exo1 mutant into cells defective for Rad53, Mec1, Chk1, Rad9, or Tel1. Remarkably, we found that the exo1 mutant completely suppressed the instability of a rad53 mutant, based on both measured levels of dicentric chromosomes and the frequency of mixed colonies (Table 1), and that the exo1 mutant demonstrated a modest increase in instability (Discussion). Also consistent with the findings of Seguardo and Diffley (14), we found that the exo1 mutation did not suppress the instability of a mec1 mutant (Table 1). The observation that exo1 suppressed the instability of rad53 but not that of mec1 suggests that Rad53 and Mec1 act in two pathways to suppress inverted repeat fusion and dicentric formation. We further investigated whether suppression by an exo1 mutation might also confirm our conclusion that Rad53 and Rad9 act in separate pathways, and found that an exo1 mutation did not suppress the instability of rad9 or chk1 mutants.
Taken together, these findings provide a coherent view of checkpoint protein pathways, inverted repeat fusion, and replication fork stability. Mec1 may regulate both the Rad53 and Chk1 pathways. Rad53 then acts in one pathway that inhibits Exo1, whereas Chk1 and Rad9 act in a second pathway independent of Exo1. Dun1 lies downstream of Rad53 in a pathway independent of Exo1. In the absence of Rad53 and Exo1, Dun1 must not play a very substantial role in preventing inverted repeat fusion. How checkpoint pathways and Exo1 might regulate inverted repeat fusion is addressed in Discussion.
Role of MRX and Exo1 in Inverted Repeat Fusion.
We previously reported that a mutation in Rad50, one of the subunits of the MRX(MRN) complex, resulted in a high level of inverted repeat fusion and instability, suggesting an interaction between MRX and replication forks (10). Previous studies also have suggested an association between Rad50 (and other subunits of the MRX complex, Mre11 and Xrs2) and replication forks (31). Here we extend our earlier study and show that mutations in any of the three components of MRX—Mre11, Rad50, and Xrs2— lead to very high levels of dicentric products (increased by as much as 320-fold in an xrs2 mutant) and, correspondingly, similar very high levels of mixed colonies (73-, 100-, and 100-fold increases in mixed colonies in rad50, mre11, and xrs2 mutants, respectively; Table 1). Note that because of a clerical error, in a previous paper we mistakenly reported only a 30-fold increase in rad50 mutant instability (10). The resolution of the dicentric to the translocation is evident in all MRX single mutants as well. We next examined whether an exo1 mutation might reveal whether MRX acts in the Mec1 or Rad53/Exo1 pathways. Given the previously reported association of Exo1 with MRX proteins, we conjectured that an exo1 mutation might suppress the instability of MRX mutants. We generated an xrs2 exo1 double mutant and found that the exo1 mutation indeed caused ∼90% suppression. We speculate that the MRX proteins somehow act in conjunction with Rad53 in inhibiting Exo1 to prevent inverted repeat fusion and instability. Because tel1 mutants also exhibit instability (Table 1), and because Tel1 interacts with MRX proteins (32), we examined whether tel1 lies in the Rad53/MRX/Exo1 pathway for suppression of inverted repeat fusion. We found that tel1 and tel1 exo1 mutants have similar levels of instability, and thus Tel1 acts differently than MRX in this reaction (Discussion).
Discussion
Inverted repeats are prominent in genomes and pose a threat to its stability. Herein, we have shown they can fuse to form dicentric chromosomes, which have long been known to be inherently unstable and have been linked to various disease states (33). Furthermore, significant links between checkpoint proteins and genome stability have been identified in human and other mammalian organisms, although the mechanistic details are poorly understood (34, 35). The current study provides a more complete view of how checkpoint proteins, nearby inverted repeats, and dicentrics are linked (9). We report two major findings here. First, two pathways prevent inverted repeat fusion, presumably through their regulation of fork stability (Fig. 2). One pathway contains Rad53 and MRX proteins, both of which inhibit Exo1, and the other contains Chk1 and Rad9 and is not affected by Exo1. (Formally, Mrc1 and Tel1 also may be in the Chk1/Rad9 pathway, although that assignment awaits corroborating studies.) Both pathways are regulated by Mec1. The second major finding, related to the first, is that only low-level, as opposed to global, activity of Rad53 is needed to prevent inverted repeat fusion, presumably through regulation of fork stability.
Implicit in the interpretation of our data, and in the discussion of mechanisms that follows, is the idea that inverted repeat fusion arises from unstable replication forks. It thus follows that mutants with high frequencies of inverted repeat fusion and instability might be defective in fork stability per se. Because checkpoint pathways have been shown to affect fork stability globally, we feel confident in attributing high frequencies of inverted repeat fusion and instability to a defect in fork stability. We have not yet directly shown that an unstable fork undergoes inverted repeat fusion, however. Thus, it remains a formal possibility that in some mutants, instability might arise from some other defect in DNA metabolism rather than from fork stability per se.
Two Pathways That Regulate Replication Fork Stability Prevent Inverted Repeat Fusion.
Based on our current findings and on previous work by others, we propose two pathways prevent nearby inverted repeat fusion through preservation of fork stability (Fig. 2). Our findings agree strikingly well with the recent report from Segurado and Diffley (14), who used a different method (i.e., density shift experiments) to examine fork stability after a genome-wide insult by DNA damage. They found that DNA replication fork instability in a rad53 mutant is dependent on Exo1, whereas fork stability in mec1 mutants is not dependent on Exo1. Based on these and other results, they suggested that Mec1 regulates Rad53 in an Exo1-dependent pathway, and that Chk1 in an Exo1-independent pathway. Following what might be a locus-specific (not genome-wide) defect, we found that both mec1 and rad53 mutants have high levels of inverted repeat fusion and chromosome instability, and that these phenotypes were completely suppressed by an exo1 mutation in rad53 mutants, but not in mec1 mutants. Below we provide a simple model for how Exo1 might permit inverted repeat fusion. Interestingly, Rad53 also regulates Exo1-mediated degradation at telomeres, amid a complex array of degradation activities (36); thus, checkpoint proteins regulate Exo1-mediated degradation associated with both replication forks and telomeres. Below we also provide a simple model for how Exo1-mediated degradation associated with replication forks might permit inverted repeat fusion.
Our genetic studies support and provide further definition to checkpoint pathways that regulate fork stability. First, our data suggest that Rad9 acts through Chk1 in a pathway that does not interact with Exo1 and is parallel to the Rad53 pathway. Second, we find that the instability of the dun1 mutant is not suppressed by an exo1 mutation. This places Dun1 downstream of Rad53 (based on an earlier observation that Rad53 regulates Dun1), although Dun1 does not regulate Exo1. Lastly, we found that instability in MRX mutants is also largely dependent on Exo1 (Table 1), and thus we propose that MRX also functions to stabilize replication forks via direct or indirect inhibition of Exo1.
Our model does not include five additional regulators that demonstrate different levels of instability. We found very high levels of instability in rad17, rad24, and dpb11 mutants. Rad17 and Rad24, as members of checkpoint clamp loader and sliding clamp, are required for the function of Mec1 and would be expected to have similar phenotypes as mec1 mutants, which they do. The pathways through which the corresponding proteins act remain to be tested, although, by inference, many appear to act at the level of Mec1 function or in the Chk1/Rad9 pathway. We found very low levels of instability in mrc1 and tel1 mutants; how these proteins prevent inverted repeat fusion is unclear.
Role of Exo1 in Replication Fork Stability and Inverted Repeat Fusion.
How Mec1, Rad53, Chk1, and MRX proteins actually act at a molecular level to moderate fork stability remains largely speculative. Previous studies have shown that in damage-treated cells, defects in Mec1 and Rad53 caused the dissociation of MCM helicases and DNA polymerases from stalled replication forks (37, 38), but whether these molecular events are relevant to fusion of inverted repeats is unclear.
Exo1 is another molecule associated with Rad53 that has an affect on instability. Exo1 was recently identified as a Rad53 phosphorylation target, and Rad53-dependent phosphorylation of Exo1 might be inhibitory (39). We also found that MRX proteins somehow inhibit Exo1. A molecular explanation for this inhibition is less clear; perhaps MRX proteins regulate the structure of a stalled fork that facilitates Exo1 activity (40).
So, given the strong phenotypes of exo1 mutation, what might be the connection among Exo1 activity, inverted repeat fusion, and dicentrics? We suggest Rad53 and MRX regulate Exo1-dependent degradation at stalled forks to allow for a proper template switch reaction to enable fork recovery. We suggest that when forks stall, modest degradation by Exo1 results in proper template choice (during template switching) and fork resumption (Fig. 2B). Which strands might need to be degraded to allow proper template switching is unclear. In contrast, improper and excessive degradation by Exo1 might result in an improper template choice (of, e.g., a nearby inverted repeat sequence; Fig. 2C). Perhaps when Exo1 is not inhibited, in rad53 or MRX mutants, Exo1 degrades a newly formed duplex on the lagging strand, providing an ssDNA template where the stalled leading strand primer might anneal. Another model, involving a “chicken-foot” regressed fork structure, is shown in Fig. S1.
The mammalian Exo1 might play a similar role in events associated with replication forks and genomic instability. Recent findings suggest that after DNA damage, human Exo1 is phosphorylated, which targets it for ubiquitination and degradation (41). Furthermore, recent studies of Exo1 knockout mice also suggest that the mammalian Exo1 protein plays an important role in mutation avoidance and tumor progression (42); in our system, exo1 mutations show a modest increase in instability.
Alternative Roles of MRX and Tel1 in Suppressing Inverted Repeat Fusion.
Recently, Doksani et al. (40) reported that a double-strand break could possibly cause regression of a nearby replication fork. They inferred that MRX and Tel1 proteins prevent fork regression at sites of double-strand breaks, and that the two proteins have slightly different roles given their different mutant phenotypes. In our assay, we found a moderate level of instability in tel1 mutant cells and high instability in MRX mutant cells. One possible explanation for this instability is that in MRX- or Tel1-defective cells, a replication fork that approaches a DNA lesion (i.e., a double-strand break or perhaps some “lesion,” such as torsional stress) might regress, as suggested by Doksani et al. (40), and then undergo a faulty template switch (Fig. S1).
Global Versus Local Checkpoint Activity and Roles of Rad53, Rad9, and Mrc1.
Global activation of Rad53 requires Rad9 and Mrc1. Interestingly, we found that rad9 mrc1 mutants are far more stable than rad53 mutants. This suggests that Rad53 activity at a stalled fork does not require the Rad9 and Mrc1 mediators. We interpret this result as reflecting “global” versus “local” activity of Rad53; the global activation requires amplification and thus the role played by Rad9 or Mrc1, whereas the local activity for Rad53 to stabilize single forks does not require amplification. In support of this model, we found no increase in inverted repeat fusion in Rad53 alleles harboring mutations in an autophosphorylation domain compared with WT cells. Thus, disruption of signal amplification and global checkpoint response in these rad53 mutants, as well as in rad9 mrc1 double mutants, does not hinder Rad53’s ability to prevent repeat fusion. Thus, we suggest that Rad53 has two activation states: a low-level state that acts locally to maintain replication fork structures, and a high-level state that causes cell-wide responses (e.g., cell cycle arrest, inhibition of late-origin firing). The potential benefit of Rad53 activity acting only locally might be to prevent unwanted checkpoint regulation of normal DNA replication processes occurring elsewhere in the cell.
Materials and Methods
The yeast strains used are derivatives of the A364a strain described previously (Table S2) (9, 10). The chromosome instability assay was performed as described previously (10). Reported frequencies were determined from analysis of at least six colonies per mutant isolate. Two independently derived isolates were tested for all mutants; the average and SDs are shown. Statistical analyses (shown in bold in Table 1) were done using the Kruskal-Wallis method (43). PCR-based molecular assays were performed to detect altered chromosome intermediates, dicentric chromosomes, and translocation, as described previously (9, 10). See SI Materials and Methods for details.
Supplementary Material
Acknowledgments
We thank Richard Kolodner, Hiroyuki Araki, and Kyungjae Myung for plasmids and members of the Weinert group, especially Andrew Paek, for helpful discussions. This work is supported by a grant from Native American Cancer Research Partnership/National Cancer Institute and National Institutes of Health Grants 1R01 GM076186 (to T.W.) and GM008659-11A1 (to S.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1001938107/-/DCSupplemental.
References
- 1.Youssoufian H, Pyeritz RE. Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet. 2002;3:748–758. doi: 10.1038/nrg906. [DOI] [PubMed] [Google Scholar]
- 2.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 3.Sharp AJ, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 4.Gisselsson D, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci USA. 2000;97:5357–5362. doi: 10.1073/pnas.090013497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myung K, Kolodner RD. Suppression of genome instability by redundant S-phase checkpoint pathways in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99:4500–4507. doi: 10.1073/pnas.062702199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemoine FJ, Degtyareva NP, Lobachev K, Petes TD. Chromosomal translocations in yeast induced by low levels of DNA polymerase: A model for chromosome-fragile sites. Cell. 2005;120:587–598. doi: 10.1016/j.cell.2004.12.039. [DOI] [PubMed] [Google Scholar]
- 7.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 8.Jurka J. Repeats in genomic DNA: Mining and meaning. Curr Opin Struct Biol. 1998;8:333–337. doi: 10.1016/s0959-440x(98)80067-5. [DOI] [PubMed] [Google Scholar]
- 9.Admire A, et al. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006;20:159–173. doi: 10.1101/gad.1392506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paek AL, et al. Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev. 2009;23:2861–2875. doi: 10.1101/gad.1862709. ( www.genesdev.org) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collick A, et al. Instability of long inverted repeats within mouse transgenes. EMBO. 1996;15:1163–1171. [PMC free article] [PubMed] [Google Scholar]
- 12.Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst) 2009;8:1038–1046. doi: 10.1016/j.dnarep.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 13.McClintock B. The stability of broken ends of chromosomes in Zea mays. Genetics. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Segurado M, Diffley JF. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008;22:1816–1827. doi: 10.1101/gad.477208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Majka J, Burgers PM. Clamping the Mec1/ATR checkpoint kinase into action. Cell Cycle. 2007;6:1157–1160. doi: 10.4161/cc.6.10.4221. [DOI] [PubMed] [Google Scholar]
- 16.Lydall D, Weinert T. G2/M checkpoint genes of Saccharomyces cerevisiae: Further evidence for roles in DNA replication and/or repair. Mol Gen Genet. 1997;256:638–651. doi: 10.1007/s004380050612. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Elledge SJ. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics. 2002;160:1295–1304. doi: 10.1093/genetics/160.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 1998;148:989–1005. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez Y, et al. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–360. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- 20.Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994;8:2401–2415. doi: 10.1101/gad.8.20.2401. [DOI] [PubMed] [Google Scholar]
- 21.Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 22.Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase epsilon form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- 23.Vialard JE, Gilbert CS, Green CM, Lowndes NF. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 1998;17:5679–5688. doi: 10.1093/emboj/17.19.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez Y, et al. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- 25.Alcasabas AA, et al. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- 26.Usui T, Petrini JH. The Saccharomyces cerevisiae 14-3-3 proteins Bmh1 and Bmh2 directly influence the DNA damage-dependent functions of Rad53. Proc Natl Acad Sci USA. 2007;104:2797–2802. doi: 10.1073/pnas.0611259104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz MF, et al. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell. 2002;9:1055–1065. doi: 10.1016/s1097-2765(02)00532-4. [DOI] [PubMed] [Google Scholar]
- 28.Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–1767. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blankley RT, Lydall D. A domain of Rad9 specifically required for activation of Chk1 in budding yeast. J Cell Sci. 2004;117:601–608. doi: 10.1242/jcs.00907. [DOI] [PubMed] [Google Scholar]
- 30.Gardner R, Putnam CW, Weinert T. RAD53, DUN1 and PDS1 define two parallel G2/M checkpoint pathways in budding yeast. EMBO J. 1999;18:3173–3185. doi: 10.1093/emboj/18.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tittel-Elmer M, Alabert C, Pasero P, Cobb JA. The MRX complex stabilizes the replisome independently of the S phase checkpoint during replication stress. EMBO J. 2009;28:1142–1156. doi: 10.1038/emboj.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakada D, Matsumoto K, Sugimoto K. ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev. 2003;17:1957–1962. doi: 10.1101/gad.1099003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka H, Yao MC. Palindromic gene amplification—an evolutionarily conserved role for DNA inverted repeats in the genome. Nat Rev Cancer. 2009;9:216–224. doi: 10.1038/nrc2591. [DOI] [PubMed] [Google Scholar]
- 34.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 35.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 36.Jia X, Weinert T, Lydall D. Mec1 and Rad53 inhibit formation of single-stranded DNA at telomeres of Saccharomyces cerevisiae cdc13-1 mutants. Genetics. 2004;166:753–764. doi: 10.1534/genetics.166.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cobb JA, et al. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005;19:3055–3069. doi: 10.1101/gad.361805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucca C, et al. Checkpoint-mediated control of replisome–fork association and signalling in response to replication pausing. Oncogene. 2004;23:1206–1213. doi: 10.1038/sj.onc.1207199. [DOI] [PubMed] [Google Scholar]
- 39.Morin I, et al. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008;27:2400–2410. doi: 10.1038/emboj.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doksani Y, et al. Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell. 2009;17(137):247–258. doi: 10.1016/j.cell.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Shemerly M, Janscak P, Hess D, Jiricny J, Ferrari S. Degradation of human exonuclease 1b upon DNA synthesis inhibition. Cancer Res. 2005;65:3604–3609. doi: 10.1158/0008-5472.CAN-04-4069. [DOI] [PubMed] [Google Scholar]
- 42.Wei K, et al. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003;17:603–614. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583–621. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.