

Abstract
Although commonplace in human disease genetics, genome-wide association (GWA) studies have only relatively recently been applied to plants. Using 32 phenotypes in the inbreeding crop barley, we report GWA mapping of 15 morphological traits across ∼500 cultivars genotyped with 1,536 SNPs. In contrast to the majority of human GWA studies, we observe high levels of linkage disequilibrium within and between chromosomes. Despite this, GWA analysis readily detected common alleles of high penetrance. To investigate the potential of combining GWA mapping with comparative analysis to resolve traits to candidate polymorphism level in unsequenced genomes, we fine-mapped a selected phenotype (anthocyanin pigmentation) within a 140-kb interval containing three genes. Of these, resequencing the putative anthocyanin pathway gene HvbHLH1 identified a deletion resulting in a premature stop codon upstream of the basic helix-loop-helix domain, which was diagnostic for lack of anthocyanin in our association and biparental mapping populations. The methodology described here is transferable to species with limited genomic resources, providing a paradigm for reducing the threshold of map-based cloning in unsequenced crops.
Keywords: genetic variation, small grain cereals, colinearity
The convergence of high-throughput genotyping platforms with the development of appropriate statistical methods has meant that the impasse in successful implementation of association mapping in humans has now been largely overcome, with genome-wide association (GWA) scans reporting both known and novel loci influencing common disease risk in humans (1, 2). This success has led to the application of association mapping approaches in plant species (reviewed in ref. 3), where attention has predominantly focused on candidate genes or previously cloned loci (4–6). Of the relatively few GWA scans performed in plants (7–10), only one study, in which the underlying gene was not known a priori, has achieved resolution to the level of putative causative variant (using maize inbred lines) (11). Unlike maize, the majority of the world's important crops do not yet possess genome sequence assemblies. Here, we use the inbreeding cereal crop barley (Hordeum vulgare ssp. vulgare L.) to investigate the feasibility of GWA mapping to candidate polymorphism resolution in an unsequenced large-genome crop species.
In association mapping studies, detection of significant association relies predominantly on genetic marker coverage, the number of individuals studied, and linkage disequilibrium (LD) between causative and linked polymorphisms (12). Although genetic stratification in the majority of human studies is low (13), inbreeding crops such as barley commonly display highly complex population structure because of their primarily inbreeding reproductive strategy, population history, and close kinship (14). The resulting elevation of long-range LD can lead to increased frequency of false-positive associations during association analyses (15). However, if robust statistical correction for the effects of population substructure/kinship can be used, high LD should permit successful GWA scans using relatively low marker densities (16). Here, we validate this assumption, first by an in silico estimation of statistical power and then by successful GWA mapping of 15 morphological traits. Fine-mapping of one of these identified a candidate gene of biological relevance with an exonic insertion/deletion (InDel) causing a premature stop codon perfectly correlated with the nonfunctional allele in our association and biparental mapping populations.
Results
Genetic Markers, Population Substructure, and Correction of False-Positive Associations.
Using publicly available barley expressed sequence tags (ESTs), we recently developed and validated a 1,536-feature SNP array, averaging 1.4 markers/cM across the ∼1,100-cM genome (14, 17), which represents the most comprehensive resource of its kind currently available in barley. This was used to genotype a collection of 500 cultivars selected from UK registration trials over the past 20 y. Markers with minor allele frequency <0.1 or genotyping success rate ≤0.95 were removed from the dataset as were cultivars with a success rate ≤0.84. The low level of heterozygous genotypes observed (0.8%) is consistent with the inbreeding nature of barley, and these data points were excluded from subsequent analysis. The final dataset consisted of 490 cultivars (Table S1) and 1,111 markers (mean nucleotide diversity = 0.41; mean, median, and mode distance between markers = 1.0, 0.5, and 0.0 cM, respectively; 5.7% markers ≥4-cM spacing), with a call rate of 0.997.
We first investigated genetic substructure within the association panel. Principal component analysis showed 24% of the genetic variation can be described by the first two components (Fig. 1A); overlaying phenotypic information for the two major agronomic classes (ear row number and seasonal growth habit) suggests that these categorical trait combinations are largely responsible for the major genetic divisions observed (Fig. 1A and SI Text, Genetic Stratification). The extent of genetic stratification was further investigated using a Bayesian approach to assign fractional membership of cultivars to a number of historical subpopulations (K) using the program STRUCTURE (18–20) (Fig. 1A). Analysis of the likelihood of various models (LnP[D] within the range K = 2–20; burnin = 250,000; Markov Chain Monte Carlo iterations = 1,000,000) indicates the optimum number of subpopulations is ∼10 (Fig. 1A). To account for the strong genetic stratification observed, we investigated various methodologies (SI Text, Correction of Genetic Stratification), finding a mixed linear regression model (21) with coefficients of kinship estimated using a matrix of between-individual genetic correlation to perform best (22).
Fig. 1.

Investigation of genetic substructure. (A) Principle component analysis of UK cultivars and barley progenitor lines (n = 620). Phenotypic combinations for row number (2 = two-row, 6 = six-row) and seasonal growth habit (S, spring; W, winter) are indicated. Varietal membership to the 10 subpopulations identified using STRUCTURE are overlaid. (B) Decay of pair-wise marker LD over increasing genetic distance (cM) before (Left) and after (Right) correction using the mixed linear model. Off-chromosome comparisons are shown at 200 cM. The Bonferroni corrected P = 0.05 significance threshold is indicated. (C) Naïve and corrected GWA analysis of seasonal growth habit illustrating the extent of confounding present. Arrow indicates the significance threshold. (D) Predicted experimental power to detect a trait controlled by 1, 2, and 10 loci (h2 = 0.9) over genetic distance (±cM). Power is measured as the proportion of simulations in which at least one causative locus was detected (q value ≤ 0.1). Error bars denote ±1 SE.
Because identification of marker trait associations relies on detection of significant LD after correction for spurious signal caused by population genealogy, we investigated the extent of pair-wise marker associations with and without statistical correction for confounding (Fig. 1B) (a justification of the use of P values from the mixed model for binary data is given in SI Text, Corrected Between-Marker LD and Fig. S1). Strikingly, we find that, for uncorrected analysis, 35% of interchromosomal associations between marker pairs are significant (−log10 P ≥ 4.35). Furthermore, significant intrachromosomal LD is evident across the full length of chromosomes (mean distance between significant marker pairs = 40.2 cM, median = 30.7 cM). After adjustment using the mixed model, this is reduced to <10 cM (mean = 1.2 cM, median = 0.6 cM), with the proportion of significant interchromosomal associations controlled to just 0.1%. The specificity of the control achieved using the mixed model is shown by the suppression of off-chromosome and long-distance association while retaining signal at shorter genetic distances. The extreme levels of confounding encountered are illustrated by naïve analysis of a selected trait (seasonal growth habit), where 72% of all markers return significant associations. This is reduced to 1% after correction using the mixed model (Fig. 1C).
Statistical Power.
We modeled the power to detect 1, 2, and 10 independent loci distributed randomly across the genome (100, 100, and 375 permutations, respectively), with a heritability (h2) of 0.5 and 0.9 (Fig. 1D and Fig. S2). Using the mixed model to correct for genetic substructure, simulations based on a trait controlled by one locus predict that our experimental design has a high probability (≥0.92 for both values of h2) of detecting significant (q value ≤ 0.1) associations within windows of ≤8 cM. This compares favorably with correction using genomic control, where the power to detect a single locus within the same genetic interval is 0.46 (h2 = 0.5) and 0.55 (h2 = 0.9) and falls dramatically in the two-locus model (0.18, h2 = 0.5; 0.23, h2 = 0.9). For a 10-locus trait, even when considering a genetic interval of 20 cM, power to detect one or more loci after correction with the mixed model is low (0.25, h2 = 0.5; 0.58, h2 = 0.9), whereas power after correction with genomic control is effectively zero (≤0.01 for all scenarios investigated). To report only the most robust marker trait associations in the experimental dataset, we used a Bonferroni corrected P ≤ 0.05 threshold (−log10 P ≥ 4.35) to account for multiple testing. To simplify the results reported here, we set our discovery criterion to be more than or equal to two significant markers within a 4-cM interval.
GWA Mapping and Marker Enrichment.
We analyzed 32 morphological traits (Table S2) scored to varying degrees of coverage within the association panel (Fig. S3). Initially, uncorrected association analyses between each SNP and the phenotype were implemented. For all traits, the expectation of strong confounding because of a heavily structured population was realized in the observed excess of associations when tested without correction (Fig. 1C and Fig. S4). Subsequent analysis using the mixed model to correct for population substructure identified 18 genomic locations associated (5 ≤ −log10 P ≤ 113) with 15 traits (Fig. 2 and Table S3). Analysis of quantile–quantile plots (23) of expected and observed associations indicate that, although power to detect significant associations is retained, efficient correction for the extensive genetic substructure observed has been largely achieved (Fig. S4). The majority of traits with significant associations seemed to identify a single genetic locus: seasonal growth habit (chromosome 1H), grain lateral nerve spiculation (2H), awn anthocyanin coloration, awn anthocyanin intensity, auricle anthocyanin coloration, auricle anthocyanin intensity, lemma nerve anthocyanin intensity (identifying overlapping regions on 2H), grain aleurone color (4H), hairiness of leaf sheath (4H), rachilla hair type (5H), ear attitude (5H), and grain ventral furrow hair (6H). The ear morphology characters sterile spikelet attitude and ear row number both identified two regions of association (1H and 2H; 3H and 4H, respectively). Traits for which significant associations were detected had a mean heritability of h2 = 0.58, whereas the proportion of phenotypic variation (VP) accounted for by peak markers was between 0.08 and 0.84 (mean VP = 0.41) (Table S4). Overall, 17 traits (mean h2 = 0.18) failed to return significant associations satisfying our discovery criteria (Fig. S5). To investigate the potential for improved mapping resolution, we exploited colinearity between cereal genomes (24) to develop additional genetic markers within target genomic regions. Using sequence homology between barley ESTs and the rice (Oryza sativa) genome, 49 gene-specific markers were developed, and their genetic map locations were determined (Table S5), representing the addition of 3–18 genes within the majority of significant chromosomal regions identified. Resequencing these amplicons in a subset of 90 varieties identified 195 polymorphisms over ∼22 kb of genetic sequence (176 SNPs, 13 InDels, and 6 simple sequence repeats (SSRs)). The inclusion of additional polymorphisms in association analyses within this subset identified 14 genes with associations at least as significant as the peak marker identified in the original GWA scan (Table S5).
Fig. 2.

GWA scans of the 15 traits with significant associations (P < 0.05, Bonferroni corrected; indicated by a dashed line). Barley chromosomes 1H to 7H are shown.
Fine-Mapping the ANTHOCYANINLESS 2 Locus.
Because higher mapping resolution was found to be generally achievable, we selected one phenotype (anthocyanin pigmentation) for fine-mapping. Studies of anthocyanin biosynthesis have reported a Mendelian locus on chromosome 2H termed ANTHOCYANINLESS 2 (ANT2) (SI Text, Mendelian Loci Previously Identified in Biparental Mapping Populations). To support the hypothesis that our GWA scans identified ANT2, we developed a doubled haploid mapping population (n = 209) between two cultivars included in our association panel that differ for the presence (Retriever) and absence (Saffron) of anthocyanin pigmentation (subsequently referred to as red and white varieties, respectively) and genotyped this population using our 1,536-feature SNP array. Anthocyanin pigmentation was mapped as a single dominant Mendelian trait that cosegregated with markers 11_21007 and 11_21175 at 98.82 cM on the long arm of chromosome 2H, lying within the chromosomal interval identified in the association panel and collinear with the previously mapped ANT2 locus (25).
On the grounds that the ANT2 locus encodes a master switch for anthocyanin production, with the recessive allele resulting in abolition of anthocyanin expression, we then used a composite phenotype with two character states: absence of anthocyanin coloration in all recorded tissues (awns, auricles, and lemma nerves) or presence in one or more of these structures (Fig. 3A). GWA analysis found the genetic interval controlling this trait to lie between 93.5 and 103.67 cM on chromosome 2H, with the peak association (−log10 P = 51.7, marker 11_21175) at 96.82 cM (Fig. S6A). Sliding-window LD analysis around the ANT2 locus shows a putative selective sweep of 17 cM (D’ = 1) in white varieties (Fig. S6C). Towards developing additional genetic markers around the ANT2 locus, we investigated the extent of macrocolinearity between the genetic map of barley chromosome 2H and the physical maps of rice chromosome 4 and brachypodium (Brachypodium distachyon) chromosome 5 (Fig. S6B and Table S6A). Using these comparative analyses, we developed genotypic assays for six additional barley genes (HvOs04g47010, HvOs04g47020, HvOs04g47080, HvOs04g47110, HvOs04g47120, and HvOs04g47170) close to the most significant markers identified during GWA analysis (Fig. 3B) and applied these across the complete panel. Subsequent association analysis shows that the ANT2 locus is defined within a ≤0.57-cM interval by recombination events distal to HvOs04g47110 (conserved hypothetical protein) and proximal to HvOs04g47020 (genetic modifier) (Fig. 3 C and D). A contiguous barley physical map encompassing the flanking markers was constructed, and the minimum tiling path was sequenced (bacterial artificial chromosome (BAC) clones 77O02, 739E22, and 274B17; GenBank accession HM163343). The 260-kb sequenced interval contains 11 genes, of which 8 are located at collinear positions in one or more related cereal genomes (Fig. 3B and Table S6B). Within the sequenced contig, three gene models were identified between the flanking markers, including a strong candidate gene encoding a protein containing a basic helix-loop-helix (bHLH) DNA-binding domain (Fig. 3 B and E), a feature common among transcription factors known to regulate pigment synthesis in other plant species (26). Phylogenetic analysis of bHLH proteins from the anthocyanin pigmentation pathways of petunia, antirrhinum, maize, and arabidopsis as well as their rice homologs shows that the barley ANT2 candidate gene belongs to a clade containing bHLH proteins encoded by genes at the R/B loci (Fig. S7A) previously found to control anthocyanin pigmentation in maize (27). Semiquantitative RT-PCR found HvbHLH1 to be expressed in the target tissues of both Saffron and Retriever (Fig. S7B). Sequencing HvbHLH1 from −343 to +4,628 bp in a subset of 90 cultivars (GenBank accessions HM370298–HM370387) identified 69 polymorphisms arranged in four haplotypes, with haplotype 1 exclusive to white varieties, whereas haplotypes 2–4 were associated with anthocyanin coloration in one or more tissues. The identified polymorphisms include eight synonymous and four nonsynonymous variants as well as a 16-bp deletion within exon 6 that results in truncation of the predicted protein upstream of the bHLH domain (Fig. 3 E and F). Subsequent genotyping in the complete association panel established that the 16-bp deletion occurred in all cultivars lacking anthocyanin pigmentation but was absent in cultivars in which anthocyanin is expressed in one or more tissues (Fig. 3D). We also found the deletion to perfectly cosegregate with white ant2 alleles in our biparental mapping population.
Fig. 3.

Fine-mapping of ANT2. (A) Example of anthocyanin expression in tissues from selected “red” (Retriever) and “white” (Saffron) varieties. (B) The sequenced physical map of the barley ANT2 region aligned to colinear regions of rice, sorghum, and Brachypodium. Barley predicted genes: Expressed Protein 1 (EP1), Acyl-CoA thioesterase (ACA), Conserved Hypothetical Protein (CHP), Basic Helix-loop-helix protein 1 (HvbHLH1), Genetic Modifier (GM), Expressed Protein 2 (EP2), No Apical Meristem (NAM), and Cis-Zeatin O-Glucosyltransferase (Cis-ZOG). Barley BAC clones from left to right are 274B17, 739E22, and 77O02 (GenBank accession HM163343). The positions of BAC end sequences and the transposon-based marker 739E22-1 are indicated by black and green circles, respectively. (C) Haplotype frequencies in “red” and “white” varieties. Awn coloration is illustrated. (D) GWA scan for the trait anthocyanin expression per se. The two peak markers are highlighted in red. (E) Structure of the ANT2 candidate gene, HvbHLH1. Exons are denoted by black rectangles; the region encoding the predicted bHLH domain is highlighted in blue. The position of the premature stop codon in the white allele is indicated by an asterisk. (F) Partial DNA sequence of HvbHLH1 exon 6 illustrating the position of the 16-bp InDel. The 10-bp repeat sequences flanking the deletion are underlined; the probability (p) that these locations are by chance is indicated.
Origins of the White ant2 Allele.
Sequence analysis suggests that the white ant2 allele is a mutated form of the wild-type red allele, caused by a 16-bp exonic deletion in HvbHLH1 that results in a truncated predicted protein. This model is supported by the presence of identical 10-bp sequence motifs that flank the deletion (P repeat sequences are located by chance = 0.00044), a hallmark of illegitimate recombination after double-stranded DNA break repair (28) (Fig. 3F). To investigate the geographic origin of the white allele, we screened 117 wild barley accessions and 471 predominantly European landraces (distinct locally adapted populations that predate formal crop improvement) for the 16-bp InDel in HvbHLH1. Although not found in wild barley, 20 landraces possessed the deletion, of which 13 are located within Italy (Fig. S7C).
Discussion
Before reporting the results of any GWA scan, it is essential to understand the limitations of experimental design (12). Such an approach affords prior knowledge of the experimental power and precision that is likely to be achieved. This is normal in human studies but is infrequently applied in plants, representing a serious omission with implications for the reliability of the associations reported (for an example of concerns regarding reported associations in plants, see ref. 3). In this study, a priori assessments showed that marker density was sufficient to identify significant pair-wise marker associations after correction for genetic substructure, whereas power calculations predicted our experimental design to readily detect highly heritable traits controlled by a small number of loci. These experimental parameters were validated in practice, with GWA scans identifying significant associations for 47% of the traits investigated; between one and three significant loci were detected per trait (SI Text, Interpretation of GWA Peaks). Of these, the genetic map locations of associations identified in nine traits have been previously corroborated in biparental mapping populations, where they map as Mendelian loci (SI Text, Mendelian Loci Previously Identified in Biparental Mapping Populations). Although relatively few genes have been map-based cloned in barley, four associations map to regions spanning cloned genes controlling related phenotypes (SI Text, Comparison of GWAs with Previously Map-Based Cloned Genes). Low estimates of heritability for the 17 traits for which no significant associations were identified (mean h2 = 0.18) are likely to have contributed to this failure. Furthermore, 10 of these 17 traits are scored on a more continuous scale (seven or more character states) and therefore, are assumed to be controlled by multiple loci. Our power calculations indicate that only a small proportion of loci-controlling polygenic traits are likely to be detected in an experiment of this scale. These findings should inform future experimental design for plant GWA studies, which have predominantly used smaller populations and marker numbers than described here.
In contrast to human studies, where genetic stratification is generally low, we show successful GWA mapping in the presence of extremely high population substructure. There has been much debate about the most efficient way to correct for genetic stratification in association studies. An increasingly held view is that correction using Q alone is often inadequate, especially where complex patterns of kinship are present (3, 10), and that correction using Q +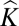 is more effective (3). However, determining the appropriate Q matrix is computationally intensive and therefore, is impractical when dealing with large datasets. Although it has been commonplace to set negative entries of
is more effective (3). However, determining the appropriate Q matrix is computationally intensive and therefore, is impractical when dealing with large datasets. Although it has been commonplace to set negative entries of 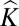 = 0, in our definition,
= 0, in our definition, 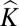 represents a correlation coefficient measuring excess/lack of between-individual allele sharing compared with that expected by chance; thus, negative kinship values are interpretable and provide valuable information about population structure. Accordingly, we use
represents a correlation coefficient measuring excess/lack of between-individual allele sharing compared with that expected by chance; thus, negative kinship values are interpretable and provide valuable information about population structure. Accordingly, we use 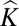 alone, arguing that this captures most of the population substructure (22). Our demonstration that type 1 error rates are close to nominal levels when applying the linear mixed model to binary traits provides statistical justification for the detection of Mendelian loci using this approach.
alone, arguing that this captures most of the population substructure (22). Our demonstration that type 1 error rates are close to nominal levels when applying the linear mixed model to binary traits provides statistical justification for the detection of Mendelian loci using this approach.
Despite the exceptionally high levels of long-range LD in barley (29), we show that adequate levels of marker saturation for GWA analysis are yet to be achieved. Indeed, additional marker development and fine-mapping of anthocyanin pigmentation identified sufficient recombination to define a physical interval containing just three genes. This region included HvbHLH1, whose predicted protein shows homology to bHLH proteins known to control anthocyanin regulation in petunia (AN1 and JAF13) (30), arabidopsis (TT8) (31), and maize (R and B) (27). Population-based resequencing identified an exonic deletion predicted to knockout the bHLH domain, showing a gene of biological relevance with a plausible mutation that shows perfect association with the absence of anthocyanin (and providing breeders with an easily applied diagnostic marker). The lower haplotype frequency and elevated LD associated with the white ant2 allele are consistent with its mutation from a red allele and seem to have occurred because of nonhomologous recombination, a process increasingly thought to play an important role in plant and human genome dynamics (28, 32). Although not found in wild barley, the white allele is present at low frequency (4%) in landraces. Geographical clustering suggests that it arose in Italy and persisted at low frequency until wider deployment at the onset of modern breeding at the end of the 19th century. Accordingly, the recent introduction of a single white HvbHLH1 haplotype by early breeders is likely to have been from a very restricted germplasm pool, helping to explain the putative selective sweep observed around the ANT2 locus.
Hierarchical approaches to association mapping in humans, animals, and plants have been suggested, in which a primary panel consisting of individuals with high LD is used for coarse mapping followed by fine-mapping in a second panel of individuals with low LD (13, 16, 33). Of course, a two-tier approach is not possible if the trait of interest is not segregating at sufficient frequency in the secondary panel. However, the recent history of artificial out-crossing between inbred barley cultivars provides a pseudo-outbreeding population (14) with extended haplotype blocks, within which it is relatively straightforward to detect where recombination has taken place. Encouragingly, we show that even using relatively small populations (<500), detectable recombination may often be sufficient to resolve GWA analyses to manageable physical intervals in primary association panels. This is an important observation, because candidate genes and functional polymorphisms may not always be obvious and therefore, require precise fine-mapping before being considered for further investigation. The genomic location of the barley ANT2 candidate gene HvbHLH1, less than 100 kb from one of the three peak markers identified during GWA analysis, shows the feasibility of GWA scans in cereal crops to effectively genome land, a process that can only improve with the availability of additional genetic markers and larger association panels.
Materials and Methods
Germplasm and Genotyping.
We selected 500 UK barley cultivars from entries to national registration trials between 1993 and 2005 (Table S1). DNA was extracted from leaf tissue using the Nucleoplex Automated DNA Isolation kit (Tepnel). A set of 1,536 EST-based SNPs were genotyped using GoldenGate BeadArray technology (Illumina) as previously described (14). Additional genotyping was conducted as described in SI Text, Additional Gene-Based Markers using primers listed in Table S5. The doubled haploid mapping population was derived from F1 progeny from a cross between cultivars Saffron and Retriever using microspore culture (34).
Phenotypes.
Historical phenotypic data (Table S2) collected during varietal registration were obtained from records curated at NIAB (http://www.niab.com/). Traits were scored over two seasons according to International Union for the Protection of New Varieties of Plants (UPOV) protocols (http://www.upov.int/). Traits with a fill of <200 cultivars were removed, leaving 32 traits for analyses.
Colinearity, Phylogeny, and Geographic Plots.
We used a previously described barley consensus genetic map (35). Putative homologs of barley HarvEST U32 Unigenes (http://harvest.ucr.edu/) corresponding to the 1,536 SNPs assayed were identified by BLASTX analysis of rice gene models (MSU rice genome annotation, Release 6.1) using a cutoff value of 7.0 e−5 and plotted using Microsoft Excel. Phylogenetic analysis was conducted using the PHYLIP package v3.5 (36) as described in SIText, Phylogenetic Analysis. Geodata were plotted using ArcGIS v.10 (ESRI).
BAC Analysis and RT-PCR.
Clones from barley BAC libraries constructed from the cultivar Morex (ACPFG) were identified by quantitative (qPCR) screening of pooled DNAs (SI Text, BAC Library Screening) and integrated into the ongoing construction of the barley physical map. Selected BAC clones were sequenced using the 454 GS-FLX platform (Roche), and sequence annotation was performed using the TriAnnot pipeline (37) with additional manual annotation. RT-PCR methodology is described in SI Text, RT-PCR.
Statistical Analysis.
Principal component analysis was performed using GenStat v.8 (VSN International) on a similarity matrix created using a simple matching coefficient. We used a Bayesian MCMC approach implemented in the program STRUCTURE (18–20) v2 to estimate the membership probability of each cultivar to a number of hypothetical founding subpopulations (K). To avoid overestimation of subpopulation divergence (20), a subset of 307 genome-wide genetic markers with ≥2-cM spacing was selected for this analysis. K was estimated using the admixture model with correlated allele frequencies modeled with a burnin of 2.5 × 105 cycles followed by 106 cycles with duplicate runs between K = 2–20; each returned matrices (Q) of fractional subpopulation membership for each cultivar. Agreement between duplicates was assessed as previously described (29). A mixed linear regression model was also applied, which accounts for multiple levels of relatedness because of historical population substructure and kinship (38). We used the efficient mixed model association approach (21), implemented in R v 2.9.0 (http://www.R-project.org/), using previously described software (22). Relatedness between two individuals was estimated as pair-wise correlation based on standardized (subtract mean and divide by SD) genotypes. SNPHAP v1.3 (http://www-gene.cimr.cam.ac.uk/clayton/software/) was used to infer missing genotypic data using sliding windows of 30 adjacent markers with 20 marker overlaps. The significance of GWA scans was estimated using a Bonferroni corrected 0.05 P value (−log10 P = 4.35). Power was estimated with significance determined using q value (http://cran.r-project.org/), which implements the method described in ref. 39. Genomic control (40, 41) was implemented by dividing the 1 df test statistic for association by the ratio of the observed median of the test statistic to its expected median under the null. Analyses of association without adjustment for population substructure were by 1 df χ2 or t tests. Experimental power, h2, and VP were estimated as described in SI Text, Estimations of Statistical Power, Heritability, and Phenotypic Variation. The probability that short sequence repeat motifs flank deletion breakpoints within HvbHLH1 were estimated as previously described (28).
Supplementary Material
Acknowledgments
We thank J. DeYoung for SNP genotyping, the NIAB Agricultural Food Crops team for phenotypic data, and Eurofins MWG Operon for BAC sequencing. This work was supported by Defra, the Scottish Government, and the Biotechnology and Biological Sciences Research Council through Sustainable Arable LINK Program Grant 302/BB/D522003/1, Association Genetics of UK Elite Barley, and the NIAB Trust.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. HM370298–HM370387 and HM163343).
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010179107/-/DCSupplemental.
Contributor Information
Chris Booer, Brewing Research International, Surrey, United Kingdom;.
Steve Pike, Calibre Control International, Warington, United Kingdom;.
Graeme Hamilton, Molson Coors Brewing Company (UK), Burton-on-Trent, United Kingdom;.
Graham Jellis, Home-Grown Cereals Authority, Kenilworth, United Kingdom;.
Nigel Davies, The Maltsters Association of Great Britain, Newark, United Kingdom;.
Anne Ross, Mylnefield Research Services Ltd., Invergowrie, Dundee, Scotland, United Kingdom;.
Paul Bury, Syngenta Seeds Ltd, Market Stainton, Market Rasen, United Kingdom;.
Rodney Habgood, Nickerson (UK) Ltd., Rothwell, Market Rasen, United Kingdom;.
Steve Klose, LS Plant Breeding (SERASEM), Cambridge, United Kingdom;.
Dominique Vequaud, Secobra Recherches, Maule, France;.
Therese Christerson, Svaloff Weibull AB, Svalöv, Sweden;.
James Brosnan, The Scotch Whiskey Research Institute, Edinburgh, Scotland, United Kingdom;.
Adrian Newton, Scottish Crop Research Institute, Invergowrie, Dundee, Scotland, United Kingdom;.
Joanne Russell, Scottish Crop Research Institute, Invergowrie, Dundee, Scotland, United Kingdom;.
Paul Shaw, Scottish Crop Research Institute, Invergowrie, Dundee, Scotland, United Kingdom;.
Rosemary Bayles, National Institute of Agricultural Botany, Cambridge, United Kingdom;.
Minghui Wang, University of Birmingham, Birmingham, United Kingdom..
References
- 1.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCarthy MI, et al. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 3.Myles S, et al. Association mapping: Critical considerations shift from genotyping to experimental design. Plant Cell. 2009;21:2194–2202. doi: 10.1105/tpc.109.068437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thornsberry JM, et al. Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet. 2001;28:286–289. doi: 10.1038/90135. [DOI] [PubMed] [Google Scholar]
- 5.Salvi S, et al. Conserved noncoding genomic sequences associated with a flowering-time quantitative trait locus in maize. Proc Natl Acad Sci USA. 2007;104:11376–11381. doi: 10.1073/pnas.0704145104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stracke S, et al. Association mapping reveals gene action and interactions in the determination of flowering time in barley. Theor Appl Genet. 2009;118:259–273. doi: 10.1007/s00122-008-0896-y. [DOI] [PubMed] [Google Scholar]
- 7.Aranzana MJ, et al. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 2005;1:e60. doi: 10.1371/journal.pgen.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kraakman AT, Niks RE, Van den Berg PM, Stam P, Van Eeuwilk FA. Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics. 2004;168:435–446. doi: 10.1534/genetics.104.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao K, et al. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 2007;3:e4. doi: 10.1371/journal.pgen.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atwell S, et al. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010;465:627–631. doi: 10.1038/nature08800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belo A, et al. Whole genome scan detects an allelic variant of fad2 associated with increased oleic acid levels in maize. Mol Genet Genomics. 2008;279:1–10. doi: 10.1007/s00438-007-0289-y. [DOI] [PubMed] [Google Scholar]
- 12.Mackay I, Powell W. Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 2007;12:57–63. doi: 10.1016/j.tplants.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg NA, et al. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rostoks N, et al. Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci USA. 2006;103:18656–18661. doi: 10.1073/pnas.0606133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balding DJ. A tutorial on statistical methods for population association studies. Nat Rev Genet. 2006;7:781–791. doi: 10.1038/nrg1916. [DOI] [PubMed] [Google Scholar]
- 16.Waugh R, Jannink J-L, Muehlbauer GJ, Ramsay L. The emergence of whole genome association scans in barley. Curr Opin Plant Biol. 2009;12:218–222. doi: 10.1016/j.pbi.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 17.Close TJ, et al. Development and implementation of high-throughput SNP genotyping in barley. BMC Genomics. 2009 doi: 10.1186/1471-2164-10-582. 10.1186/1471-2164-10-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. Am J Hum Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang HM, et al. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Astle W, Balding DJ. Population structure and cryptic relatedness in genetic association studies. Statist Sci. 2009;24:451–471. [Google Scholar]
- 23.Weir BS, Hill WG, Cardon LR, SNP Consortium Allelic association patterns for a dense SNP map. Genet Epidemiol. 2004;27:442–450. doi: 10.1002/gepi.20038. [DOI] [PubMed] [Google Scholar]
- 24.Devos KM. Updating the ‘crop circle.’. Curr Opin Plant Biol. 2005;8:155–162. doi: 10.1016/j.pbi.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Lahaye T, et al. High-resolution genetic and physical mapping of the Rar1 locus in barley. Theor Appl Genet. 1998;97:526–534. [Google Scholar]
- 26.Sweeney MT, Thomson MJ, Pfeil BE, McCouch S. Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell. 2006;18:283–294. doi: 10.1105/tpc.105.038430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goff SA, Cone KC, Chandler VL. Functional analysis of the transcriptional activator encoded by the maize B gene: Evidence for a direct functional interaction between two classes of regulatory proteins. Genes Dev. 1992;6:864–875. doi: 10.1101/gad.6.5.864. [DOI] [PubMed] [Google Scholar]
- 28.Cockram J, Mackay IJ, O'Sullivan DM. The role of double-stranded break repair in the creation of phenotypic diversity at cereal VRN1 loci. Genetics. 2007;177:2535–2539. doi: 10.1534/genetics.107.074765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cockram J, et al. Association mapping of partitioning loci in barley. BMC Genet. 2008 doi: 10.1186/1471-2156-9-16. 10.1186/1471-2156-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spelt C, Quattrocchio F, Mol J, Koes R. ANTHOCYANIN1 of petunia controls pigment synthesis, vacuolar pH, and seed coat development by genetically distinct mechanisms. Plant Cell. 2002;14:2121–2135. doi: 10.1105/tpc.003772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baudry A, et al. TT2, TT8 and TTG1 synergistically specify the expression of BANYLUS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J. 2004;39:336–380. doi: 10.1111/j.1365-313X.2004.02138.x. [DOI] [PubMed] [Google Scholar]
- 32.Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karlsson EK, et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat Genet. 2007;39:1321–1328. doi: 10.1038/ng.2007.10. [DOI] [PubMed] [Google Scholar]
- 34.Kasha KJ, et al. An improved in vitro technique for isolated microspore culture of barley. Euphytica. 2004;120:379–385. [Google Scholar]
- 35.Thiel T, et al. Evidence and evolutionary analysis of ancient whole-genome duplication in barley predating the divergence from rice. BMC Evol Biol. 2009 doi: 10.1186/1471-2148-9-209. 10.1186/1471-2148-9-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Felsenstein J. PHYLIP—phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 37.Sabot F, et al. Updating of transposable element annotations from large wheat genomic sequences reveals diverse activities and gene associations. Mol Genet Genomics. 2005;274:119–130. doi: 10.1007/s00438-005-0012-9. [DOI] [PubMed] [Google Scholar]
- 38.Yu J, et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet. 2006;38:203–208. doi: 10.1038/ng1702. [DOI] [PubMed] [Google Scholar]
- 39.Storey JD, Tibshirani R. Statistical significance for genome wide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 41.Reich DE, Goldstein DB. Detecting association in a case-control study while correcting for population stratification. Genet Epidemiol. 2001;20:4–16. doi: 10.1002/1098-2272(200101)20:1<4::AID-GEPI2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.