

Abstract
Time-resolved single-molecule biophysical experiments yield data that contain a wealth of dynamic information, in addition to the equilibrium distributions derived from histograms of the time series. In typical force spectroscopic setups the molecule is connected via linkers to a readout device, forming a mechanically coupled dynamic network. Deconvolution of equilibrium distributions, filtering out the influence of the linkers, is a straightforward and common practice. We have developed an analogous dynamic deconvolution theory for the more challenging task of extracting kinetic properties of individual components in networks of arbitrary complexity and topology. Our method determines the intrinsic linear response functions of a given object in the network, describing the power spectrum of conformational fluctuations. The practicality of our approach is demonstrated for the particular case of a protein linked via DNA handles to two optically trapped beads at constant stretching force, which we mimic through Brownian dynamics simulations. Each well in the protein free energy landscape (corresponding to folded, unfolded, or possibly intermediate states) will have its own characteristic equilibrium fluctuations. The associated linear response function is rich in physical content, because it depends both on the shape of the well and its diffusivity—a measure of the internal friction arising from such processes as the transient breaking and reformation of bonds in the protein structure. Starting from the autocorrelation functions of the equilibrium bead fluctuations measured in this force clamp setup, we show how an experimentalist can accurately extract the state-dependent protein diffusivity using a straightforward two-step procedure.
Keywords: optical tweezer, spectral analysis, fluctuation–dissipation relation, coordinate-dependent diffusion
Force spectroscopy of single biomolecules relies most commonly on atomic force microscope (AFM) (1–7) or optical tweezer (8–13) techniques. By recording distance fluctuations under applied tension, these experiments serve as sensitive probes of free energy landscapes (8–11, 13), and structural transformations associated with ligand binding or enzymatic activity (3, 6, 7). All such studies share an unavoidable complication: The signal of interest is the molecule extension as a function of time, but the experimental output signal is an indirect measure like the deflection of the AFM cantilever or the positions of beads in optical traps. The signal is distorted through all elements in the system, which in addition typically include polymeric handles such as protein domains or double-stranded DNA, which connect the biomolecule to the cantilever or bead. As shown in the case of an RNA hairpin in an optical tweezer (14), handle fluctuations lead to nontrivial distortions in equilibrium properties like the energy landscape as well as dynamic quantities like folding/unfolding rates. If an accurate estimate of the biomolecule properties is the goal, then one needs a systematic procedure to subtract the extraneous effects and recover the original signal from experimental time-series data.
Static deconvolution, which operates on the equilibrium distribution functions of connected objects, is a well-known statistical mechanics procedure and has been successfully applied to recover the free energy landscapes of DNA hairpins (9, 10) and more recently of a leucine zipper domain (13). In contrast, for dynamic properties of the biomolecule, no comprehensive deconvolution method exists. Handles and beads have their own dissipative characteristics and will tend to suppress the high-frequency fluctuations of the biomolecule and as a result distort the measured power spectrum. In the context of single-molecule pulling experiments, theoretical progress has been made in accounting for handle/bead effects on the observed unfolding transition rates (15–18). However, the full intrinsic fluctuation spectrum, as encoded in the time-dependent linear response function, has remained out of reach. The current work presents a systematic dynamic deconvolution procedure that fills this gap, providing a way to recover the linear response of a biomolecule integrated into a mechanical dissipative network. We work in the constant force ensemble as appropriate for optical force clamp setups with active feedback mechanisms (19, 20) or passive means (12, 21). Although our theory is general and applies to mechanical networks of arbitrary topology, we illustrate our approach for the specific experimental setup of ref. 13: a protein attached to optically trapped beads through dsDNA handles. The only inputs required by our theory are the autocorrelation functions of the bead fluctuations in equilibrium. We demonstrate how the results from two different experimental runs—one with the protein, and one without—can be combined to yield the protein linear response functions.
We apply this two-step procedure on a force clamp setup simulated through Brownian dynamics and verify the accuracy of our dynamic deconvolution method. Knowledge of mechanical linear response functions forms the basis of understanding viscoelastic material properties; the protein case is particularly interesting because every folding state, i.e., each well in the free energy landscape, will have its own spectrum of equilibrium fluctuations and hence a distinct linear response function. Two key properties determine this function: the shape of the free energy around the minimum, and the local diffusivity. The latter has contributions both from solvent drag and the effective roughness of the energy landscape—internal friction due to molecular torsional transitions and the formation and rupture of bonds between different parts of the peptide chain. The diffusivity profile is crucial for getting a comprehensive picture of protein folding kinetics, and arguably it is just as important as the free energy landscape itself for very fast folding proteins (22–24). Our dynamic deconvolution theory provides a promising route to extract this important protein characteristic from future force clamp studies.
Results and Discussion
Force Clamp Experiments and Static Deconvolution.
As a representative case, in this paper we consider the double trap setup shown in Fig. 1A, which typically involves two optically trapped polystyrene beads of radius 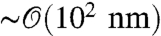 , two double-stranded DNA handles, each
, two double-stranded DNA handles, each 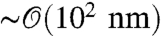 , attached to a protein in the center (13). For fixed trap positions and sufficiently soft trapping potentials, the entire system will be in equilibrium at an approximately constant tension F. We are interested in a force regime (F ≳ 10 pN in the system under consideration) where the handles are significantly stretched in the direction parallel to the applied force (chosen as the z axis), and rotational fluctuations of the handle-bead contact points are small. Because the experimental setup is designed to measure the z separation of the beads as a function of time, we focus entirely on the dynamic response of the system along the z direction. However, the methods below can be easily generalized to the transverse response as well. Though we consider only a passive measurement system in our analysis, an active feedback loop that minimizes force fluctuations can also be incorporated, as an additional component with its own characteristic dynamic response (with the added complication that the response of the feedback mechanism would have to be independently determined).
, attached to a protein in the center (13). For fixed trap positions and sufficiently soft trapping potentials, the entire system will be in equilibrium at an approximately constant tension F. We are interested in a force regime (F ≳ 10 pN in the system under consideration) where the handles are significantly stretched in the direction parallel to the applied force (chosen as the z axis), and rotational fluctuations of the handle-bead contact points are small. Because the experimental setup is designed to measure the z separation of the beads as a function of time, we focus entirely on the dynamic response of the system along the z direction. However, the methods below can be easily generalized to the transverse response as well. Though we consider only a passive measurement system in our analysis, an active feedback loop that minimizes force fluctuations can also be incorporated, as an additional component with its own characteristic dynamic response (with the added complication that the response of the feedback mechanism would have to be independently determined).
Fig. 1.

(A) Double optical tweezer force clamp setup for the study of equilibrium protein dynamics, with soft traps approximating a constant tension F. (B) To define linear response functions, consider an individual component at tension F. A small additional oscillatory force fL exp(-iωt) applied at the left end leads to endpoint oscillations with amplitudes zL = Jself,L(ω)fL and zR = Jcross(ω)fL, which defines the self- and cross-response functions. (C) Two objects X and Y connected in series behave as a composite object XY whose response functions can be derived through simple rules (Eq. 2) from the individual X and Y response functions. (D) Schematic representation of the optical tweezer setup consisting of beads (B), double-stranded DNA handles (H), and protein (P), with connecting springs.
To set the stage for our dynamic deconvolution theory, we first illustrate the static deconvolution for two objects X and Y connected in series under constant tension F, e.g., a protein and a handle. Let 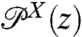 and
and 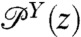 be the constant-force probability distributions for each of these objects having end-to-end distance z. The total system end-to-end distribution is given by
be the constant-force probability distributions for each of these objects having end-to-end distance z. The total system end-to-end distribution is given by 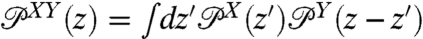 . In terms of the Fourier-transformed distributions, this can be stated simply through the ordinary convolution theorem,
. In terms of the Fourier-transformed distributions, this can be stated simply through the ordinary convolution theorem, 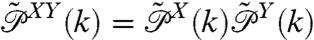 . If
. If 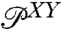 is derived from histograms of the experimental time series, and if
is derived from histograms of the experimental time series, and if 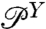 can be estimated independently (either from an experiment without the protein, or through theory), then we can invert the convolution relation to solve for the protein distribution
can be estimated independently (either from an experiment without the protein, or through theory), then we can invert the convolution relation to solve for the protein distribution 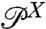 and thus extract the folding free energy landscape. A similar approach works for multiple objects in series or in parallel.
and thus extract the folding free energy landscape. A similar approach works for multiple objects in series or in parallel.
Dynamic Response Functions.
Before we consider dynamic networks, we define the linear response of a single object under constant stretching force F along the z direction, as shown in Fig. 1B. Imagine applying an additional small oscillatory force fL exp(-iωt) along the z axis to the left end. The result will be small oscillations zL exp(-iωt) and zR exp(-iωt) of the two ends around their equilibrium positions. The complex amplitudes zL and zR are related to fL through linear response: zL = Jself,L(ω)fL, zR = Jcross(ω)fL, defining the self-response function Jself,L(ω) of the left end and the cross-response function Jcross(ω). If the oscillatory force is applied instead at the right end, the response takes the form zR = Jself,R(ω)fR, zL = Jcross(ω)fR. Note that because the object is in general asymmetric, Jself,L(ω) and Jself,R(ω) are distinct functions. However, there is only a single cross-response [in the absence of time-reversal breaking effects such as magnetic fields (25)]. For the purposes of dynamic deconvolution of a network, these three response functions contain the complete dynamical description of a given component and are all we need. It is convenient to define the end-to-end response function Jee(ω), with zR - zL = Jee(ω)f, where the oscillatory force f exp(-iωt) is applied simultaneously to both ends of the object in opposite directions. This response turns out to be a linear combination of the other functions: Jee = Jself,L + Jself,R - 2Jcross.
As an illustration we take the simplest, nontrivial example: two spheres with different mobilities μL and μR connected by a harmonic spring of stiffness k. In water we are typically in the low Reynolds number regime and an overdamped dynamical description is appropriate. If an oscillating force of amplitude fL is applied to the left sphere, its velocity will oscillate with the amplitude -iωzL = μL(fL + k[zR - zL]), whereas the velocity amplitude of the right sphere is given by -iωzR = -μRk[zR - zL]. Using the above definitions of the response functions we obtain
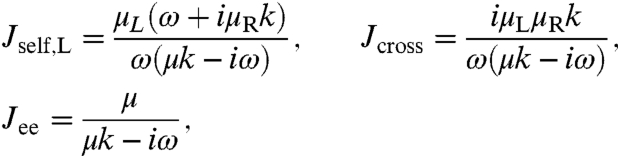 |
[1] |
where μ = μL + μR. By symmetry Jself,R is the same as Jself,L with subscripts L and R interchanged. The end-to-end response Jee has a standard Lorentzian form. For more realistic force transducers, such as semiflexible polymers, Jee will later be written as a sum of Lorentzians reflecting the polymer normal modes. Note that when k = 0, and the spheres no longer interact, Jself,L = iμL/ω, the standard result for a diffusing sphere, and Jcross = 0, as expected, because there is no force transmission from one sphere to the other.
Though all the linear response functions are defined in terms of an external oscillatory force, in practice one does not need to actually apply such a force to determine the functions experimentally. As described in the two-step deconvolution procedure below, one can extract them from measurements that are far easier to implement in the lab, namely by calculating autocorrelation functions of equilibrium fluctuations.
Dynamic Convolution of Networks.
Based on the notion of self and cross-response functions, we now consider the dynamics of composites. We explicitly display the convolution formulas for combining two objects in series and in parallel; by iteration the response of a network of arbitrary topology and complexity can thus be constructed. As shown in Fig. 1C, assume we have two objects X and Y connected by a spring. X is described by response functions 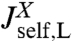 ,
, 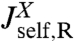 , and
, and 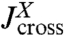 , and we have the analogous set for Y. The internal spring is added for easy evaluation of the force acting between the objects; it is eliminated at the end by sending its stiffness to infinity. We would like to know the response functions of the composite XY object,
, and we have the analogous set for Y. The internal spring is added for easy evaluation of the force acting between the objects; it is eliminated at the end by sending its stiffness to infinity. We would like to know the response functions of the composite XY object, 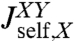 ,
, 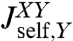 , and
, and 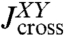 , where the X and Y labels correspond to left and right ends, respectively. The rules (with the full derivation in SI Text) read
, where the X and Y labels correspond to left and right ends, respectively. The rules (with the full derivation in SI Text) read
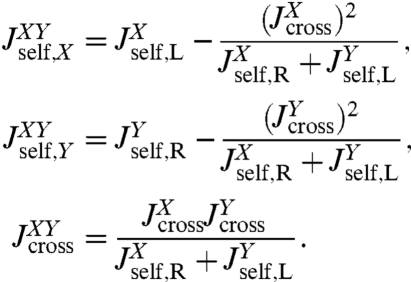 |
[2] |
The rules for connecting two objects in parallel are more straightforward and read 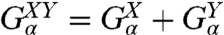 , where α is any one of the function categories (self, cross, or end-to-end), and G denote the inverse response functions. One particularly relevant realization for parallel mechanical pathways are long-range hydrodynamic coupling effects, which experimentally act between beads and polymer handles in the force clamp setup. We derive the parallel rule and show an example hydrodynamic application in SI Text. For simplicity, however, we will concentrate in our analysis on serial connections. To proceed, if we set X = H and Y = B, we can obtain the response functions of the composite handle-bead (HB) object,
, where α is any one of the function categories (self, cross, or end-to-end), and G denote the inverse response functions. One particularly relevant realization for parallel mechanical pathways are long-range hydrodynamic coupling effects, which experimentally act between beads and polymer handles in the force clamp setup. We derive the parallel rule and show an example hydrodynamic application in SI Text. For simplicity, however, we will concentrate in our analysis on serial connections. To proceed, if we set X = H and Y = B, we can obtain the response functions of the composite handle-bead (HB) object, 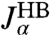 , if we know the response functions of the bead and handle separately. Our full system in Fig. 1D is just the protein sandwiched between two HB components (oriented such that the handle ends of each HB are attached to the protein). The total system response functions (denoted by “2HB + P”) in terms of the individual protein and HB functions result by iterating the pair convolution in Eq. 2 twice. In particular, the end-to-end response
, if we know the response functions of the bead and handle separately. Our full system in Fig. 1D is just the protein sandwiched between two HB components (oriented such that the handle ends of each HB are attached to the protein). The total system response functions (denoted by “2HB + P”) in terms of the individual protein and HB functions result by iterating the pair convolution in Eq. 2 twice. In particular, the end-to-end response 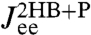 is given by
is given by
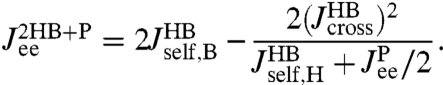 |
[3] |
This is a key relation, because we show below that both 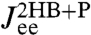 and the three HB response functions
and the three HB response functions 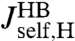 ,
, 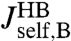 , and
, and  can be derived from force clamp experimental data. Hence Eq. 3 allows us to estimate the unknown protein response function
can be derived from force clamp experimental data. Hence Eq. 3 allows us to estimate the unknown protein response function 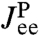 .
.
We note a striking similarity to the signal processing scenario (26), where the output of a linear time-invariant (LTI) network (e.g., an RLC electric circuit) is characterized through a “transfer function.” For such networks, combination rules in terms of serial, parallel, and feedback loop motifs exist. The result for 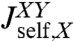 in Eq. 2 can be seen in a similar light: The first term is the self-response
in Eq. 2 can be seen in a similar light: The first term is the self-response 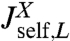 of object X, which is independent of the presence of object Y. The rational function in the second term is the “feedback” due to X interacting with Y. As expected, if the cross-response
of object X, which is independent of the presence of object Y. The rational function in the second term is the “feedback” due to X interacting with Y. As expected, if the cross-response 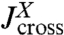 connecting the two ends of X is turned off, this feedback disappears. In analogy to the transfer function theory for LTI systems, our convolution rules form a comprehensive basis to describe the response of an arbitrarily complicated network inside a force clamp experiment. And like the transfer functions that arise out of LTI feedback loops, the convolution of interacting components consists of a nonlinear combination of the individual response functions. The rational functions due to the feedback of mechanical perturbations across the connected elements are nontrivial, but can be exactly accounted for via iteration of the convolution rules.
connecting the two ends of X is turned off, this feedback disappears. In analogy to the transfer function theory for LTI systems, our convolution rules form a comprehensive basis to describe the response of an arbitrarily complicated network inside a force clamp experiment. And like the transfer functions that arise out of LTI feedback loops, the convolution of interacting components consists of a nonlinear combination of the individual response functions. The rational functions due to the feedback of mechanical perturbations across the connected elements are nontrivial, but can be exactly accounted for via iteration of the convolution rules.
Two-Step Dynamic Deconvolution Yields Protein Dynamic Properties.
To illustrate our theory, we construct a two-step procedure to analyze the experimental system in Fig. 1A, with the ultimate goal of determining dynamic protein properties in the force clamp. Under a constant force F, the protein extension will fluctuate around a mean corresponding to a folded or unfolded state. Though it has been demonstrated that under appropriately tuned forces the protein can show spontaneous transitions between folded and unfolded states, we for the moment neglect this more complex scenario. (In SI Text we analyze simulation results for a protein exhibiting a double-well free energy, where the two states can be analyzed independently by pooling data from every visit to a given well; the same idea can be readily extended to analyze time-series data from proteins with one or more intermediate states.) We consider the protein dynamics as diffusion of the reaction coordinate 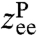 (the protein end-to-end distance) in a free energy landscape
(the protein end-to-end distance) in a free energy landscape 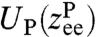 . The end-to-end response function
. The end-to-end response function 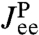 reflects the shape of UP around the local minimum, and the internal protein friction (i.e., the local mobility), which is the key quantity of interest. The simplest example is a parabolic well at position
reflects the shape of UP around the local minimum, and the internal protein friction (i.e., the local mobility), which is the key quantity of interest. The simplest example is a parabolic well at position 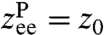 , namely
, namely 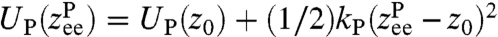 . If we assume the protein mobility μP is approximately constant within this state, the end-to-end response is given by
. If we assume the protein mobility μP is approximately constant within this state, the end-to-end response is given by
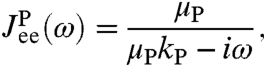 |
[4] |
which has the same Lorentzian form as the harmonic two-sphere end-to-end response in Eq. 1. Depending on the resolution and quality of the experimental data, more complex fitting forms may be substituted, including anharmonic corrections and nonconstant diffusivity profiles (an example of these is given in the two-state protein case analyzed in SI Text). However, for practical purposes Eq. 4 is a good starting point.
An experimentalist seeking to determine 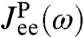 would carry out the following two-step procedure:
would carry out the following two-step procedure:
First step: Make a preliminary run using a system without the protein (just two beads and two handles, as illustrated in Fig. 2A). As described in the Materials and Methods, time derivatives of autocorrelation functions calculated from the bead position time series can be Fourier-transformed to directly give  and
and 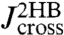 . The convolution rules in Eq. 2 relate
. The convolution rules in Eq. 2 relate  and
and 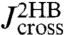 to the bead/handle response functions,
to the bead/handle response functions,  and
and  , which via another application of Eq. 2 are related to the response functions of a single bead and a single handle. The bead functions
, which via another application of Eq. 2 are related to the response functions of a single bead and a single handle. The bead functions 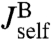 and
and 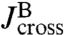 depend solely on known experimental parameters (Materials and Methods), leaving only the handle functions
depend solely on known experimental parameters (Materials and Methods), leaving only the handle functions 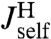 and
and  as unknowns in the convolution equations. Choosing an appropriate fitting form, determined by polymer dynamical theory (see Materials and Methods), we can straightforwardly determine
as unknowns in the convolution equations. Choosing an appropriate fitting form, determined by polymer dynamical theory (see Materials and Methods), we can straightforwardly determine 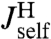 and
and  .
.
Fig. 2.

The first step in the deconvolution procedure, needed to determine the handle-bead response JHB: analysis of the optical tweezer system without the protein. (A, Top to Bottom) A Brownian dynamics simulation snapshot; schematic representation of the system; after the first convolution step handles and beads are grouped into composite handle-bead (HB) objects; after the second convolution step the full system (“2HB”) constitutes a single object. (B) Part of the simulation time series for the total system end-to-end distance 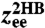 . The time series yields the equilbrium probability distribution
. The time series yields the equilbrium probability distribution 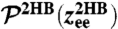 shown at Right. (C, Top) The MSD functions
shown at Right. (C, Top) The MSD functions 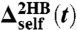 and
and 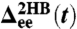 (Eq. 5) calculated from the simulation; (Bottom) the time-domain response functions
(Eq. 5) calculated from the simulation; (Bottom) the time-domain response functions 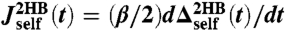 ,
, 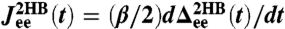 . Simulation results (symbols) are numerical derivatives of the curves (Top). The solid lines are a 5-exponential fit to the simulation results. (D, Left) The real and imaginary parts of the J2HB self and end-to-end response functions. Simulation results (symbols) are just the Fourier transforms of the multiexponential fits in C. Theoretical fitting results according to Eq. 2 and based on HB functions JHB are shown as solid lines. (Right) The HB response functions, as determined by the theoretical fitting to the full system data.
. Simulation results (symbols) are numerical derivatives of the curves (Top). The solid lines are a 5-exponential fit to the simulation results. (D, Left) The real and imaginary parts of the J2HB self and end-to-end response functions. Simulation results (symbols) are just the Fourier transforms of the multiexponential fits in C. Theoretical fitting results according to Eq. 2 and based on HB functions JHB are shown as solid lines. (Right) The HB response functions, as determined by the theoretical fitting to the full system data.
Second step: Make a production run with the protein. Eq. 3 relates the resulting end-to-end response 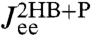 , extracted from the experimental data, to the response of the protein alone
, extracted from the experimental data, to the response of the protein alone 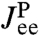 . Because the first step yielded the composite handle-bead functions
. Because the first step yielded the composite handle-bead functions 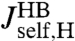 ,
, 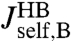 , and
, and  that appear in Eq. 3, the only unknown is
that appear in Eq. 3, the only unknown is 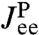 . We can thus solve for the parameters μP and kP that appear in Eq. 4.
. We can thus solve for the parameters μP and kP that appear in Eq. 4.
This two-step procedure can be repeated at different applied tensions, revealing how the protein properties (i.e., the intramolecular interactions that contribute to the diffusivity μP) depend on force. Even analyzing the unfolded state of the protein might yield interesting results: Certain forces might be strong enough to destroy the tertiary structure, but not completely destabilize the secondary structure, which could transiently refold and affect μP.
Simulations Validate the Deconvolution Technique.
To demonstrate the two-step deconvolution procedure in a realistic context, we perform Brownian dynamics simulations mimicking a typical force clamp experiment: Two beads that undergo rotational and translational fluctuations are trapped in 3D harmonic potentials and connected to two semiflexible polymers that are linked together via a potential function that represents the protein folding landscape (see Materials and Methods for details). We ignore hydrodynamic effects that can easily be accounted for through parallel coupling pathways, as mentioned above.
We begin with the first step of the deconvolution procedure. A snapshot of the simulation system, two handles and two beads without a protein, is shown in Fig. 2A. A representative segment of the 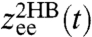 time series is shown in Fig. 2B. Equilibrium analysis of the time series yields the end-to-end distribution
time series is shown in Fig. 2B. Equilibrium analysis of the time series yields the end-to-end distribution 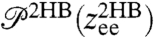 , which is useful for extracting static properties of the protein like the free energy landscape: When the protein is added to the system, the total end-to-end distribution is just a convolution of the 2HB and protein distributions. (The asymmetry of
, which is useful for extracting static properties of the protein like the free energy landscape: When the protein is added to the system, the total end-to-end distribution is just a convolution of the 2HB and protein distributions. (The asymmetry of 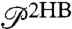 seen in Fig. 2B arises from the semiflexible nature of the handles.) As described in Materials and Methods, we use the time series to calculate self and end-to-end mean square displacement (MSD) curves
seen in Fig. 2B arises from the semiflexible nature of the handles.) As described in Materials and Methods, we use the time series to calculate self and end-to-end mean square displacement (MSD) curves  and
and 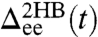 (Fig. 2C) whose derivatives are proportional to the time-domain response functions
(Fig. 2C) whose derivatives are proportional to the time-domain response functions 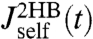 and
and 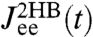 . The multiexponential fits to these functions are illustrated in Fig. 2C, and their analytic Fourier transforms plotted in the left column of Fig. 2D. We thus have a complete dynamical picture of the 2HB system response. However, in order to use Eq. 3 to extract the protein response, we first have to determine the handle-bead response functions
. The multiexponential fits to these functions are illustrated in Fig. 2C, and their analytic Fourier transforms plotted in the left column of Fig. 2D. We thus have a complete dynamical picture of the 2HB system response. However, in order to use Eq. 3 to extract the protein response, we first have to determine the handle-bead response functions 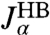 . Although a general fit of
. Although a general fit of 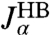 is possible, it is useful to apply the knowledge about the bead parameters and symmetry properties of the handle response. The handle parameters (the set
is possible, it is useful to apply the knowledge about the bead parameters and symmetry properties of the handle response. The handle parameters (the set 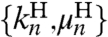 in Materials and Methods Eq. 7) are the only unknowns in the three HB response functions:
in Materials and Methods Eq. 7) are the only unknowns in the three HB response functions: 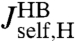 ,
, 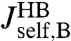 , and
, and  . Note that the handle-bead object is clearly asymmetric, so the self-response will be different at the handle (H) and bead (B) end. Convolving two HB objects according to Eq. 2 and fitting the handle parameters to the simulation results for
. Note that the handle-bead object is clearly asymmetric, so the self-response will be different at the handle (H) and bead (B) end. Convolving two HB objects according to Eq. 2 and fitting the handle parameters to the simulation results for 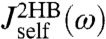 and
and 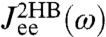 leads to the excellent description shown as solid lines on the left in Fig. 2D. The handle parameters derived from this fitting completely describe the HB response functions
leads to the excellent description shown as solid lines on the left in Fig. 2D. The handle parameters derived from this fitting completely describe the HB response functions 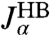 , shown in the right column of Fig. 2D. The HB response curves reflect their individual components: There is a low frequency peak/plateau in the imaginary/real part of the HB self-response, related to the slow relaxation of the bead in the trap. The higher frequency contributions are due to the handles, and as a result they are more prominent in the self-response of the handle end than of the bead end. There is a similarly nontrivial structure in the end-to-end response, due to the complex interactions between the handle normal modes and the fluctuations of the trapped bead (see SI Text for more details).
, shown in the right column of Fig. 2D. The HB response curves reflect their individual components: There is a low frequency peak/plateau in the imaginary/real part of the HB self-response, related to the slow relaxation of the bead in the trap. The higher frequency contributions are due to the handles, and as a result they are more prominent in the self-response of the handle end than of the bead end. There is a similarly nontrivial structure in the end-to-end response, due to the complex interactions between the handle normal modes and the fluctuations of the trapped bead (see SI Text for more details).
The double-HB end-to-end distribution in Fig. 2B and the HB response functions in Fig. 2D are all we need to know about the optical tweezer system: The equilibrium end-to-end distribution and linear response of any object that we now put between the handles can be reconstructed. We will illustrate this using a toy model of a protein. In our simulations for the second step, we use a parabolic potential UP with 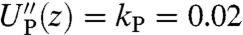 kBT/a2, and a fixed mobility μP = 0.05μ0. Here a = 1 nm and μ0 = 1/6πηa, where η is the viscosity of water. This leads to the single-Lorentzian response function
kBT/a2, and a fixed mobility μP = 0.05μ0. Here a = 1 nm and μ0 = 1/6πηa, where η is the viscosity of water. This leads to the single-Lorentzian response function 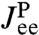 of Eq. 4. If this exact theoretical form of
of Eq. 4. If this exact theoretical form of 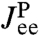 is convolved with the HB response functions from the first step according to Eq. 3, we get the result in Fig. 3: very close agreement with
is convolved with the HB response functions from the first step according to Eq. 3, we get the result in Fig. 3: very close agreement with 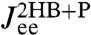 directly derived from the simulated time-series data. For comparison we also plot the separate end-to-end responses of the protein alone (P) and the double-HB setup without a protein (2HB). As expected
directly derived from the simulated time-series data. For comparison we also plot the separate end-to-end responses of the protein alone (P) and the double-HB setup without a protein (2HB). As expected 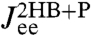 differs substantially from both of these as correctly predicted by the convolution theory. The effect of adding handles and beads to the protein is to shift the peak in the imaginary part of the total system response to lower frequencies. Additionally, we see in
differs substantially from both of these as correctly predicted by the convolution theory. The effect of adding handles and beads to the protein is to shift the peak in the imaginary part of the total system response to lower frequencies. Additionally, we see in 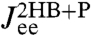 the contributions of the handle and bead rotational motions, which are dominant at higher frequencies. The sensitivity of the theoretical fit is shown in the insets of Fig. 3: Zooming in on the maxima of
the contributions of the handle and bead rotational motions, which are dominant at higher frequencies. The sensitivity of the theoretical fit is shown in the insets of Fig. 3: Zooming in on the maxima of 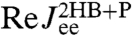 and
and 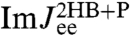 , we plot the true theoretical prediction (red/blue curves) and results with μP shifted away from the true value (thin pink/cyan curves). In fact, if kP and μP are taken as free parameters, numerical fitting to the simulation
, we plot the true theoretical prediction (red/blue curves) and results with μP shifted away from the true value (thin pink/cyan curves). In fact, if kP and μP are taken as free parameters, numerical fitting to the simulation 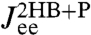 yields accurate values of μP = 0.050μ0 and kP = 0.0199 kBT/a2. Examples of successful deconvolution with other values of the intrinsic protein parameters are given in the double-well free energy analysis in SI Text.
yields accurate values of μP = 0.050μ0 and kP = 0.0199 kBT/a2. Examples of successful deconvolution with other values of the intrinsic protein parameters are given in the double-well free energy analysis in SI Text.
Fig. 3.

Real (Top) and imaginary (Bottom) parts of the total end-to-end response 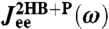 of an optical tweezer system with the protein modeled as a single parabolic potential well (kP = 0.02 kBT/a2, μP = 0.05μ0). Symbols are simulation results, and the solid line is the theoretical prediction, based on the convolution of the protein response
of an optical tweezer system with the protein modeled as a single parabolic potential well (kP = 0.02 kBT/a2, μP = 0.05μ0). Symbols are simulation results, and the solid line is the theoretical prediction, based on the convolution of the protein response 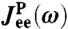 with the HB response functions JHB of Fig. 2D according to Eq. 3. For comparison,
with the HB response functions JHB of Fig. 2D according to Eq. 3. For comparison, 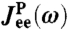 (dashed line) and
(dashed line) and 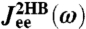 (dot-dashed line) are also included. (Insets) To show the sensitivity of the theoretical fitting, zoomed-in sections of
(dot-dashed line) are also included. (Insets) To show the sensitivity of the theoretical fitting, zoomed-in sections of 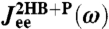 near the maxima of the real (Top) and imaginary (Bottom) components. Both simulation (symbols) and theoretical (blue/red curve) results are plotted. The thin pink/cyan curves are theoretical results with μP different from the true value: (Left to Right) μP = 0.01, 0.03, 0.07, and 0.09μ0.
near the maxima of the real (Top) and imaginary (Bottom) components. Both simulation (symbols) and theoretical (blue/red curve) results are plotted. The thin pink/cyan curves are theoretical results with μP different from the true value: (Left to Right) μP = 0.01, 0.03, 0.07, and 0.09μ0.
In practice, any theoretical approach must take into consideration instrumental limitations: Most significantly, there will be a minimum possible interval ts between data collections, related to the time resolution of the measuring equipment. The deconvolution theory can always be applied in the frequency range up to ωs = 1/ts. Whatever physical features of any component in the system that fall within this range can be modeled and extracted without requiring inaccessible knowledge of fluctuation modes above the frequency cutoff ωs. In SI Text, we illustrate this directly on the toy protein discussed above, coarse-graining the simulation time series to 0.01-ms intervals, mimicking the equipment resolution used in ref. 13. The characteristic frequency of the protein within the tweezer setup falls within the cutoff, and hence our two-step deconvolution procedure can still be applied to yield accurate best-fit results for the protein parameters. SI Text also includes a discussion of other experimental artifacts—white noise, drift, and effective averaging of the time series on the time scale ts—and shows how to adapt the procedure to correct for these effects.
Conclusion
Dynamic deconvolution theory allows us to extract the response functions of a single component from the overall response of a multicomposite network. The theory is most transparently formulated in the frequency domain and provides the means to reverse the filtering influence of all elements that are connected to the component of interest. From the extracted single-component response function, dynamic properties such as the internal mobility or friction can be directly deduced. At the heart of our theory stands the observation that the response of any component in the network is completely determined by three functions, namely the cross-response and the two self-responses, which are in general different at the two ends. The response of any network can be predicted by repeated iteration of our convolution formulas for serial and parallel connections. Self-similar or more complicated network topologies, as occur in viscoelastic media, can thus be treated as well. We demonstrate the application of our deconvolution theory for a simple mechanical network that mimics a double-laser-tweezer setup, but the underlying idea is directly analogous to the signal processing rules that describe other scalar dynamic networks, such as electrical circuits or chemical reaction pathways in systems biology (27). We finally point out that dynamic convolution also occurs in FRET experiments on proteins where polymeric linkers and conformational fluctuations of fluorophores, as well as the internal fluorescence dynamics, modify the measured dynamic fluctuation spectrum (28–30). The experimental challenge in the future will thus be to generate time-series data for single biomolecules with a sufficient frequency range in order to perform an accurate deconvolution. For this a careful matching of the relevant time and spatial scales of the biomolecule under study and the corresponding scales of the measuring device (handles as well as beads) is crucial, for which our theory provides the necessary guidance.
Materials and Methods
Determining the Total System Response from the Experimental Time Series.
A key step in the experimental analysis is to obtain the system response functions Jself(ω) and Jcross(ω) from the raw data (which can either be the double handle-bead system with or without a protein). These data consist of two time series zB,L(t) and zB,R(t) for the left/right bead positions from which we calculate the MSD functions
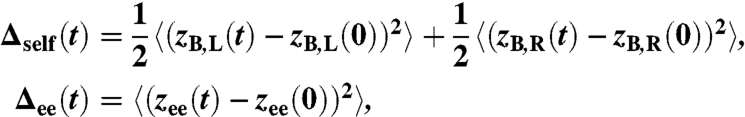 |
[5] |
where zee(t) = zB,R(t) - zB,L(t), and we have averaged the self-MSD of the two endpoints because they are identical by symmetry. Calculating the MSD functions is equivalent to finding the autocorrelation of the time series: For example, if Ree(t) = 〈zee(t)zee(0)〉 is the end-to-end autocorrelation, the MSD Δee(t) is simply given by Δee(t) = 2(Ree(0) - Ree(t)).
From the fluctuation-dissipation theorem (25), the time-domain response functions Jself(t) and Jee(t) are related to the derivatives of the MSD functions: Jself(t) = (β/2)dΔself(t)/dt, Jee(t) = (β/2)dΔee(t)/dt, where β = 1/kBT. To get the Fourier-space response, the time-domain functions can be numerically fit to a multiexponential form; for example, 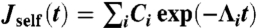 . In our simulation examples typically four to five exponentials are needed for a reasonable fit. Once the parameters Ci and Λi are determined, the expression can be exactly Fourier-transformed to give the frequency-domain response function,
. In our simulation examples typically four to five exponentials are needed for a reasonable fit. Once the parameters Ci and Λi are determined, the expression can be exactly Fourier-transformed to give the frequency-domain response function, 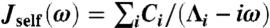 . An analogous procedure is used to obtain Jee(ω). The cross-response follows as Jcross = Jself - Jee/2. The power spectrum associated with a particular type of fluctuation, for example, the end-to-end spectrum Ree(ω) (defined as the Fourier transform of the autocorrelation), is just proportional to the imaginary part of the corresponding response function: Ree(ω) = (2kBT/ω)ImJee(ω).
. An analogous procedure is used to obtain Jee(ω). The cross-response follows as Jcross = Jself - Jee/2. The power spectrum associated with a particular type of fluctuation, for example, the end-to-end spectrum Ree(ω) (defined as the Fourier transform of the autocorrelation), is just proportional to the imaginary part of the corresponding response function: Ree(ω) = (2kBT/ω)ImJee(ω).
Bead Response Functions.
The response functions of the beads in the optical traps are the easiest to characterize, because they depend on quantities that are all known by the experimentalist: the trap stiffness ktrap, bead radius R, mobility μB = 1/6πηR, and rotational mobility μrot = 1/8πηR3. Here η is the viscosity of water. For each bead the three response functions can be defined as described above, with the two “endpoints” being the handle-attachment point on the bead surface (zS) and the bead center (zB). The latter point is significant because this position is what is directly measured by the experiment. For the case of large F, where the rotational diffusion of the bead is confined to small angles away from the z axis, the response functions are
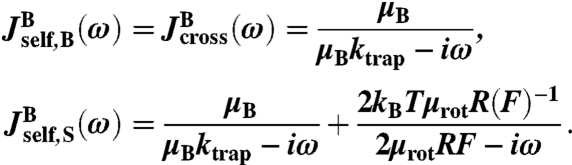 |
[6] |
The second term in 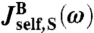 describes the contribution of the bead rotational motion, which has a characteristic relaxation frequency 2μrotRF. This term is derived in SI Text, though it can also be found from an earlier theory of rotational Brownian diffusion in uniaxial liquid crystals (31).
describes the contribution of the bead rotational motion, which has a characteristic relaxation frequency 2μrotRF. This term is derived in SI Text, though it can also be found from an earlier theory of rotational Brownian diffusion in uniaxial liquid crystals (31).
Handle Response Functions.
The double-stranded DNA handles are semiflexible polymers whose fluctuation behavior in equilibrium can be decomposed into normal modes. We do not need the precise details of this decomposition, beyond the fact that by symmetry these modes can be grouped into even and odd functions of the polymer contour length, and that they are related to the linear response of the polymer through the fluctuation-dissipation theorem. From these assumptions, the handle response functions have the following generic form (a fuller description can be found in SI Text):
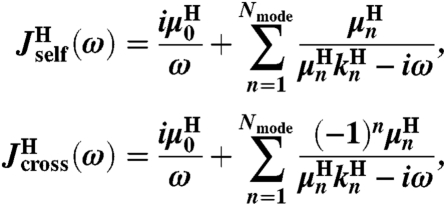 |
[7] |
for some set of 2Nmode + 1 parameters 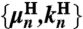 . Note that because the handles are symmetric objects, the self-response of each endpoint is the same function
. Note that because the handles are symmetric objects, the self-response of each endpoint is the same function 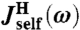 . The mobilities
. The mobilities 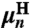 and elastic coefficients
and elastic coefficients 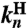 encode the normal mode characteristics, with the mode relaxation times
encode the normal mode characteristics, with the mode relaxation times 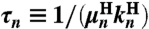 ordered from largest (n = 1) to smallest (n = Nmode). The parameter
ordered from largest (n = 1) to smallest (n = Nmode). The parameter 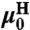 is the center-of-mass mobility of the handle along the force direction. Simple scaling expressions for the zeroth and first mode parameters in terms of physical polymer parameters as well as the connection between the expressions in Eq. 1 and Eq. 7 are given in SI Text. (These same expressions, with a smaller persistence length lp, could describe a completely unfolded, noninteracting, polypeptide chain at high force.) In practice, the high-frequency cutoff Nmode can be kept quite small (i.e., Nmode = 4) to describe the system over the frequency range of interest.
is the center-of-mass mobility of the handle along the force direction. Simple scaling expressions for the zeroth and first mode parameters in terms of physical polymer parameters as well as the connection between the expressions in Eq. 1 and Eq. 7 are given in SI Text. (These same expressions, with a smaller persistence length lp, could describe a completely unfolded, noninteracting, polypeptide chain at high force.) In practice, the high-frequency cutoff Nmode can be kept quite small (i.e., Nmode = 4) to describe the system over the frequency range of interest.
Numerical Inversion of Convolution Equations.
Care must be taken in manipulating Fourier-space relationships such as Eq. 3. Directly inverting such equations generally leads to numerical instabilities due to noise and singularities. In our case, we can avoid direct inversion because the forms of the component functions are known beforehand (i.e., Eq. 6 for the beads, Eq. 7 for the handles, and Eq. 4 for the protein). Thus when we model the response of the double handle-bead system with a protein, 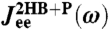 , we end up through Eqs. 2 and 3 with some theoretical function
, we end up through Eqs. 2 and 3 with some theoretical function 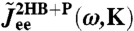 , where K is the set of unknown parameters related to the components. Because
, where K is the set of unknown parameters related to the components. Because 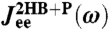 is known as a function of ω from the experimental time-series, we find K by minimizing the goodness-of-fit function
is known as a function of ω from the experimental time-series, we find K by minimizing the goodness-of-fit function 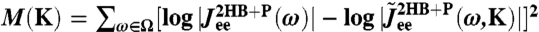 , where Ω is a logarithmically spaced set of frequencies, up to the cutoff frequency ωs = 1/ts determined by the time resolution ts of the measuring equipment. This is equivalent to simultaneously fitting the real and imaginary parts of our system response on a log–log scale.
, where Ω is a logarithmically spaced set of frequencies, up to the cutoff frequency ωs = 1/ts determined by the time resolution ts of the measuring equipment. This is equivalent to simultaneously fitting the real and imaginary parts of our system response on a log–log scale.
Simulations.
In our Brownian dynamics simulations each handle is a semiflexible bead-spring chain of 25 beads of radius a = 1 nm, every bead having mobility μ0 = 1/6πηa. The handle persistence length is lp = 20a. The harmonic springs used to connect all components together (including the beads making up the handles) have stiffness γ = 300 kBT/a2. The beads have radius R = 50a, and the traps have strength ktrap = 0.00243 kBT/a2, which corresponds to 0.01 pN/nm. The traps are positioned such that the average force in equilibrium F ≈ 3 kBT/a = 12.35 pN.
To capture the essential features of protein dynamics, we construct a simple toy model. The protein is characterized by two vectors: a center-of-mass position 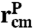 and an end-to-end separation
and an end-to-end separation 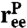 . Both
. Both 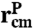 and the transverse components of
and the transverse components of 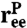 obey standard Langevin dynamics with a mobility μcm = μ⊥ = 0.12μ0. The internal dynamics of protein fluctuations is modeled through the longitudinal end-to-end component
obey standard Langevin dynamics with a mobility μcm = μ⊥ = 0.12μ0. The internal dynamics of protein fluctuations is modeled through the longitudinal end-to-end component 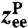 , subject to a potential
, subject to a potential 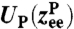 and a mobility μP. The transverse components
and a mobility μP. The transverse components 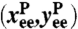 feel a harmonic potential with spring constant k⊥ = 1.5 kBT/a2.
feel a harmonic potential with spring constant k⊥ = 1.5 kBT/a2.
The simulation dynamics are implemented through a discretized free-draining Langevin equation with time step Δt = 3 × 10-4τ, where the time unit τ = a2/kBTμ0. Data are collected every 1,000 time steps. Typical simulation times are 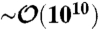 steps, with averages collected from ≈20 independent runs for each system considered.
steps, with averages collected from ≈20 independent runs for each system considered.
Supplementary Material
Acknowledgments.
We thank the Feza Gürsey Institute for computational resources provided by the Gilgamesh cluster. This work was supported by Deutsche Forschungsgemeinschaft within Grant SFB 863.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010476107/-/DCSupplemental.
References
- 1.Carrion-Vazquez M, et al. Mechanical and chemical unfolding of a single protein: A comparison. Proc Natl Acad Sci USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oesterhelt F, et al. Unfolding pathways of individual bacteriorhodopsins. Science. 2000;288:143–146. doi: 10.1126/science.288.5463.143. [DOI] [PubMed] [Google Scholar]
- 3.Wiita AP, et al. Probing the chemistry of thioredoxin catalysis with force. Nature. 2007;450:124–130. doi: 10.1038/nature06231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khatri BS, Kawakami M, Byrne K, Smith DA, McLeish TCB. Entropy and barrier-controlled fluctuations determine conformational viscoelasticity of single biomolecules. Biophys J. 2007;92:1825–1835. doi: 10.1529/biophysj.106.097709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greene DN, et al. Single-molecule force spectroscopy reveals a stepwise unfolding of Caenorhabditis elegans giant protein kinase domains. Biophys J. 2008;95:1360–1370. doi: 10.1529/biophysj.108.130237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puchner EM, et al. Mechanoenzymatics of titin kinase. Proc Natl Acad Sci USA. 2008;105:13385–13390. doi: 10.1073/pnas.0805034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junker JP, Ziegler F, Rief M. Ligand-dependent equilibrium fluctuations of single calmodulin molecules. Science. 2009;323:633–637. doi: 10.1126/science.1166191. [DOI] [PubMed] [Google Scholar]
- 8.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 9.Woodside MT, et al. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science. 2006;314:1001–1004. doi: 10.1126/science.1133601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodside MT, et al. Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc Natl Acad Sci USA. 2006;103:6190–6195. doi: 10.1073/pnas.0511048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenleaf WJ, Frieda KL, Foster DAN, Woodside MT, Block SM. Direct observation of hierarchical folding in single riboswitch aptamers. Science. 2008;319:630–633. doi: 10.1126/science.1151298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen YF, Blab GA, Meiners JC. Stretching submicron biomolecules with constant-force axial optical tweezers. Biophys J. 2009;96:4701–4708. doi: 10.1016/j.bpj.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebhardt JCM, Bornschlögl T, Rief M. Full distance-resolved folding energy landscape of one single protein molecule. Proc Natl Acad Sci USA. 2010;107:2013–2018. doi: 10.1073/pnas.0909854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyeon C, Morrison G, Thirumalai D. Force-dependent hopping rates of RNA hairpins can be estimated from accurate measurement of the folding landscapes. Proc Natl Acad Sci USA. 2008;105:9604–9609. doi: 10.1073/pnas.0802484105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manosas M, Ritort F. Thermodynamic and kinetic aspects of RNA pulling experiments. Biophys J. 2005;88:3224–3242. doi: 10.1529/biophysj.104.045344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudko OK, Hummer G, Szabo A. Intrinsic rates and activation free energies from single-molecule pulling experiments. Phys Rev Lett. 2006;96:108101. doi: 10.1103/PhysRevLett.96.108101. [DOI] [PubMed] [Google Scholar]
- 17.Manosas M, et al. Force unfolding kinetics of RNA using optical tweezers. II. Modeling experiments. Biophys J. 2007;92:3010–3021. doi: 10.1529/biophysj.106.094243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudko OK, Hummer G, Szabo A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc Natl Acad Sci USA. 2008;105:15755–15760. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang MJ, Asbury CL, Shaevitz JW, Block SM. An automated two-dimensional optical force clamp for single molecule studies. Biophys J. 2002;83:491–501. doi: 10.1016/S0006-3495(02)75185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nambiar R, Gajraj A, Meiners JC. All-optical constant-force laser tweezers. Biophys J. 2004;87:1972–1980. doi: 10.1529/biophysj.103.037697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenleaf WJ, Woodside MT, Abbondanzieri EA, Block SM. Passive all-optical force clamp for high-resolution laser trapping. Phys Rev Lett. 2005;95:208102. doi: 10.1103/PhysRevLett.95.208102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cellmer T, Henry ER, Hofrichter J, Eaton WA. Measuring internal friction of an ultrafast-folding protein. Proc Natl Acad Sci USA. 2008;105:18320–18325. doi: 10.1073/pnas.0806154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Best RB, Hummer G. Coordinate-dependent diffusion in protein folding. Proc Natl Acad Sci USA. 2010;107:1088–1093. doi: 10.1073/pnas.0910390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hinczewski M, von Hansen Y, Dzubiella J, Netz RR. How the diffusivity profile reduces the arbitrariness of protein folding free energies. J Chem Phys. 2010;132:245103. doi: 10.1063/1.3442716. [DOI] [PubMed] [Google Scholar]
- 25.Landau L, Lifshitz E. Statistical Physics, Part 1. Oxford, UK: Pergamon Press; 1980. [Google Scholar]
- 26.Oppenheim AV, Willsky AS, Nawab SH. Signals & Systems. 2nd Ed. Upper Saddle River, NJ: Prentice-Hall; 1996. [Google Scholar]
- 27.Muzzey D, Gomez-Uribe CA, Mettetal JT, van Oudenaarden A. A systems-level analysis of perfect adaptation in yeast osmoregulation. Cell. 2009;138:160–171. doi: 10.1016/j.cell.2009.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gopich IV, Szabo A. Decoding the pattern of photon colors in single-molecule FRET. J Phys Chem B. 2009;113:10965–10973. doi: 10.1021/jp903671p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung HS, Louis JM, Eaton WA. Distinguishing between protein dynamics and dye photophysics in single-molecule FRET experiments. Biophys J. 2010;98:696–706. doi: 10.1016/j.bpj.2009.12.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung HS, et al. Extracting rate coefficients from single-molecule photon trajectories and FRET efficiency histograms for a fast-folding protein. J Phys Chem A. (in press) [DOI] [PMC free article] [PubMed]
- 31.Szabo A. Theory of polarized fluorescent emission in uniaxial liquid-crystals. J Chem Phys. 1980;72:4620–4626. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.