

Abstract
Recent laboratory studies have demonstrated that isoprene oxidation products can partition to atmospheric aerosols by reacting with condensed phase sulfuric acid, forming low-volatility organosulfate compounds. We have identified organosulfate compounds in free tropospheric aerosols by single particle mass spectrometry during several airborne field campaigns. One of these organosulfates is identified as the sulfate ester of IEPOX, a second generation oxidation product of isoprene. The patterns of IEPOX sulfate ester in ambient data generally followed the aerosol acidity and NOx dependence established by laboratory studies. Detection of the IEPOX sulfate ester was most sensitive using reduced ionization laser power, when it was observed in up to 80% of particles in the tropical free troposphere. Based on laboratory mass calibrations, IEPOX added > 0.4% to tropospheric aerosol mass in the remote tropics and up to 20% in regions downwind of isoprene sources. In the southeastern United States, when acidic aerosol was exposed to fresh isoprene emissions, accumulation of IEPOX increased aerosol mass by up to 3%. The IEPOX sulfate ester is therefore one of the most abundant single organic compounds measured in atmospheric aerosol. Our data show that acidity-dependent IEPOX uptake is a mechanism by which anthropogenic SO2 and marine dimethyl sulfide emissions generate secondary biogenic aerosol mass throughout the troposphere.
Keywords: acid-catalyzed particle phase reactions, epoxides, free troposphere, secondary organic aerosol
Organic compounds constitute a significant fraction of global aerosol mass in the troposphere (1–5). Much of this organic material is thought to be of biogenic origin, especially in heavily vegetated areas such as the southeastern United States (6) and the Amazon basin (7). Recent analyses have shown that conversion of biogenic volatile organic compounds (VOCs) to aerosol material can be promoted by anthropogenic pollution (6, 8), and chamber studies have revealed a number of biogenic aerosol formation mechanisms in both polluted and clean environments. Nevertheless, models based on aerosol yields from oxidation of biogenic compounds in chamber experiments significantly under predict observed aerosol mass in the troposphere (9, 10).
Oxidation products from isoprene have been shown to be significant contributors to organic aerosol mass globally (11–14). The yield of secondary organic aerosol (SOA) mass from isoprene oxidation is strongly dependent on the concentration of NOx during gas-phase reaction and the acidity of the aerosol phase (15–17). Ambient observations and chamber experiments give clear evidence that isoprene oxidation products form organosulfate compounds in the aerosol phase (18–20). Paulot et al. (21) demonstrated that isoprene photooxidation under low-NOx conditions can generate large concentrations of gas-phase epoxydiols that then react with acidic sulfate aerosols to form organosulfates (22). The increase in aerosol mass due to epoxydiol uptake was quantified in a chamber study (22), and detailed reaction kinetics have recently been reported (23–25).
In this study, we report airborne measurements of aerosol organosulfates. Using the National Oceanic and Atmospheric Administration (NOAA) Particle Ablation by Laser Mass Spectrometry (PALMS) single particle mass spectrometer, we have identified an organosulfate compound formed from reactive uptake of IEPOX (2,3-epoxy-2-methyl-1,4-butanediol, C5H10O3), a gas-phase epoxydiol generated at high yield from isoprene oxidation (21). The PALMS instrument is sensitive to this compound and does not suffer from the nearly complete degradation of organosulfates observed in other online aerosol mass spectrometers (26). Observations of the IEPOX sulfate ester product are discussed for a variety of tropospheric environments, ranging from the Arctic to the tropics and from ground level to the stratosphere. By calibrating the PALMS organosulfate signal to aerosol growth in controlled reactive uptake experiments, the contribution of this isoprene-SOA pathway to tropospheric aerosol mass is determined.
Results
Laboratory Calibrations.
Laboratory experiments were performed to identify and calibrate epoxydiol product signatures in particle mass spectra. We measured the reactive uptake of synthesized BEPOX (2,3-epoxy-1,4-butanediol, C4H8O2), a compound nearly identical to the isoprene-derived β-IEPOX but without the methyl group alpha to the epoxide ring (SI Text and Fig. S1). BEPOX vapor was mixed with nebulized aerosols containing either acidic or neutralized sulfate in a glass reaction vessel at low relative humidity. Aerosol particles exiting the vessel were monitored by PALMS and a white light optical particle counter (OPC).
BEPOX reaction with acidic seed aerosols resulted in accumulation of reaction products in the aerosol phase. At the highest BEPOX concentrations, the particle mode size shifted from 700 to 895 nm, doubling the original aerosol volume (Fig. 1A). Because seed aerosols were not pure sulfuric acid, this observation suggests that epoxide uptake is not always sulfuric acid-limited. Acid is necessary for epoxide ring opening, and aqueous sulfate is lost through formation of the sulfate ester. However, further uptake of BEPOX can proceed via oligomer formation, which only requires an acid catalyst and does not consume sulfate (22). BEPOX may also condense directly onto aerosols with an aqueous layer and/or those already rich in BEPOX products (22, 27). Neutralized seed aerosols showed no detectable evidence of BEPOX reaction for exposure times up to 200 s at the highest BEPOX concentrations (Fig. 1B).
Fig. 1.

Aerosol size distributions of BEPOX uptake experiments. (A) Reactive uptake of BEPOX onto 700-nm acidic sulfate aerosols. (B) Negligible (< 1%) partitioning to neutralized aerosol, (NH4)2SO4, under the highest BEPOX concentrations. The dashed line is the unreacted seed aerosol, and colors correspond to different BEPOX exposures (sample temperature, % of total flow sent through sample, reaction time).
A PALMS negative ion mass spectrum of an acidic sulfate aerosol particle that reacted with gas-phase BEPOX is shown in Fig. S2. The major reaction product occurs at a mass-to-charge ratio (m/z) of 201. Isotopic signatures and a high accuracy mass analysis (see next section) confirm that this ion is composed of organic carbon and one sulfur atom and is consistent with the empirical formula 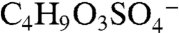 . This ion was positively identified under similar reaction conditions in other mass spectral studies (22) as the sulfate ester product of BEPOX and sulfuric acid,
. This ion was positively identified under similar reaction conditions in other mass spectral studies (22) as the sulfate ester product of BEPOX and sulfuric acid, 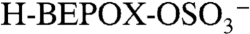 . Based on this evidence, we identify the PALMS m/z 201 signal as BEPOX sulfate ester monomer. A possible alternative assignment for m/z 201 is the ion cluster,
. Based on this evidence, we identify the PALMS m/z 201 signal as BEPOX sulfate ester monomer. A possible alternative assignment for m/z 201 is the ion cluster, 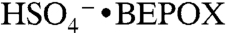 , but formation of this ion is not probable because the
, but formation of this ion is not probable because the 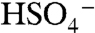 ion and organic species do not cluster efficiently in the instrument. Accordingly, the 201 ion was not observed in particles composed of equal amounts ammonium sulfate and BEPOX. A substantial fraction of the sulfate ester ion fragments into nonspecific organic carbon (
ion and organic species do not cluster efficiently in the instrument. Accordingly, the 201 ion was not observed in particles composed of equal amounts ammonium sulfate and BEPOX. A substantial fraction of the sulfate ester ion fragments into nonspecific organic carbon (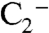 and C2H-) and sulfate (
and C2H-) and sulfate (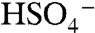 ) signals. Positive ion spectra were rich in peaks up to m/z 150, with additional peaks at higher masses for particles with BEPOX mass loadings above 30%. This spectral pattern is commonly observed for aerosols with high polymeric character such as polystyrene latex spheres. Also observed beginning at 18% mass were the sulfated BEPOX dimer (22) at m/z 305 and the
) signals. Positive ion spectra were rich in peaks up to m/z 150, with additional peaks at higher masses for particles with BEPOX mass loadings above 30%. This spectral pattern is commonly observed for aerosols with high polymeric character such as polystyrene latex spheres. Also observed beginning at 18% mass were the sulfated BEPOX dimer (22) at m/z 305 and the 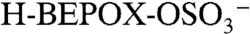 ion with an additional sulfuric acid at m/z 299, which is an ion cluster analogous to
ion with an additional sulfuric acid at m/z 299, which is an ion cluster analogous to 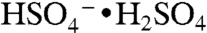 . Minor components in the mass spectrum are further discussed in SI Text.
. Minor components in the mass spectrum are further discussed in SI Text.
The PALMS BEPOX sulfate ester signal at negative m/z 201 was calibrated to the mass of BEPOX that accumulated on acidic aerosols. Calibrations were performed for both high and low ionization laser power spectra (Fig. S3). PALMS has a higher sensitivity to this compound than many organic species because the sulfate group forms a very stable negative ion. At reduced laser powers, fragmentation was less severe, giving a lower mass detection limit on a per particle basis. At any given sulfate ester mass loading, the greater sensitivity in low power mode will increase the fraction of particles with a detectable BEPOX sulfate ester signal. For PALMS to detect a signal using high (low) laser power, the particle must contain approximately > 0.1%( > 0.01%) BEPOX by mass. For the remainder of the article we report the aerosol mass attributed to the epoxides BEPOX and IEPOX based on the abundance of the respective epoxide sulfate ester signals at m/z = 201 or 215.
Identification of IEPOX Products in Atmospheric Particles.
Fig. 2A is a mass spectrum of a 1.1 μm diameter particle recorded at 3 km altitude showing an organosulfate signature at m/z 215. Several lines of evidence support the identification of this peak as the molecular ion of the sulfate ester product of isoprene-derived IEPOX and sulfuric acid, 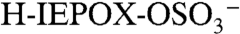 (IEPOX = β-IEPOX + δ-IEPOX) (21). Ambient particles with m/z 215 have many of the same spectral patterns and fragments as the laboratory BEPOX product spectra. The m/z 215 signal is more frequently detected in regions of biogenic VOC emissions, high acidity, and low NOx as discussed further below. This ion has never been generated in the laboratory by mixtures of sulfate, nitrate, and dozens of organic species, indicating that it results from unique chemistry and is not simply a general marker for large organic molecules. Finally, a high accuracy mass analysis was performed to identify the atomic components of m/z 215 and other peaks in PALMS mass spectra (Fig. 2B). Noninteger mass defects for each ion were determined and then averaged over many spectra (see Fig. S4 for details). The IEPOX sulfate ester (
(IEPOX = β-IEPOX + δ-IEPOX) (21). Ambient particles with m/z 215 have many of the same spectral patterns and fragments as the laboratory BEPOX product spectra. The m/z 215 signal is more frequently detected in regions of biogenic VOC emissions, high acidity, and low NOx as discussed further below. This ion has never been generated in the laboratory by mixtures of sulfate, nitrate, and dozens of organic species, indicating that it results from unique chemistry and is not simply a general marker for large organic molecules. Finally, a high accuracy mass analysis was performed to identify the atomic components of m/z 215 and other peaks in PALMS mass spectra (Fig. 2B). Noninteger mass defects for each ion were determined and then averaged over many spectra (see Fig. S4 for details). The IEPOX sulfate ester (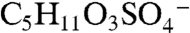 ) would have a mass defect of +0.025. The other likely candidate ion at m/z 215 is
) would have a mass defect of +0.025. The other likely candidate ion at m/z 215 is 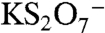 , which has a much lower mass defect of -0.119 due to the potassium atom. Observed mass defects of +0.027 to +0.041 in ambient spectra indicate that the 215 ion does not contain potassium and is instead abundant in oxygen and hydrogen atoms. The high accuracy mass analysis confirms the identification of m/z 201 as the BEPOX sulfate ester in laboratory studies and m/z 215 as the IEPOX sulfate ester in tropospheric aerosols.
, which has a much lower mass defect of -0.119 due to the potassium atom. Observed mass defects of +0.027 to +0.041 in ambient spectra indicate that the 215 ion does not contain potassium and is instead abundant in oxygen and hydrogen atoms. The high accuracy mass analysis confirms the identification of m/z 201 as the BEPOX sulfate ester in laboratory studies and m/z 215 as the IEPOX sulfate ester in tropospheric aerosols.
Fig. 2.

Organosulfate species in single particle mass spectra. (A) Ambient particle spectrum from the TC4 campaign recorded at 3 km altitude over Central America showing prominent IEPOX sulfate ester monomer and dimer signals. The * indicates masses that in MS/MS studies corresponded to organosulfates from isoprene (18, 19). (B) Chemical identification of BEPOX and IEPOX uptake products and other organosulfate ions in negative ion mass spectra using a high accuracy mass analysis (Fig. S4). Ions are identified by comparing exact mass defects for candidate species (black bars) with values derived from laboratory and free tropospheric aerosol spectra.
Other Organosulfate Signatures in Ambient Particle Spectra.
Several other peaks observed in ambient mass spectra may indicate isoprene-derived organosulfate species. Peaks marked with a star in Fig. 2A correspond to masses where isoprene-derived organosulfate compounds were observed together with the IEPOX sulfate ester using offline mass spectrometry techniques in previous chamber studies and ground level samples (18, 19). Some peaks are observed by PALMS in both the laboratory BEPOX experiments and ambient aerosols (e.g., m/z 139 and 169). High accuracy mass analysis suggests the empirical formulas 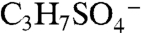 and
and 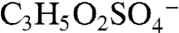 for 139, and
for 139, and 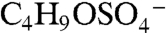 and
and 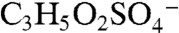 for 169. The former correspond to probable fragmentation products of BEPOX sulfate ester and organosulfates of similar structure, and the latter are consistent with the sulfate esters of glycolaldehyde and methylglyoxal (18, 19).
for 169. The former correspond to probable fragmentation products of BEPOX sulfate ester and organosulfates of similar structure, and the latter are consistent with the sulfate esters of glycolaldehyde and methylglyoxal (18, 19).
The m/z 155 negative ion is a common but unidentified peak in ambient aerosol spectra. In the atmosphere 155 is mostly observed in aerosols containing acidic sulfate, and isotope signatures indicate the presence of sulfur. Its presence correlates with the molecular ion of IEPOX sulfate ester, but it is not produced from BEPOX laboratory spectra. Fragmentation of the m/z 215 IEPOX sulfate ester at the center of the carbon chain could potentially form the 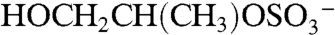 ion at m/z 155 (empirical formula
ion at m/z 155 (empirical formula 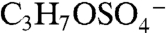 ). The same fragmentation of the BEPOX sulfate ester would form an ion at m/z 141, but this ion is not observed in BEPOX spectra. High accuracy mass analysis constrains the 155 ion to
). The same fragmentation of the BEPOX sulfate ester would form an ion at m/z 141, but this ion is not observed in BEPOX spectra. High accuracy mass analysis constrains the 155 ion to 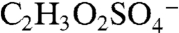 rather than the IEPOX fragment. Therefore, the 155 peak may be due to glyoxal sulfate (18, 20) or glycolic acid sulfate (28). These species have been previously detected during isoprene oxidation under high NOx conditions in the presence of acidic aerosol (18) and in neutralized aerosol mixed with glyoxal under UV light (28). This ion was not observed by PALMS in laboratory aerosol generated from mixtures of acidic sulfate and glyoxal. We conclude that the m/z 155 peak is not directly related to IEPOX sulfate ester but appears to be the result of parallel chemistry involving production of organosulfates either from other biogenic VOCs or from isoprene but by different reaction pathways than those that produce IEPOX.
rather than the IEPOX fragment. Therefore, the 155 peak may be due to glyoxal sulfate (18, 20) or glycolic acid sulfate (28). These species have been previously detected during isoprene oxidation under high NOx conditions in the presence of acidic aerosol (18) and in neutralized aerosol mixed with glyoxal under UV light (28). This ion was not observed by PALMS in laboratory aerosol generated from mixtures of acidic sulfate and glyoxal. We conclude that the m/z 155 peak is not directly related to IEPOX sulfate ester but appears to be the result of parallel chemistry involving production of organosulfates either from other biogenic VOCs or from isoprene but by different reaction pathways than those that produce IEPOX.
Atmospheric Measurements and Implications
Distributions of Isoprene Oxidation Products in Tropospheric Aerosol.
In this section we discuss observations of the isoprene-derived IEPOX sulfate ester in atmospheric particles during several aircraft campaigns. Aerosol particles with IEPOX sulfate ester were identified by spectra where the m/z 215 peak was detected above the noise level and was not obscured by interferences and neighboring peaks. Only particles classified as internal mixtures of sulfate and organic carbon (typically the predominant aerosol type) were investigated for the IEPOX sulfate ester signatures because other particle types such as sea salt and mineral dust often have many interfering ion peaks at high masses.
We concentrate our analysis on two regions. First, during the summer 2004 ITCT/NEAQS campaign (http://www.esrl.noaa.gov/csd/2004/) the NOAA P-3 aircraft probed tropospheric air over the eastern United States downwind of urban and industrial pollution sources as well as regions with strong biogenic VOC emissions. These data include simultaneous measurements of gas-phase isoprene and other relevant species. Second, tropical aerosols were measured during three aircraft campaigns based in Costa Rica (http://www.espo.nasa.gov/missions.php): Pre-AVE (Northern Hemisphere, winter 2004) and CR-AVE (winter 2006), sampling up to 19 km aboard the NASA WB-57, and TC4 (summer 2007), up to 13 km altitude aboard the NASA DC-8.
Southeastern United States.
Fig. 3A is a flight track map showing the frequency of IEPOX sulfate ester occurrence in PALMS particle mass spectra during the NEAQS campaign. The IEPOX sulfate ester was detected in 0.6% of particles throughout the mission and was evenly distributed among urban outflow, remote continental, and offshore regions. This signature was not enhanced during the SOA formation events downwind of urban areas documented previously (29, 30). By far the highest abundance (4.8% of particles at high laser power) was during the southern half of a flight from Florida to New Hampshire (July 5, 2004) where the aircraft flew downwind of biogenic VOC sources and anthropogenic SO2 emissions in the southeastern United States.
Fig. 3.

IEPOX sulfate ester levels during the ITCT/NEAQS aircraft campaign, summer 2004. (A) Flight track map of the NOAA P-3, where points are 3 min averages, size indicates frequency of IEPOX sulfate ester detected in single particle mass spectra, and color indicates aircraft altitude. Further details of flight segments 1, 2, and 3 are given in Fig. S5. (B) Correlation between IEPOX sulfate ester in single particles and gas-phase methyl vinyl ketone (MVK) + methacrolein for acidic and neutralized aerosols at two NOx levels. Points are averages of 15–250 particle spectra.
The relationship between isoprene emissions, aerosol acidity, and IEPOX sulfate ester abundance is generalized in Fig. 3B. When concentrations of gas-phase methyl vinyl ketone and methacrolein (two principal oxidation products of isoprene) exceeded 1.2 ppbv under very low NOx conditions, observations of IEPOX sulfate ester in acidic sulfate aerosols increased sharply. Under the highest isoprene influence, more than 70% of acidic aerosol particles contained detectable amounts of IEPOX sulfate ester. Higher NOx levels shift isoprene oxidation branching ratios away from IEPOX (21), and in controlled chamber studies the IEPOX sulfate ester was not detected in aerosols under high NOx conditions (16, 22). This NOx dependence is observed here in the ambient particle data: For NOx > 1 ppbv, the IEPOX sulfate ester was not observed until MVK + methacrolein levels exceeded 2 ppbv. Aerosols with neutralized sulfate did not contain IEPOX sulfate ester under any precursor loading. Fig. 3B agrees with previous laboratory and ground level studies, demonstrating that both aerosol chemical composition and pollution levels are critical to tropospheric aerosol mass production via the IEPOX pathway.
Tropics.
The vertical distribution of the IEPOX sulfate ester in the tropical atmosphere is shown in Fig. 4A. Of the three tropical campaigns presented here, TC4 spanned the broadest geographic coverage, including the Caribbean and Pacific sides of Central America and both low and high altitude segments over the Amazon jungle. As a TC4 mission average, the IEPOX sulfate ester was detected in 0.5% of tropospheric aerosols using full laser power, similar to the average detection frequency over North America. IEPOX sulfate esters were most abundant directly over and downwind of Central and South American and were least abundant in the Caribbean upwind of continental regions. During TC4, mass spectra were also recorded using reduced ionization laser power, where PALMS is significantly more sensitive to m/z 215 (Fig. S3). In low power mode 12.1% of all tropospheric particle spectra showed an IEPOX sulfate ester signal, and above 5 km the TC4 regional average increased to 30–80%.
Fig. 4.

Altitude profiles of average aerosol composition for several aircraft campaigns (lines) and a summertime ground site in Atlanta (point). IEPOX sulfate ester observations (A) tend to follow acidic sulfate profiles (B) except in the stratosphere, > 17 km. (C) Percent of aerosol mass attributed to IEPOX. All except the CR-AVE data are from regions 1–3 days downwind of isoprene sources. Solid lines and point: bottom axis; dashed lines: top axis.
The fraction of particles containing acidic sulfate is plotted in Fig. 4B. For the CR-AVE and TC4 campaigns, the sulfate ester curves generally track sulfate acidity except in the stratosphere, suggesting that for these studies partitioning of IEPOX to aerosols was limited by acidity rather than isoprene emissions. The maxima for both the acidity and sulfate ester curves (high power spectra) occur at several km altitude in the free troposphere. Highly acidic sulfate aerosols were not typically found in the continental boundary layer, and consequently, IEPOX did not strongly partition to aerosols near the isoprene source. During a boundary layer flight leg over the Colombian jungle, isoprene concentrations were 3–4 ppbv, and the level of gas-phase isoprene oxidation products (ISOPOOH + IEPOX) was relatively high (21). Despite intense precursor loadings, the occurrence of IEPOX sulfate ester in aerosols was below the mission average -0% of particles at full laser power and 9.3% at low power—due to a low abundance of acidic aerosols and a relatively short time since emission. Once isoprene and derivatives are lofted above the boundary layer, IEPOX can more efficiently partition to the acidic sulfate aerosols found in the free troposphere.
Free tropospheric air encountered during the Pre-AVE campaign was heavily influenced by convection over the Amazon jungle from 2–3 d prior (31). Amazonian convection generated a large volume of organic-rich aerosols throughout the troposphere from both direct injection of boundary layer particles as well as accumulation of secondary material aloft. Although fewer than 20% of aerosols contained acidic sulfate, IEPOX sulfate ester was observed more frequently (up to 40%) than for other tropical campaigns, often in particles with no acidic signature. Similarly, acidic particles were rare above 7 km during TC4, where IEPOX sulfate ester was most frequently observed using low power. In these cases it is not clear whether very high isoprene loadings allowed IEPOX to partition to mostly neutralized particles in the jungle canopy or whether these aerosols became neutralized after IEPOX uptake. One possibility is that aerosol sulfuric acid was fully titrated by reaction with IEPOX and other compounds to form organosulfates, so that initially acidic aerosols did not produce the sulfate acidity signature in PALMS spectra. Although this sulfate titration effect was not observed even upon substantial BEPOX loadings in laboratory experiments, laboratory aerosols were predominantly sulfate in contrast to Pre-AVE where sulfate was a minor component. It remains unclear why a predominant fraction of the free tropospheric aerosols encountered during Pre-AVE that contained IEPOX sulfate ester appeared fully neutralized. Because convection played a stronger role in defining the properties of the Pre-AVE troposphere, aqueous reactions of SO2, ammonia, and isoprene products may have generated these aerosols through different isoprene reaction mechanisms (32).
Unlike TC4 and Pre-AVE where continental isoprene sources were nearby, the CR-AVE troposphere was characterized by long range transport across the Pacific. During the 5–10 d journey, a highly acidic sulfate aerosol population was generated from maritime convective lofting of dimethyl sulfide (DMS) and other sulfur precursors (31). IEPOX uptake would only occur once these precursors had oxidized and generated acidic aerosols. Isoprene oxidation and subsequent loss of the gas-phase products ISOPOOH and IEPOX via OH reaction requires more than 1 photolytic day (21). Tens of percent of the original IEPOX could remain several transport days downstream of continental isoprene sources to react with newly formed acidic aerosols. The CR-AVE case is important in that, unlike the southeastern United States environment, isoprene-derived aerosol formation did not require anthropogenic emissions but instead resulted from the combination of biogenic VOC emissions and biogenic DMS. This case also demonstrates that isoprene can generate aerosol material well downwind of emission regions and that these aerosols can be transported globally.
Other Regions.
No IEPOX sulfate ester signals were observed at full laser power in the tropical lower stratosphere (17–19 km), a region characterized by upward mass flux across the tropopause. Although during CR-AVE stratospheric sulfate aerosols were highly acidic, they did not accumulate organic mass as they ascended and aged, suggesting that gas-phase organic precursors in general had been removed at lower altitudes. Somewhat surprisingly, although up to 40% of lowermost stratospheric aerosols originated in the troposphere (31), these particles did not have detectable IEPOX signatures.
In the Alaskan Arctic during April 2008 most of the aerosol mass was transported from northern Asia when isoprene emissions were low. The IEPOX sulfate ester was detected in only 0.01% of the particles despite the frequent presence of acidic sulfate. During the same campaign, 0.8% of aerosols in the free troposphere near Florida had detectable amounts of sulfate ester, similar to the summertime North American average. IEPOX sulfate ester was also not observed at full laser power in the Houston area during wintertime when isoprene emissions were low. Within tropical volcanic plumes that contained acidic sulfate aerosol, IEPOX sulfate ester frequency was 1.5–3.0%, up to six times the TC4 mission average.
Mass Estimates.
The fraction of total aerosol mass attributed to IEPOX can be estimated by applying laboratory calibrations to atmospheric IEPOX sulfate ester signals. The calibrations showed that the PALMS instrument is 2–50 times more sensitive to epoxydiol sulfate esters when operating in low laser power mode (Fig. S3). For particles smaller than about 0.5 μm diameter, the IEPOX signal at high laser power usually falls below the detection limit, leading to an underestimate of the IEPOX mass fraction. In general, most particles did not have organosulfate signals above detection limits, but many particles contained tens of % by mass IEPOX (Fig. S6).
Fig. 4C shows vertical profiles of IEPOX mass for the tropical campaigns and the southeastern United States. Although some data was collected at both laser powers on several missions, only TC4 had enough sampling time at low laser power for reliable mass estimates. During TC4, IEPOX comprised as much as 20% of aerosol mass at altitudes where acidic sulfate was most abundant and > 5% throughout most of the free troposphere. The mission average, which included low altitude regions of neutralized sulfate aerosol, was about 2%. Comparing these values to those determined at high laser power during TC4, we find that the high laser power mass estimates for other missions presented below should be regarded as lower limits and could be underestimated by factors of 10–20 (2–5 for Pre-AVE).
The mass fractions for other campaigns shown in Fig. 4C are calculated using high laser power spectra only. Results from the NEAQS July 5 flight, as well as at the Atlanta Supersite ground site in 1999, show that, in the summertime southeastern United States, IEPOX comprised 1–3% of the total aerosol mass below 2 km (Fig. 4C). The NEAQS campaign average for eastern North America was 0.3%. IEPOX mass loadings downwind of Amazonian convection in Pre-AVE were 1% averaged over the troposphere with values of 3–14% at the level of maximum convective outflow. For the CR-AVE remote tropical case, a dominant population of high-acidity particles compensated for distant isoprene emissions to yield an IEPOX aerosol mass fraction of 0.4% in the free troposphere and sporadically up to 1–2%.
Total aerosol loadings varied by orders of magnitude over the cases presented here: from 0.01–0.1 μg m-3 in the tropical upper troposphere to 1–50 μg m-3 in the midlatitude lower troposphere. Table 1 gives absolute aerosol mass estimates for IEPOX by applying the data in Fig. 4C to measurements of submicron aerosol mass. Free tropospheric mass loadings of IEPOX ranged from < 1 ng m-3 in remote tropical regions to tens of ng m-3 downwind of isoprene sources. In the southeastern United States, large summertime biogenic VOC fluxes combined with anthropogenic sulfur emissions to generate 81 ng m-3 aloft during 2004 and more than 900 ng m-3 at ground level in Atlanta during 1999. Using electrospray ionization mass spectrometry, Chan et al. (27) report much lower mass concentrations of 7–64 ng m-3 during a 2008 Atlanta ground study. This apparent disagreement may be due to differences in aerosol acidity between the 1999 and 2008 studies. In 1999 sulfate aerosol was not fully neutralized on average (33) and was highly acidic (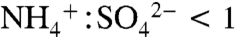 ) for about half of the daytime hours. Data from the United States Environmental Protection Agency (EPA) Speciation Trends Network reveal a strongly increasing trend in sulfate aerosol neutralization: From 2001 to 2008, the summertime
) for about half of the daytime hours. Data from the United States Environmental Protection Agency (EPA) Speciation Trends Network reveal a strongly increasing trend in sulfate aerosol neutralization: From 2001 to 2008, the summertime 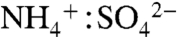 ratio in eastern Atlanta increased at rate of 0.50 per decade (see the public database of the EPA Speciation Trends Network for Dekalb, GA, at http://www.epa.gov/ttn/amtic/amticpm.html).
ratio in eastern Atlanta increased at rate of 0.50 per decade (see the public database of the EPA Speciation Trends Network for Dekalb, GA, at http://www.epa.gov/ttn/amtic/amticpm.html).
Table 1.
Submicron aerosol mass (ng m-3) attributed to IEPOX at relevant altitude ranges
| TC4*tropics | Pre-AVE*tropics | CR-AVE*tropics | NEAQS (July 5)†southeastern US | Atlanta Supersite‡southeastern US | ||||||
| Downwind of Central American isoprene sources, some convection | Heavily impacted by Amazonian convection | Long-range transport, distant isoprene sources | Downwind of strong isoprene sources | Strong local isoprene sources | ||||||
| 0–3 km | 0.03 | 0.58§ | 7–17 km | 32 | 7–17 km | 0.42 | 0–2 km | 84 | ground site | 910 |
| 3–12 km | 0.05 | 1.2§ | 17–19 km | 0 | 17–19 km | 0 | 2–4 km | 0 | ||
*An average aerosol density of 1.2 g cm-3 is assumed to convert measured volume to mass.
†Aerosol mass for the July 5 flight is derived from Aerodyne Aerosol Mass Spectrometer data and may be anomalously low by up to 50% due to inlet transmission inefficiency (34).
‡Total aerosol burden is from PM2.5 measurements (35), where the large majority of aerosol mass was submicron.
§Using reduced laser power for higher sensitivity. All other values using full laser power.
Atmospheric Implications.
There are several observations of isoprene-derived organosulfates at ground sites in the eastern United States (18, 27, 36), but quantification is so far restricted to a few offline techniques. Our airborne mass spectrometry data demonstrates that acid-catalyzed uptake of IEPOX is effective in a variety of tropospheric environments and that the IEPOX sulfate ester product is one of the most abundant single organic compounds in atmospheric aerosol. IEPOX mass loadings can vary by orders of magnitude depending on biogenic VOC emissions and NOx levels and are particularly sensitive to aerosol acidity. The most frequent sulfate ester observations and highest overall mass fractions were found 1–3 d downwind of isoprene sources. In the tropics IEPOX reactive uptake contributed 1–20% of the tropospheric aerosol mass where continental convection was active. Particulate IEPOX levels in wintertime midlatitudes and springtime Arctic were more than 50 times lower than typical midlatitude levels, whereas aerosols in the remote tropical troposphere far from isoprene sources had consistently nonzero abundance. In several regions IEPOX uptake appeared to be acid-limited, suggesting that emissions of biogenic and anthropogenic sulfur rather than VOCs can dictate secondary organic aerosol generation in the atmosphere via this pathway. Global models estimate that low-NOx aerosol production from isoprene contributes from one to three quarters of SOA mass (14). Although this acid-catalyzed epoxide mechanism is capable of producing significant aerosol mass, an acidity parameter is not currently implemented in global models of isoprene SOA generation (14).
The production of secondary organic aerosol material via IEPOX reactive uptake is conceptually similar to tropospheric ozone production in that it proceeds via a combination of biogenic VOCs and anthropogenic pollution, in this case SO2 emissions. Through this pathway, SO2 emission control strategies may lead to a decrease in biogenic aerosol loadings. Limited data suggest this may have occurred in Atlanta over the last decade. On the other hand, the remote CR-AVE case, where acidic, sulfate-rich aerosol particles were generated from marine sources, demonstrates that secondary organic aerosol material is also produced via the IEPOX pathway without anthropogenic input.
Materials and Methods
PALMS Single Particle Mass Spectrometer.
The NOAA PALMS instrument (5) detects individual aerosol particles (Dp > 200 nm) by light scattering and then pulses a UV excimer laser (λ = 193 nm) to vaporize and ionize each particle. Ions are extracted into a time-of-flight mass spectrometer to generate a complete positive or negative mass spectrum per particle. The percent of aerosol mass attributed to IEPOX uptake was calculated by applying the laboratory calibration to average mass spectral intensities. The mass calibration in Fig. S3 is applied to all particle sizes. An underlying assumption is that the composition of sulfate-organic aerosols is homogeneous over the detected size range. To estimate absolute IEPOX mass, the IEPOX mass fraction was applied to submicron aerosol volume (30, 31) (data publicly available at the NASA ESPO database at http://espoarchive.nasa.gov/archive/arcs/tc4/; see SI Text) at 10 min intervals and then averaged over each campaign. Sulfate acidity signatures in PALMS spectra have been calibrated to 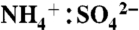 ratio using ambient particles (31). Aerosol sulfate is considered acidic here for
ratio using ambient particles (31). Aerosol sulfate is considered acidic here for 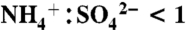 and neutralized for
and neutralized for 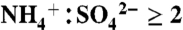 . Aircraft campaigns: ITCT/NEAQS (Intercontinental Transport and Chemical Transformation/New England Air Quality Study), Pre-AVE (Pre-Aura Validation Experiment), CR-AVE (Costa Rica Aura Validation Experiment), TC4 (Tropical Composition, Cloud, and Climate Coupling).
. Aircraft campaigns: ITCT/NEAQS (Intercontinental Transport and Chemical Transformation/New England Air Quality Study), Pre-AVE (Pre-Aura Validation Experiment), CR-AVE (Costa Rica Aura Validation Experiment), TC4 (Tropical Composition, Cloud, and Climate Coupling).
BEPOX Uptake Experiments.
BEPOX was synthesized according to Surratt et al. (22). Nebulized aerosols were mixed with BEPOX vapor inside a 7.5 cm i.d. flowing reaction vessel at 295 K. BEPOX concentrations (not monitored) were varied by passing dry air over crystals maintained at a constant temperature from 273–338 K. Neutralized and acidic aerosols generated from dilute (NH4)2SO4 and NH4HSO4/H2SO4 (1–2.75∶1) solutions were dried to < 10% RH and size selected using a differential mobility analyzer before entering the reaction vessel. Generated aerosols also contained 30% organic carbon mass on average. Residence times in the reaction vessel were 45–300 s. Aerosol mass was determined from OPC scattering intensities using BEPOX densities and refractive indices of 1.0 g cm-3 and 1.42 based on epoxides of similar structure (37), and values for NH4HSO4/H2SO4 of 1.62 g cm-3 and 1.45 (38).
Gas-Phase Measurements.
VOC concentrations during the ITCT/NEAQS 2004 campaign were measured using proton transfer reaction mass spectrometry (PTR-MS) (29). Methyl vinyl ketone and methacrolein are isomeric, and only the sum of their abundances was determined. NOx concentrations were measured using a NO2 photolysis-NO chemiluminescence technique (39).
Supplementary Material
Acknowledgments.
We are grateful to Jerome Brioude for FLEXPART analysis of isoprene emissions, to Ann Middlebrook for AMS data from the ITCT/NEAQS 2004 campaign and PALMS data from the 1999 Atlanta Supersite, and to Jason Surratt for valuable input. This work was supported by NOAA base funding, NOAA climate change programs, NASA Earth Science Program Office for aircraft deployments, and the Camille and Henry Dreyfus Postdoctoral Program in Environmental Chemistry. S.M. Murphy acknowledges a National Research Council Research Associateship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012561107/-/DCSupplemental.
References
- 1.Kanakidou M, et al. Organic aerosol and global climate modelling: A review. Atmos Chem Phys. 2005;5:1053–1123. [Google Scholar]
- 2.Heald CL, et al. Total observed organic carbon (TOOC) in the atmosphere: A synthesis of north american observations. Atmos Chem Phys. 2008;8:2007–2025. [Google Scholar]
- 3.Yu S, Bhave PV, Dennis RL, Mathur R. Seasonal and regional variations of primary and secondary organic aerosols over the continental United States: Semi-empirical estimates and model evaluation. Environ Sci Technol. 2007;41:4690–4697. doi: 10.1021/es061535g. [DOI] [PubMed] [Google Scholar]
- 4.Hallquist M, et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos Chem Phys. 2009;9:5155–5236. [Google Scholar]
- 5.Murphy DM, et al. Single-particle mass spectrometry of tropospheric aerosol particles. J Geophys Res-Atmos. 2006;111:D23S32. [Google Scholar]
- 6.Goldstein AH, Koven CD, Heald CL, Fung IY. Biogenic carbon and anthropogenic pollutants combine to form a cooling haze over the southeastern United States. Proc Natl Acad Sci USA. 2009;106:8835–8840. doi: 10.1073/pnas.0904128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin ST, et al. Sources and properties of amazonian aerosol particles. Rev Geophys. 2010;48:RG2002. [Google Scholar]
- 8.Weber RJ, et al. A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States. J Geophys Res-Atmos. 2007;112:D13302. [Google Scholar]
- 9.Heald CL, et al. A large organic aerosol source in the free troposphere missing from current models. Geophys Res Lett. 2005;32:L18809. [Google Scholar]
- 10.de Gouw JA, et al. Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002. J Geophys Res-Atmos. 2005;110:D16305. [Google Scholar]
- 11.Claeys M, et al. Formation of secondary organic aerosols through photooxidation of isoprene. Science. 2004;303:1173–1176. doi: 10.1126/science.1092805. [DOI] [PubMed] [Google Scholar]
- 12.Edney EO, et al. Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States. Atmos Environ. 2005;39(29):5281–5289. [Google Scholar]
- 13.Henze DK, Seinfeld JH. Global secondary organic aerosol from isoprene oxidation. Geophys Res Lett. 2006;33:L09812. [Google Scholar]
- 14.Carlton AG, Wiedinmyer C, Kroll JH. A review of secondary organic aerosol (SOA) formation from isoprene. Atmos Chem Phys. 2009;9:4987–5005. [Google Scholar]
- 15.Kroll JH, Ng NL, Murphy SM, Flagan RC, Seinfeld JH. Secondary organic aerosol formation from isoprene photooxidation. Environ Sci Technol. 2006;40:1869–1877. doi: 10.1021/es0524301. [DOI] [PubMed] [Google Scholar]
- 16.Surratt JD, et al. Effect of acidity on secondary organic aerosol formation from isoprene. Environ Sci Technol. 2007;41:5363–5369. doi: 10.1021/es0704176. [DOI] [PubMed] [Google Scholar]
- 17.Chan AWH, et al. Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation. Atmos Chem Phys. 2010;10:7169–7188. [Google Scholar]
- 18.Surratt JD, et al. Organosulfate formation in biogenic secondary organic aerosol. J Phys Chem A. 2008;112:8345–8378. doi: 10.1021/jp802310p. [DOI] [PubMed] [Google Scholar]
- 19.Surratt JD, et al. Evidence for organosulfates in secondary organic aerosol. Environ Sci Technol. 2007;41:517–527. doi: 10.1021/es062081q. [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Gonzalez Y, et al. Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(−) electrospray ionization mass spectrometry. J Mass Spectrom. 2008;43:371–382. doi: 10.1002/jms.1329. [DOI] [PubMed] [Google Scholar]
- 21.Paulot F, et al. Unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science. 2009;325:730–733. doi: 10.1126/science.1172910. [DOI] [PubMed] [Google Scholar]
- 22.Surratt JD, et al. Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proc Natl Acad Sci USA. 2010;107:6640–6645. doi: 10.1073/pnas.0911114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minerath EC, Elrod MJ. Assessing the potential for diol and hydroxy sulfate ester formation from the reaction of epoxides in tropospheric aerosols. Environ Sci Technol. 2009;43:1386–1392. doi: 10.1021/es8029076. [DOI] [PubMed] [Google Scholar]
- 24.Minerath EC, Schultz MP, Elrod MJ. Kinetics of the reactions of isoprene-derived epoxides in model tropospheric aerosol solutions. Environ Sci Technol. 2009;43:8133–8139. doi: 10.1021/es902304p. [DOI] [PubMed] [Google Scholar]
- 25.Eddingsaas NC, VanderVelde DG, Wennberg PO. Kinetics and products of the acid-catalyzed ring-opening of atmospherically relevant butyl epoxy alcohols. J Phys Chem A. 2010;114:8106–8113. doi: 10.1021/jp103907c. [DOI] [PubMed] [Google Scholar]
- 26.Farmer DK, et al. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc Natl Acad Sci USA. 2010;107:6670–6675. doi: 10.1073/pnas.0912340107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan MN, et al. Characterization and quantification of isoprene-derived epoxydiols in ambient aerosol in the southeastern United States. Environ Sci Technol. 2010;44:4590–4596. doi: 10.1021/es100596b. [DOI] [PubMed] [Google Scholar]
- 28.Galloway MM, et al. Glyoxal uptake on ammonium sulphate seed aerosol: Reaction products and reversibility of uptake under dark and irradiated conditions. Atmos Chem Phys. 2009;9:3331–3345. [Google Scholar]
- 29.de Gouw JA, et al. Sources of particulate matter in the northeastern United States in summer: 1. Direct emissions and secondary formation of organic matter in urban plumes. J Geophys Res-Atmos. 2008;113:D08301. [Google Scholar]
- 30.Brock CA, et al. Sources of particulate matter in the northeastern United States in summer: 2. Evolution of chemical and microphysical properties. J Geophys Res-Atmos. 2008;113:D08302. [Google Scholar]
- 31.Froyd KD, et al. Aerosol composition of the tropical upper troposphere. Atmos Chem Phys. 2009;9:4363–4385. [Google Scholar]
- 32.Ervens B, et al. Secondary organic aerosol yields from cloud-processing of isoprene oxidation products. Geophys Res Lett. 2008;35:L02816. [Google Scholar]
- 33.Weber R. Short-term temporal variation in PM2.5 mass and chemical composition during the Atlanta Supersite Experiment, 1999. J Air Waste Manage. 2003;53:84–91. doi: 10.1080/10473289.2003.10466123. [DOI] [PubMed] [Google Scholar]
- 34.Bahreini R, et al. Design and operation of a pressure-controlled inlet for airborne sampling with an aerodynamic aerosol lens. Aerosol Sci Tech. 2008;42:465–471. [Google Scholar]
- 35.Solomon P, et al. Comparison of integrated samplers for mass and composition during the 1999 Atlanta Supersites project. J Geophys Res-Atmos. 2003;108:8423–8433. [Google Scholar]
- 36.Altieri KE, Turpin BJ, Seitzinger SP. Oligomers, organosulfates, and nitrooxy organosulfates in rainwater identified by ultra-high resolution electrospray ionization FT-ICR mass spectrometry. Atmos Chem Phys. 2009;9:2533–2542. [Google Scholar]
- 37.Lide DR, editor. CRC Handbook of Chemistry and Physics, 2009–2010. 90th Ed. Boca Raton, FL: CRC Press; 2009. [Google Scholar]
- 38.Tang IN, Munkelwitz HR. Water activities, densities, and refractive-indexes of aqueous sulfates and sodium-nitrate droplets of atmospheric importance. J Geophys Res-Atmos. 1994;99:18801–18808. [Google Scholar]
- 39.Ryerson TB, Williams EJ, Fehsenfeld FC. An efficient photolysis system for fast-response NO2 measurements. J Geophys Res-Atmos. 2000;105:26447–26461. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.