

Abstract
“Head-to-head” terpene synthases catalyze the first committed steps in sterol and carotenoid biosynthesis: the condensation of two isoprenoid diphosphates to form cyclopropylcarbinyl diphosphates, followed by ring opening. Here, we report the structures of Staphylococcus aureus dehydrosqualene synthase (CrtM) complexed with its reaction intermediate, presqualene diphosphate (PSPP), the dehydrosqualene (DHS) product, as well as a series of inhibitors. The results indicate that, on initial diphosphate loss, the primary carbocation so formed bends down into the interior of the protein to react with C2,3 double bond in the prenyl acceptor to form PSPP, with the lower two-thirds of both PSPP chains occupying essentially the same positions as found in the two farnesyl chains in the substrates. The second-half reaction is then initiated by the PSPP diphosphate returning back to the Mg2+ cluster for ionization, with the resultant DHS so formed being trapped in a surface pocket. This mechanism is supported by the observation that cationic inhibitors (of interest as antiinfectives) bind with their positive charge located in the same region as the cyclopropyl carbinyl group; that S-thiolo-diphosphates only inhibit when in the allylic site; activity results on 11 mutants show that both DXXXD conserved domains are essential for PSPP ionization; and the observation that head-to-tail isoprenoid synthases as well as terpene cyclases have ionization and alkene-donor sites which spatially overlap those found in CrtM.
Keywords: triterpene, X-ray crystallography, drug discovery, staphyloxanthin, quinuclidine
Head-to-head terpene synthases catalyze the first committed steps in the biosynthesis of sterols and carotenoid pigments: the C1′-2,3 condensation of two isoprenoid diphosphates to form a cyclopropylcarbinyl diphosphate (1, 2), followed by ring opening to form squalene, dehydrosqualene, or phytoene. In humans and in many pathogenic yeasts, fungi, and protozoa, as well as in plants, the isoprenoid diphosphate is farnesyl diphosphate (FPP) and the initial product is the C30 isoprenoid, presqualene diphosphate (PSPP). As implied by its name, PSPP is then converted (by the same enzyme as used in the condensation reaction, squalene synthase, SQS) to squalene which, after epoxidation, is cyclized to lanosterol (3), as shown in Fig. 1. Lanosterol then undergoes numerous additional reactions, resulting in formation of sterols, key cell membrane components. As such, squalene synthase inhibitors are of interest as antiparasitics, in particular against Trypanosoma cruzi (4) and Leishmania spp. (5), the causative agents of Chagas disease and the leishmaniases. In plants, the related enzyme phytoene synthase (PSY) catalyzes the condensation of two C20 isoprenoid diphosphate (geranylgeranyl diphosphate, GGPP) molecules (6) to form prephytoene diphosphate (PPPP) that, after ring opening, forms phytoene, which is then converted to carotenoid pigments (7) (Fig. 1). In the bacterium Staphylococcus aureus, the initial step in formation of the carotenoid pigment staphyloxanthin (STX) is carried out by condensation of two FPP molecules by the enzyme dehydrosqualene synthase (CrtM), again initially forming PSPP (8), which is then converted by the same enzyme to dehydrosqualene, which is further transformed to STX (8). Because STX is a so-called virulence factor for S. aureus (9), inhibiting its formation is of interest in the context of developing new routes to antiinfective therapies (10).
Fig. 1.

Schematic illustration of the reactions catalyzed by head-to-head terpene synthases: CrtM, SQS, and PSY. All reactions involve an initial C1′-2,3 cyclopropanation step. The end products of the biosynthetic pathways are highly varied and include sterols (cholesterol, ergosterol) and carotenoids (staphyloxanthin, β-carotene).
Given the key role of the head-to-head tri- and tetraterpene synthases in sterol and carotenoid biosynthesis, there has been remarkably little work reported on their three-dimensional structures. There has been one report of the structure of human SQS with a bound inhibitor (11), but relatively little mechanistic information was obtained because the inhibitor was not obviously substrate, intermediate, or product-like. In our group, we reported the X-ray crystallographic structure of CrtM from S. aureus (10). There were two substrate-analog inhibitor binding sites (sites S1 and S2), but determining which represented the prenyl donor (the “allylic” FPP that ionizes to form the 1′ carbocation) and which represented the prenyl acceptor (that provides the C2,3 alkene group) was not attempted because the 1′-2,3 distances for both possible assignments were ∼5 Å. In this paper, we provide structural and mechanistic models for head-to-head terpene synthase activity, and inhibition, based on nine X-ray structures; studies of site-directed mutagenesis based on both structure and bioinformatics; chemical reactivity; and comparisons with head-to-head terpene synthases and terpene cyclases.
Results and Discussion
Structure of PSPP Bound to CrtM.
The overall reactions catalyzed by CrtM, SQS, and PSY are shown in Fig. 1 and involve ionization of one isoprenoid diphosphate molecule to form a 1′ carbocation, which then undergoes nucleophilic attack by the C2,3 double bond in the second isoprenoid diphosphate to form, after proton loss, PSPP or PPPP. After a second ionization, these molecules then undergo a complex series of rearrangements (12) thought to involve cyclopropyl and cyclobutyl species to form either primarily dehydrosqualene (with CrtM; or SQS in the absence of NADPH); squalene (with SQS, in the presence of NADPH), or phytoene (with PSY). But which site houses the cation (donor), and which is the alkene (acceptor)? How do PSPP and dehydrosqualene (DHS) bind to CrtM (or SQS)? And where do the diphosphates ionize? In our work on CrtM (10), we used the nonreactive substrate analog, S-thiolo-farnesyl diphosphate (FSPP), to define two prenyl diphosphate binding sites: S1 and S2. The structure obtained (PDB ID code 2ZCP) is shown in Fig. 2A from which it can be seen that the C1′-C2,3 distances for the two possible mechanistic models (i.e., S1 = donor or S1 = acceptor) are very similar (5–5.4 Å) making it impossible to make reliable donor–acceptor site assignments based solely on this metric.
Fig. 2.

Crystallographic results for CrtM with a bound substrate-like inhibitor FSPP and the intermediate PSPP. (A) Bound FSPP (PDB code ID 2ZCP), from ref. 10. (B) The 2Fo-Fc map for PSPP contoured at 1σ (green) and 3σ (red) (PDB code ID 3ADZ). (C) Superposition of PSPP (PDB ID code 3ADZ) on FSPP in CrtM. (D) Close-up view of PSPP-FSPP superposition. Arrows indicate movements of the C1′, C2, and C3 atoms.
We therefore produced a CrtM mutant (Y129A) that, based on sequence alignment with rat SQS, corresponds to Y171 in the rat protein, which is known to be unreactive (13). The Y129A CrtM mutant was indeed unreactive, with either FPP (0% activity) or PSPP (7% activity, Fig. S1) as substrate, indicating that both the first-half (PSPP formation) as well as the second-half (PSPP to dehydrosqualene) reactions were inhibited, which enabled us to obtain diffraction quality crystals of a CrtM/PSPP complex. Our initial CrtM/PSPP crystals diffracted to 2.4 Å (full crystallographic data and structure refinement details are given in Table S1), and using the CrtM-FSPP structure (PDB ID code 2ZCP) minus ligands as a template, we obtained the electron density results shown in Fig. S2A (PDB ID code 3LGZ). We then obtained a second structure (electron density shown in Fig. 2B) with improved (1.89 Å) resolution at a higher [Mg2+] (PDB ID code 3ADZ; full crystallographic data and structure refinement details are given in Table S1; crystallization details are in SI Methods), Fig. 2C. Both structures (which have a 0.3-Å protein Cα and 0.7-Å ligand rmsd) are shown superimposed in Fig. S2C. In addition, we obtained a third PSPP structure using wild-type CrtM [crystallized in the presence of 0.2 M sodium tartrate (SI Fig. S2 B and D, and Table S1)], which had a 0.18, 0.45-Å protein and 0.19, 0.8-Å ligand rmsd versus the two mutant structures, indicating all three structures are virtually identical.
What is immediately obvious from these structures, shown (in cyan) superimposed on the CrtM/FSPP structure (in green and yellow) in Fig. 2C, is that (i) the diphosphate (PPi) group seen in the S1 FSPP site is no longer present, indicating that the FPP in this site is the prenyl donor (which ionizes to form the 1′-carbocation); (ii) the S1 FPP C1′ moves down by ∼2.5 Å to form C1′ in PSPP, while (iii) the S2 FPP C2 and C3 move up by ∼1.5 Å, to form C2,3 in the cyclopropane ring of the PSPP product; (iv) the S2 PPi group moves “up” somewhat (on average, the non-H atoms move by ∼1.8 Å), but overall this group remains very close to its original position; and (v) the lower two-thirds of the PSPP side chains hardly move at all with respect to their positions in FSPP, only a ∼1.2-Å rmsd for 27 carbon atoms (0.7 Å for 25 carbons). Also of interest is the observation that the PSPP side chain in S1 is highly “bent” and, although it appears shorter, is actually the longer one (11 versus 9 contiguous carbons), whereas the S2 chain is quite straight, occupying the same site as the biphenyl ring-containing inhibitors reported previously (10). These results strongly support a “first-half” reaction mechanism in which FPP in S1 ionizes to form the primary carbocation which then moves down to react with the C2,3 double bond in the FPP in S2 to form (after H+ abstraction), PSPP, with the highly conserved Asp residues in the first DXXXD domain being essential for catalysis (Fig. S1). To further test this mechanistic proposal, we next investigated the structure and activity of a series of S-thiolo-diphosphates interacting with CrtM.
Structure and Activity of Substrate Analogs.
In our previous work on CrtM, we used S-thiolo-FSPP as an unreactive substrate analog (10). The lack of activity of other S-thiolo-isoprenoid diphosphates as allylic/cationic prenyl donors has been studied extensively by Phan et al. (14) who found that, e.g., S-thiolo-dimethylallyl diphosphate (DMAPP) inhibits farnesyl diphosphate synthase (FPPS) (because it does not ionize, in the allylic site, S1), whereas S-thiolo-isopentenyl-diphosphate (IPP) inhibits FPPS, because it forms S-thiolo-geranyl diphosphate (GPP), which again cannot ionize, in S1 (14). Plus, Hosfield et al. (15) have shown that S-thiolo-DMAPP binds to the allylic (S1) site in FPPS, as does S-thiolo-FPP in geranylgeranyl diphosphate synthase (GGPPS) (16). These findings lead to the idea that other S-thiolo-isoprenoid diphosphates might be interesting probes of CrtM structure and function.
In order to see how FSPP might inhibit the conversion of FPP to PSPP, and then dehydrosqualene, we carried out a series of CrtM inhibition experiments. Remarkably, we found that the rate of the CrtM reaction (monitored in this case via PPi release) increased with FSPP concentration (Fig. 3A). When we assayed for DHS production, we found that its formation was being inhibited (Fig. 3B), so it seemed that FSPP must be reacting with FPP to form a nonreactive S-thiolo-PSPP, in much the same way that S-thiolo-IPP can react with DMAPP in FPPS to form S-thiolo-GPP (14). To verify formation of S-thiolo-PSPP, we isolated the product of the reaction chromatographically, then carried out a reductive cleavage (17) and obtained the mass spectrometry results shown in Fig. S3, which confirm formation of the C30H50S product, presqualene thiol.
Fig. 3.

Effects of FSPP on CrtM catalysis and a GGSPP inhibitor structure. (A) Rate of PPi generation increases with FSPP concentration (error bars are from duplicate data points, R2 = 0.85). (B) Rate of dehydrosqualene formation decreases with FSPP concentration (duplicate, R2 = 0.78). These results indicate “dead-end” formation of S-thiolo-PSPP, as confirmed by mass spectrometry. (C) The 2Fo-Fc map (contoured at 0.8σ and 3σ) of GGSPP bound to F26A CrtM. (D) GGSPP (green and yellow) binds to both S1, S2; would clash with F26 (in red) if present; FSPPs are in cyan.
To investigate this topic in more detail, we next investigated the reactions between farnesyl and geranylgeranyl diphosphates and S-thiolo diphosphates [representative liquid chromatography (LC)-MS results are shown in Figs. S4 and S5 and are summarized in Table S2]. The results can be summarized as follows: Only FPP (in the S1 site) can ionize, whereas FPP, GGPP, FSPP, or S-thiolo-geranylgeranyl diphosphate (GGSPP) (in S2) can all provide the alkene, prenyl acceptor, in WT CrtM. So, longer chains (C20) can fit in S2, but not in S1, because we find no activity (PPi release) with GGPP as substrate. However, in an F26A mutant, GGPP actually reacts to form phytoene (18), because the bulky F26 (at the “bottom” of the S1 binding site) that blocks GGPP binding to S1 is removed, and the reaction can proceed. If this idea is correct, it should be possible to bind two molecules of GGSPP to the F26A CrtM mutant. This is indeed the case, as shown in Fig. 3 C and D where we see that the S1 side chain in the CrtM/GGSPP complex (PDB ID code 3AE0; Table S1) would clash with F26, shown in red (if it were present). These results are all consistent with S1 being the ionization site for FPP in PSPP formation.
A Catalytic Model for Dehydrosqualene Synthase.
These results all support the initial reaction model shown in Fig. 4 (and Fig. S6) in which the S1 FPP ionizes to form the 1′-carbocation, Fig. 4 A and B. This carbocation then moves down into the interior of the protein to react with C2,3 in the S2 FPP, Figs. 2D and 4B, with remarkably little change in the conformation of the lower two-thirds of the FSPP and PSPP hydrocarbon chains (Fig. 2C). The second PPi in PSPP then moves up (Fig. 4C) into the S1 site to interact with the three Mg2+, and the cyclopropylcarbinyl cation so formed (Fig. 4 D and E) then rearranges, as proposed (12), forming DHS (Fig. 4F), which we find binds to the surface pocket shown in Fig. 5 A (PDB ID code 3NRI) (full crystallographic data and structure refinement details are given in Table S1; crystallization details are in SI Methods). Chemical structures are shown for clarity in Fig. 4G. The alternate model (Fig. 4H), in which the S2 site FPP ionizes initially, results in the longer (11 contiguous carbons) PSPP side chain occupying the S2 product site, which is not seen experimentally.
Fig. 4.

Schematic illustration of the conversion of FPP to dehydrosqualene catalyzed by CrtM, based on the crystallographic results. (A) Bound FPP in S1 ionizes. (B) Carbocation reacts with S2 FPP alkene group. (C) PSPP PPi in S2 moves to S1 site. (D) PSPP PPi ionizes. (E and F) Carbocation rearranges and after proton loss forms final product, dehydrosqualene. (G and H) Two possible condensation reactions, in S1, S2. Only that shown in G is consistent with the crystallographic results.
Fig. 5.

Dehydrosqualene structure, important residues in catalysis, and PSPP ionization model. (A) The 2Fo-Fc map for dehydroqualene (1σ, magenta) bound to CrtM surface pocket, superimposed on PSPP (in cyan). (B) Key catalytic residues, D48, D52, and R45 are essential for second-half reaction and indicate PPi ionization in S1. (C) Proposed movement of PSPP PPi into S1 for ionization; PSPP is in gray and the proposed ionization conformation of PSPP is in cyan. The ionization conformation is obtained by using Glide program (19), docking to PDB ID code 2ZCP.
But which residues are involved in the second-half reaction? One possibility is that the PSPP diphosphate just ionizes in the S2 site and there is indeed one Mg2+ that interacts with the PSPP PPi group. The second possibility is that the PSPP diphosphate group could simply flip back up to the two 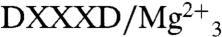 domain (Fig. 5B) to undergo this second ionization, as illustrated in the Glide (19) docking pose shown in Fig. 5C. To help distinguish between these two possibilities, we used the JPRED3 (20) and SCORECONS (21) programs to rank order the essential residues in CrtM (excluding structural Gly, Ala, and Pro), then mutated nine of these to Ala and tested their activity in both first-half (FPP to PSPP) and second-half (PSPP to DHS) reactions. Results are shown in Table S1. What is clear from these results is that Y129 as well as all of the top ranked Asp residues (D48 and D52 in the first Asp-rich region; D172 and D176 in the second Asp-rich domain; Fig. 5B) are essential for both first- and second-half reactions. This observation strongly supports the ionization of both FPP as well as PSPP in the same site—the
domain (Fig. 5B) to undergo this second ionization, as illustrated in the Glide (19) docking pose shown in Fig. 5C. To help distinguish between these two possibilities, we used the JPRED3 (20) and SCORECONS (21) programs to rank order the essential residues in CrtM (excluding structural Gly, Ala, and Pro), then mutated nine of these to Ala and tested their activity in both first-half (FPP to PSPP) and second-half (PSPP to DHS) reactions. Results are shown in Table S1. What is clear from these results is that Y129 as well as all of the top ranked Asp residues (D48 and D52 in the first Asp-rich region; D172 and D176 in the second Asp-rich domain; Fig. 5B) are essential for both first- and second-half reactions. This observation strongly supports the ionization of both FPP as well as PSPP in the same site—the 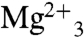 trimetal cluster observed in the FSPP structure (Fig. 2A). Based on the observation that Y129 is essential for both half-reactions, that there are no conformational changes between WT and the Y129A mutant, together with the observation that Y129 is ∼8 Å from the closest FPP, it seems that the most likely role for this highly conserved residue is to facilitate removal of the Mg2+/PPi product complex formed in both half-reactions from the enzyme.
trimetal cluster observed in the FSPP structure (Fig. 2A). Based on the observation that Y129 is essential for both half-reactions, that there are no conformational changes between WT and the Y129A mutant, together with the observation that Y129 is ∼8 Å from the closest FPP, it seems that the most likely role for this highly conserved residue is to facilitate removal of the Mg2+/PPi product complex formed in both half-reactions from the enzyme.
Structural Basis for CrtM/SQS Inhibition by Cationic Species.
Next, we consider CrtM and SQS inhibition. One of the most potent classes of SQS inhibitor are the quinuclidines (or quinuclidinols), compounds such as BPH-651, and E5700 and ER119884 (from Eisai Pharmaceuticals) (Scheme 1), which have been found to have low nanomolar activity against both T. cruzi and human SQS (4, 5, 22), as well as potent activity against T. cruzi cell growth (5). These compounds are of interest as antiinfectives, but nothing is known structurally about how they might act. The 3-OH quinuclidine pKa is expected to be ∼10–11 (23), so these compounds are likely to bind to SQS as cationic (ammonium) species, and it seemed possible that, as with, e.g., nitrogen-containing bisphosphonate inhibitors of FPPS (24, 25), they might mimic the transition state–reactive intermediates involved in PSPP (or squalene–dehydrosqualene) formation. Although BPH-651 is only a modest CrtM inhibitor (Ki = 17.5 μM), we were able to obtain the structure of a CrtM/BPH-651 complex (full crystallographic details are given in Table S3), as well as that of a second ammonium-containing species (26) BPH-673 (Table S3), a potent SQS inhibitor which has a Ki ∼ 2 μM for CrtM inhibition. Both compounds contain biphenyl side chains and bind into the “linear” S2 site, as shown in Fig. 6 A and B. In each case, the positively charged center (blue) is located quite near the cyclopropyl ring in PSPP (BPH-651, 1-Å closest distance; BPH-673, 2 Å), with the superpositions shown in Fig. 6 A and B clearly suggesting that both cationic inhibitors act as transition state–reactive intermediate analogs which occupy the S2 site, mimicking a cyclopropyl carbocation species, rather than being isosteres for the farnesyl carbocation which forms in S1. In FPPS, the cationic or nitrogen-containing bisphosphonate class of inhibitors bind to the S1 site (18, 21, 22), but of course most of the binding energy there can be attributed to the 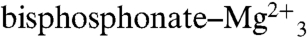 interaction. In the cationic inhibitors here, no such metal ion chelation is present, and binding is dominated by hydrophobic interactions with, potentially, the ammonium groups (and the cationic reaction intermediates) also being stabilized by the protein’s charge field, just as in FPPS (17). The Eisai SQS inhibitors (E5700, ER119884) do, however, both have an additional benzyl substituent that makes them particularly potent. So how might their bulky side chains bind? To help answer that question, we first need to determine how good a model for SQS is CrtM (and vice versa).
interaction. In the cationic inhibitors here, no such metal ion chelation is present, and binding is dominated by hydrophobic interactions with, potentially, the ammonium groups (and the cationic reaction intermediates) also being stabilized by the protein’s charge field, just as in FPPS (17). The Eisai SQS inhibitors (E5700, ER119884) do, however, both have an additional benzyl substituent that makes them particularly potent. So how might their bulky side chains bind? To help answer that question, we first need to determine how good a model for SQS is CrtM (and vice versa).
Scheme 1.

Fig. 6.

X-ray crystallographic structures of inhibitors bound to CrtM. (A) BPH-651, a 3-OH quinuclidine (PDB ID code 3ACW). (B) BPH-673 (PDB ID code 3ACX), an biphenyl/ammonium inhibitor. Both structures are shown superimposed on PSPP (PDB ID code 3ADZ, in cyan). (C) Comparison between BPH-652 bound to CrtM (orange; PDB ID code 2ZCQ) and SQS (green; PDB ID code 3LEE). The heavy atom rmsd in 2.1 Å but this distance reduces to 1.2 Å if the structures are aligned using the conserved Asp residues (or 0.6 Å for the ligands alone). The apparent “offset” may be related to an NADPH binding site in SQS. (D) BPH-702 (orange) bound to CrtM superimposed on the FSPP structure (in green, yellow). Note that the phosphonosulfonate group binds in S1, the biphenyl ether side chain in S2, while the benzyl substituent also binds in S1. (E) BPH-702 (the benzyl inhibitor, in orange) superimposed on the quinuclidine BPH-651. (F) Docking pose for the quinuclidine ER119884 (in yellow) bound to CrtM, superimposed on PSPP (in cyan).
We therefore next obtained the X-ray crystallographic structure of BPH-652, the CrtM inhibitor whose structure we reported previously (10), bound this time to human SQS (PDB ID code 3LEE; full crystallographic details are given in Table S3). Using the CrtM/SQS protein structure alignment (Fig. 6C), we find that there is a 2.0-Å rmsd between the BPH-652 ligand heavy atoms coordinates, showing close structural similarity. The rmsd reduces to 1.2 Å when the alignment is based solely on the conserved Asp residues, or 0.6 Å when the alignment is based solely on the ligands. Then we obtained the X-ray structure of an analog of BPH-652, BPH-702 (PDB ID code 3ACY), which contains the benzyl substituent found in the most potent quinuclidinol inhibitors. As can be seen in Fig. 6D, the phosphonosulfonate BPH-702 backbone binds in the S1 PPi-binding domain (seen in the FSPP structure); the biphenyl ether side chain binds in the S2 side-chain region, whereas the benzene ring in the benzyl group is located at the terminus of the S1 side-chain in the FSPP structure (Fig. 6D). When we superimpose the quinuclidinol (BPH-651) and benzyl biphenyl phosphonosulfonate (BPH-702) structures (Fig. 6E), we see that the ER119884 side chain can readily fit into the space occupied by these two compounds, and a docking pose (using Glide) (19) for this potent inhibitor (docked to the CrtM/PSPP protein structure) is shown in Fig. 6F. Also of interest is the observation that the cationic center in the quinuclidinol overlays the PSPP cyclopropane ring (Fig. 6F), suggesting that quinuclidinol may act as an isostere for a cyclopropyl carbinol carbocation reactive intermediate.
Comparisons with Head-to-Tail Terpene Synthases and Terpene Cyclases.
These results lead to the following question: Do the head-to-head, head-to tail, and terpene cyclases all have the same basic “S1, S2” structure and function? To help answer this question, we compared the CrtM/FSPP structure (PDB ID code 2ZCP) with the 3D structures of a series of other prenyl transferases: FPPS (27), GGPPS (16), epiaristolochene synthase (28), and limonene synthase (29), where the donor–acceptor site functions are well known to deduce, based on structural homology, which FPP in CrtM loses diphosphate (to form the cationic, prenyl donor), and which acts as the nucleophile or prenyl acceptor. We first aligned the head-to-tail prenyl synthase FPPS, containing zoledronate and IPP (PDB ID code 2F8Z) to the CrtM/FSPP structure using the combinatorial extension program (30). The superposition (Fig. S7A) indicates that the FSPP diphosphate in S1 (green in Fig. S7A) is close to the bisphosphonate group in the allylic (cationic) site in FPPS, whereas the IPP diphosphate (and the IPP double bond) is in S2. The same conclusion is reached with FSPP in GGPPS (Fig. S7B), the IPP molecule again being located in the S2 or prenyl acceptor site (yellow in Fig. S7B). So, these results indicate that the S2 FPP acts as the nucleophile in CrtM (and by analogy, in SQS and PSY). Similar results are found on comparison of the CrtM structure with those of the class I (ionization initiated) terpene cyclases epiaristolochene synthase (Fig. S7C) and limonene synthase (Fig. S7D) with bound inhibitors. In both cases, the phosphonate or diphosphate headgroups bind in or close to the S1 diphosphate-Mg2+ cluster seen in CrtM, whereas the side chains are in, or close to, the S2 site. Because it is clear that S1 in the cyclases has to be involved in ionization while the side chain in S2 site acts as the nucleophile, we again deduce that the S1 site in CrtM is the cationic (prenyl donor) site, whereas S2 is the (olefinic) prenyl acceptor site.
Conclusions
Overall, the results presented above are of broad general interest because they provide detailed insights into the first committed reactions in sterol and carotenoid biosynthesis: the head-to-head condensation of two prenyl diphosphates to form a cyclopropylcarbinyl diphosphate, followed by ring opening to form DHS. We conclude that FPP in the bent S1 site ionizes to form the prenyl donor carbocation which then moves into the protein’s interior to react with the S2 site FPP C2,3 double bond, forming PSPP, completing the first-half reaction. In the second-half reaction, the PSPP diphosphate then moves back to S1 to ionize. Two cationic inhibitors act as PSPP–carbocation isosteres, binding with their charge center in the same region as the cyclopropyl group, whereas their side-chain substituents bind also to the S1 side-chain site. The same S1 + S2 binding motif is seen with a potent phosphonosulfonate, and dual-site binding is likely to be the origin of the potent activity of the quinuclidinols. This information should be of interest in the context of developing CrtM/SQS inhibitors as antiinfective drugs.
Methods
FPP, FSPP, GGPP, and GGSPP were synthesized according to literature methods (14, 31). PSPP was made biosynthetically by using human SQS (hSQS). Fifty milligrams of hSQS, 10 mg of FPP, together with 20 units of baker’s yeast pyrophosphatase (Sigma-Aldrich) were dissolved in 10 mL reaction buffer (25 mM Hepes, 100 mM NaCl, 0.5 mM MgCl2, pH7.4). The reaction mixture was stirred at room temperature for 8 h, then quenched by adding 10 mM EDTA and solid NaCl. The solution was centrifuged at 4,000 × g for 20 min and the supernatant was applied to a 5-mL C8 solid-phase extraction column (SC300C8K, Western Analytical Products, Inc.). The column was washed with 10 mL reaction buffer and 10 mL 30% acetonitrile, sequentially, to remove residual enzyme, substrate, and other contaminants. PSPP was eluted using 40% acetonitrile (in water) and purity was confirmed by LC-MS/MS. S-thiolo-PSPP was prepared using a similar procedure, except that FSPP and FPP (2∶1) were used. LC/MS analyses were carried out using an Agilent LC/MSD Trap XCT Plus instrument. Compounds were separated on a 5-μm (4.6 × 150 mm) Eclipse XDB-C8 column (Agilent) using a 0–100% acetonitrile (in 25 mM NH4HCO3 buffer) gradient and monitored by using negative-ion mode electrospray ionization.
CrtM crystals were obtained as described previously (10) with some modifications and were soaked with PSPP and various inhibitors to obtain the complex structures. Full details are given in SI Methods. Structure determinations and refinements were carried out as described previously (10) with full details given in SI Methods.
Supplementary Material
Acknowledgments.
We thank Dr. Howard Robinson for collecting the hSQS X-ray data, M.-F. Hsu for technical assistance, Mr. Y.G. Gao for help with crystallization, Dr. Dolores Gonzalez-Pacanowska for providing the hSQS expression system, and R. M. Coates for a sample of DHS. This work was supported by grants from the United States Public Health Service [National Institutes of Health Grant AI-074233 (to E.O.)] and from the Academia Sinica and the National Science Council [NSC 97-3112-B-001-017 and NSC 98-3112-B-001-024 (to A.H.-J.W.)]. Portions of the research were carried out at the National Synchrotron Radiation Research Center, a national user facility supported by the NSC of Taiwan, Republic of China, and the Photon Factory in Japan. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. Use of the Life Science Collaborative Access Team Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). Data for the hSQS study were measured at beamline X29A of the National Synchrotron Light Source, where financial support comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy, and from the National Center for Research Resources of the National Institutes of Health.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank, www.pdb.org for dehydrosqualene synthase complexed with presqualene diphosphate (PDB ID codes 3LGZ, 3ADZ, 3NPR), dehydrosqualene (PDB ID code 3NRI), BPH-651 (PDB ID code 3ACW), BPH-673 (PDB ID code 3ACX), BPH-702 (PDB ID code 3ACY), and GGSPP (PDB ID code 3AE0), and for human squalene synthase complexed with BPH-652 (PDB ID code 3LEE).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010907107/-/DCSupplemental.
References
- 1.Christianson DW. Chemistry. Roots of biosynthetic diversity. Science. 2007;316:60–61. doi: 10.1126/science.1141630. [DOI] [PubMed] [Google Scholar]
- 2.Thulasiram HV, Erickson HK, Poulter CD. Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis. Science. 2007;316:73–76. doi: 10.1126/science.1137786. [DOI] [PubMed] [Google Scholar]
- 3.Thoma R, et al. Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature. 2004;432:118–122. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]
- 4.Sealey-Cardona M, et al. Kinetic characterization of squalene synthase from Trypanosoma cruzi: Selective inhibition by quinuclidine derivatives. Antimicrob Agents Chemother. 2007;51:2123–2129. doi: 10.1128/AAC.01454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandes Rodrigues JC, et al. In vitro activities of ER-119884 and E5700, two potent squalene synthase inhibitors, against Leishmania amazonensis: antiproliferative, biochemical, and ultrastructural effects. Antimicrob Agents Chemother. 2008;52:4098–4114. doi: 10.1128/AAC.01616-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Babili S, Beyer P. Golden rice—five years on the road—five years to go? Trends Plant Sci. 2005;10:565–573. doi: 10.1016/j.tplants.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham FX, Gantt E. Genes and enzymes of carotenoid biosynthesis in plants. Annu Rev Plant Physiol Plant Mol Biol. 1998;49:557–583. doi: 10.1146/annurev.arplant.49.1.557. [DOI] [PubMed] [Google Scholar]
- 8.Pelz A, et al. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J Biol Chem. 2005;280:32493–32498. doi: 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- 9.Liu GY, et al. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med. 2005;202:209–215. doi: 10.1084/jem.20050846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu CI, et al. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandit J, et al. Crystal structure of human squalene synthase. A key enzyme in cholesterol biosynthesis. J Biol Chem. 2000;275:30610–30617. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- 12.Blagg BS, Jarstfer MB, Rogers DH, Poulter CD. Recombinant squalene synthase. A mechanism for the rearrangement of presqualene diphosphate to squalene. J Am Chem Soc. 2002;124:8846–8853. doi: 10.1021/ja020411a. [DOI] [PubMed] [Google Scholar]
- 13.Gu P, Ishii Y, Spencer TA, Shechter I. Function-structure studies and identification of three enzyme domains involved in the catalytic activity in rat hepatic squalene synthase. J Biol Chem. 1998;273:12515–12525. doi: 10.1074/jbc.273.20.12515. [DOI] [PubMed] [Google Scholar]
- 14.Phan RM, Poulter CD. Synthesis of (S)-isoprenoid thiodiphosphates as substrates and inhibitors. J Org Chem. 2001;66:6705–6710. doi: 10.1021/jo010505n. [DOI] [PubMed] [Google Scholar]
- 15.Hosfield DJ, et al. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279:8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 16.Guo RT, et al. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104:10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duncan R, Drueckhammer DG. A pseudoisomerization route to aldose sugars using aldolase catalysis. J Org Chem. 1995;60:7394–7395. [Google Scholar]
- 18.Umeno D, Arnold FH. Evolution of a pathway to novel long-chain carotenoids. J Bacteriol. 2004;186:1531–1536. doi: 10.1128/JB.186.5.1531-1536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friesner RA, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 20.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valdar WS. Scoring residue conservation. Proteins. 2002;48:227–241. doi: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- 22.Urbina JA, et al. In vitro and in vivo activities of E5700 and ER-119884, two novel orally active squalene synthase inhibitors, against Trypanosoma cruzi. Antimicrob Agents Chemother. 2004;48:2379–2387. doi: 10.1128/AAC.48.7.2379-2387.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aggarwal VK, Emme I, Fulford SY. Correlation between pK(a) and reactivity of quinuclidine-based catalysts in the Baylis-Hillman reaction: Discovery of quinuclidine as optimum catalyst leading to substantial enhancement of scope. J Org Chem. 2003;68:692–700. doi: 10.1021/jo026671s. [DOI] [PubMed] [Google Scholar]
- 24.Martin MB, Arnold W, Heath HT, III, Urbina JA, Oldfield E. Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem Biophys Res Commun. 1999;263:754–758. doi: 10.1006/bbrc.1999.1404. [DOI] [PubMed] [Google Scholar]
- 25.Mao J, et al. Solid-state NMR, crystallographic, and computational investigation of bisphosphonates and farnesyl diphosphate synthase-bisphosphonate complexes. J Am Chem Soc. 2006;128:14485–14497. doi: 10.1021/ja061737c. [DOI] [PubMed] [Google Scholar]
- 26.Brown GR, et al. Phenoxypropylamines: A new series of squalene synthase inhibitors. J Med Chem. 1995;38:4157–4160. doi: 10.1021/jm00021a003. [DOI] [PubMed] [Google Scholar]
- 27.Rondeau JM, et al. Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs. ChemMedChem. 2006;1:267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]
- 28.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 29.Hyatt DC, et al. Structure of limonene synthase, a simple model for terpenoid cyclase catalysis. Proc Natl Acad Sci USA. 2007;104:5360–5365. doi: 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shindyalov IN, Bourne PE. A database and tools for 3-D protein structure comparison and alignment using the combinatorial extension (CE) algorithm. Nucleic Acids Res. 2001;29:228–229. doi: 10.1093/nar/29.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davisson VJ, et al. Phosphorylation of isoprenoid alcohols. J Org Chem. 1986;51:4768–4779. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.