

Abstract
Regulation of gene expression through translational control is a fundamental mechanism implicated in many biological processes ranging from memory formation to innate immunity and whose dysregulation contributes to human diseases. Genome wide analyses of translational control strive to identify differential translation independent of cytosolic mRNA levels. For this reason, most studies measure genes’ translation levels as log ratios (translation levels divided by corresponding cytosolic mRNA levels obtained in parallel). Counterintuitively, arising from a mathematical necessity, these log ratios tend to be highly correlated with the cytosolic mRNA levels. Accordingly, they do not effectively correct for cytosolic mRNA level and generate substantial numbers of biological false positives and false negatives. We show that analysis of partial variance, which produces estimates of translational activity that are independent of cytosolic mRNA levels, is a superior alternative. When combined with a variance shrinkage method for estimating error variance, analysis of partial variance has the additional benefit of having greater statistical power and identifying fewer genes as translationally regulated resulting merely from unrealistically low variance estimates rather than from large changes in translational activity. In contrast to log ratios, this formal analytical approach estimates translation effects in a statistically rigorous manner, eliminates the need for inefficient and error-prone heuristics, and produces results that agree with biological function. The method is applicable to datasets obtained from both the commonly used polysome microarray method and the sequencing-based ribosome profiling method.
Keywords: differential expression, RIP-CHIP, random variance model, translatomics
Regulation of gene expression is a multistep process that includes transcription, splicing, mRNA-export, -localization, -stability, and -translation. Translational control of gene expression can be achieved by modulating translation elongation, termination, or initiation, although the latter seems to be most frequent (1). Thus, differential translation usually involves a change in the number of ribosomes bound to each mRNA, leading to a change in the amount of synthesized protein per mRNA molecule and time unit. This regulation can be accomplished by a specific process, which targets individual or sets of mRNAs for regulation, or by a general process, which affects most mRNAs equally (2). Specific translational control is important in many biological processes, including cellular senescence (3) and diseases (e.g., cancer) (4).
The polysome microarray approach is the most commonly used method for studying genome wide translational control. In this approach, mRNAs associated with several ribosomes (usually > 3) are separated from mRNAs associated with fewer ribosomes and probed with microarrays. A more recent method, which circumvents some of the polysome microarray approach’s limitations (5), involves isolation and sequencing of RNA pieces that are physically protected by ribosomes (6). A critical interpretative difficulty with both methods, however, is that observed differential levels of actively translated mRNAs or protected mRNA pieces may be due to differential cytosolic mRNA levels. Correction for cytosolic mRNA level has historically been achieved by dividing actively translated mRNA levels by cytosolic mRNA levels obtained in parallel and logging the ratios. We show that these log ratios do not actually correct for cytosolic mRNA levels and that they consequently generate substantial numbers of biological false positives and false negatives. Here we propose a more sensitive and specific method for analysis of translational activity that includes analysis of partial variance (APV) linear regression to control for cytosolic mRNA levels and a variance shrinkage method for improving statistical inference.
Results and Discussion
Common Issues in Genome Wide Analysis of Translational Control.
Two types of data are produced from each sample when studying translational activity: actively translated mRNAs (“translational activity data”) and cytosolic mRNAs (“cytosolic mRNA data”) obtained in parallel. Typically, log (translational activity data/cytosolic mRNA data) ratios are calculated (i.e., logged cytosolic mRNA data is subtracted from its associated logged translational activity data) with the idea of obtaining a cytosolic mRNA data-corrected estimate of translation activity and compared between classes (Fig. 1A). Because log ratios are differences between logged values, class comparison effects are estimated from difference scores. Log difference scores can be correlated with cytosolic mRNA data, however, leading to incorrect biological conclusions. Such is the case when translational activity data and cytosolic mRNA data are uncorrelated for technical or biological reasons (Fig. 1 B and C). For example, false positives can arise from log ratios when mRNAs fail to reach the predetermined threshold for the number of ribosomes necessary to join the pool of actively translated mRNAs or when short poorly translated mRNAs, but not their paired cytosolic mRNAs, fail to achieve counts for protected mRNA pieces above the noise level. Log ratios for such mRNAs tend to produce false positives as they appear to be under translational control (Fig. 1B). False negatives can be produced by log ratios when translational activity is regulated independently of the cytosolic mRNA level (Fig. 1C). The phenomenon of a difference score (Y - Z) correlating with each of its terms (Y and Z) was first described by Pearson in 1897 (7) who labeled it “spurious correlation” because of the frequent practice of interpreting such correlations as substantive rather than artifactual. We will also refer to these correlations as spurious, although our focus will be on the inadequacy of difference scores to control for cytosolic mRNA data and as a consequence fail to correctly estimate class effects (e.g., genotype, treatment, disease). The following equation illustrates the mathematical necessity underlying the problem (8):
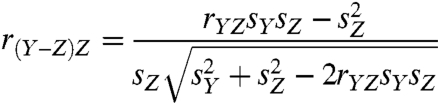 |
[1] |
where Y is a vector of translational activity data for a specific mRNA, Z is a vector of paired cytosolic mRNA data for the same mRNA, r is the Pearson correlation coefficient, and s is the sample standard deviation. When translational and cytosolic levels are uncorrelated, Eq. 1 simplifies to:
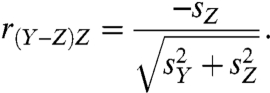 |
[2] |
Eq. 2 makes the origin of these false negatives and false positives clear; under this circumstance the correlation between the log ratios and their corresponding log cytosolic mRNA data is a function of the standard deviations of translational activity data and cytosolic mRNA data replicates. When the standard deviations are also equal, Eq. 2 yields a correlation of -0.71 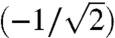 . That is, half (-0.712) of the variance associated with the log ratio would be due to cytosolic mRNA levels. A similar difficulty with log ratios arises in the more typical situation in which the correlation between translational activity data and cytosolic mRNA is nonzero. When standard deviations are also equal, Eq. 1 then simplifies to:
. That is, half (-0.712) of the variance associated with the log ratio would be due to cytosolic mRNA levels. A similar difficulty with log ratios arises in the more typical situation in which the correlation between translational activity data and cytosolic mRNA is nonzero. When standard deviations are also equal, Eq. 1 then simplifies to:
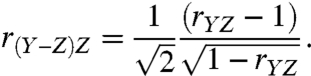 |
[3] |
When the correlation between translational activity and paired cytosolic mRNA is 0.60, Eq. 3 yields a correlation of -0.45. Thus, under various realistic scenarios, the correlation between log ratios and cytosolic mRNA is nontrivial and gives rise to biological false positives and negatives.
Fig. 1.

(A–D) Common problems when analyzing translational activity data. Data were simulated to generate two sample classes, each with five measurements of paired translational activity data (indicated as translation on each y-axis) and cytosolic mRNA data (indicated as transcription on each x-axis). The examples were generated to illustrate different scenarios of translational control analysis. For each example, a two-tailed t-test was performed comparing the sample classes using the translational activity data only or the log-ratio data (p-values as indicated). anota was also performed on each example (p-value as indicated). Lines represent the regression lines estimated by anota for the two sample categories. (E) Common spurious correlations in published datasets of translational control. Shown are boxplots of the spurious correlations that emerge between the log (translational activity data/cytosolic mRNA data) ratios and log cytosolic mRNA data. (F) Small overlap between the log-ratio approach and anota. The top 5% genes ranked by significance from the log-ratio approach and anota were collected. The overlap between the log-ratio approach and anota is visualized using Venn diagrams for the three example studies.
To examine the practical implications of problems with log ratios, we calculated the per gene correlations between logged translational activity data/cytosolic mRNA data ratios and log cytosolic mRNA data per gene across all samples in a set of published studies (9–28). The median of the per dataset medians for the spurious correlations was -0.61 with values covering almost the entire range between +1 and -1 (Fig. 1E). Although these results do not imply low data quality, they demonstrate that log ratios are inappropriate for controlling the confounding effects of cytosolic mRNA levels. It is noteworthy that using the union from the translational activity data only analysis and the log ratio approach resolves the false negative example (Fig. 1C), but leads to increased false positives (Fig. 1 A and B). Using the intersection of the translational activity data only analysis and the log ratio approach limits identification of differential translation to situations that are shown in Fig. 1D and may fail to identify instances of translational regulation that are independent of cytosolic mRNA levels (Fig. 1C). One might argue that a set of heuristic thresholds for differences at the translational activity data and cytosolic mRNA data could be used either alone or in combination with the translational activity data only or the log ratio approach. Such heuristics suffer from numerous failings, however, including the absence of objective statistical thresholds and that any choice of subjective threshold generates unknown rates of false positives and false negatives. Thus, from both theoretical and practical standpoints, current approaches are inadequate for analysis of differential translation.
Anota Outperforms Current Approaches for Analysis of Differential Translation.
APV (29) provides a more suitable analysis of translational activity data because APV-corrected translation measurements do not, by definition, show spurious correlations. We have implemented this approach in the anota (analysis of translational activity) R-package. In anota, a common slope for all sample categories is identified for each gene from the least squares linear regression of translational activity data on cytosolic mRNA data. Class comparison effects are estimated by calculating differences between sample category intercepts; the sum of squares error for these comparisons is reduced by the sum of squares associated with the covariance between the translational activity data and the cytosolic mRNA data. We found that anota potentially correctly analyzes the different scenarios outlined in Fig. 1 A–D. Anota corrects for confounding cytosolic mRNA levels (Fig. 1A), handles false positives arising from technical issues (Fig. 1B), identifies translational regulation that is independent of cytosolic mRNA levels (Fig. 1C), and identifies translational control when there is some confounding cytosolic mRNA difference (Fig. 1D). In summary, anota handles different problematic scenarios by automatically adapting the analysis to the situation at hand.
To further assess anota’s performance, we analyzed genome wide data from both the polysome microarray approach and the sequencing-based approach in three studies [Ingolia et al. (24), Kitamura et al. (21), and Otulakowski et al.(9); see Fig. 1E]. Two sample categories were identified in each study. We compared results obtained from anota with those obtained from a t-test between the log ratios of the two sample classes. Scatter plots of effects and of p-values reveal large discrepancies between the two approaches for many genes (Fig. S1). We investigated this further by classifying mRNAs as either translationally activated or inactivated and examining the overlap among the top 5% of differentially translated mRNAs (ranked by nominal p-values) between the two analytic approaches. As shown in Fig. 1F, the number of genes that were identified as translationally regulated by one method but not the other was substantially higher than the number of shared genes. Genes were then classified into one of three sets: identified by anota only, log ratio only, or by both log ratio and anota. The top two genes (ranked by p-value) from each set from the Ingolia et al. study (24) illustrate the differences between the methods (Fig. 2). Of the top two genes identified by the log ratio approach, one was of the type exemplified in Fig. 1B with cytosolic mRNA data differences combined with smaller translational activity data differences (Fig. 2, rank 2) whereas the other showed a larger difference at the cytosolic mRNA level compared to the translational activity level, leading to the conclusion that a gene with lower translational activity data is more translationally active. This could be a case of “overcorrection” and hence represents a false positive or possibly a biologically interesting finding. However, it is commonly assumed that a gene that is translationally activated would also show higher protein levels and this gene would therefore not be a primary target for follow-up studies. The top two genes that were identified by anota only showed translational regulation that was independent of cytosolic mRNA levels and accordingly belonged to the type exemplified in Fig. 1C. The genes that were identified by both anota and the log ratio approach showed translational regulation independent of cytosolic mRNA level (as exemplified in Fig. 1C) or a small difference in cytosolic mRNA level associated with a larger difference in translational activity data (as exemplified in Fig. 1D). The two other studies analyzed in Fig. 1F showed similar results (Figs. S2 and S3).
Fig. 2.

A comparison of the top two mRNAs identified as differentially translated by the log-ratio approach and by anota. Translational activity data (indicated as translation on each y-axis) and cytosolic mRNA data (indicated as transcription on each x-axis) for the top two ranked genes (by p-value) for each set (log-ratio only; anota only; and both) are plotted using a common scale for all graphs. The effects obtained from anota and from the log-ratio approach are indicated together with their corresponding p-values. The differences in means between the sample classes for the translational activity data only and the cytosolic mRNA data only are indicated as delta translation and delta transcription, respectively. Lines represent the regression lines estimated by anota for the two sample categories.
To extend the observations from Fig. 2, we compared all genes from the three sets (identified by anota only, log ratios only, or both log ratios and anota). Within each set, genes were classified further into one of three modes of regulation based on between-group changes in translational activity data and cytosolic mRNA data (shown as delta translation and delta transcription respectively, in Fig. 2). The first mode, labeled “translation > transcription,” identifies those genes for which the translational activity data difference was larger than the opposing cytosolic mRNA difference; i.e., for a gene that is translationally activated, the activation originated primarily from an activation at the translational activity level and not from a decrease at the cytosolic mRNA level (all genes identified by anota only or by both anota and log ratios in Fig. 2 fit into this group). The second mode, labeled “no translation,” identifies those genes for which there was no difference in the translational activity data; i.e., for a gene defined as activated, no increase in the translational activity data (delta translation ≤ 0) was associated with a decrease in cytosolic mRNA data (the log ratio only genes from Fig. 2 would belong to this mode). The third mode, labeled “transcription > translation,” identifies those genes that show a translational activity data difference but for which the cytosolic mRNA data difference in the opposite direction is of larger magnitude. Of these three modes, the translation > transcription mode corresponds to the examples shown in Fig. 1 C and D and represents cases when translational regulation is plausible whereas the no translation and the transcription > translation modes are variants of the example in Fig. 1B when it is unclear if there is any translational regulation. Anota primarily identified genes belonging to the preferred translation > transcription mode whereas most of the genes identified by the log ratio approach belong to the undesired no translation or transcription > translation modes (Fig. 3). Interestingly, those genes that were identified by both anota and the log ratio approach showed a distribution among the three modes similar to that of anota and thus represent a subset of genes for which the log ratio approach obtained a result similar to anota. In summary, anota outperforms the log ratio approach and can be used both for microarray-based as well as sequencing-based datasets.
Fig. 3.

A comparison of the mode of translational regulation of genes identified as differentially translated by the log-ratio approach and by anota. The top 5% of genes ranked by p-value from the three examined datasets (from Fig. 1F) were separated into translationally inactivated and translationally activated (i.e., negative and positive effects) and further divided into three sets each: those identified by log ratio only, those identified by anota only, and those identified by both log ratio and anota. The genes within each of these six sets were categorized based on their mode of regulation (the modes are further discussed in the main text): Translation > transcription (similar to Fig. 1 C and D); no translation (similar to Fig. 1B); Transcription > translation (similar to Fig. 1B). Shown is the percent of genes among the translationally activated or translationally inactivated belonging to each mode.
Improving the Performance of Anota Using Variance Shrinkage.
In genome wide studies, gene-specific variances of many genes will be greatly under or overestimated due to chance factors. It is therefore possible that some of the genes shown in Fig. 2 have been identified as a result of unrealistically low variance estimates. This characteristic of high dimensional data has produced a consensus among methodologists to favor variance shrinkage methods that adjust error variances for statistical tests by weighting gene-specific variance estimates and a variance estimate obtained from all genes in the dataset (30). We have generalized one such method, the random variance model (RVM) (31), for APV within anota (referred to as “anota RVM”). We performed an analysis identical to that shown in Fig. 2 but with the RVM-adjusted error term. The top three genes that were selected by anota RVM but not by the log ratio approach in the Ingolia et al. study (24) are shown in Fig. 4A. These genes show larger effects than those genes identified without RVM (compare Fig. 4A to Fig. 2). To illustrate the effect across all genes, we generated “volcano” plots comparing anota and anota RVM. The volcano plots from the Ingolia et al. dataset show that smaller effects are less likely to be associated with high significance when anota RVM is used (Fig. 4B). Finally, we compared the cumulative distribution function of the raw p-values and p-values that had been adjusted for multiple testing (32) from anota and anota RVM (Fig. 4C). This analysis indicated that anota RVM generated smaller p-values than anota without RVM adjustment [similar results were obtained for the other two datasets from Fig. 1F (Figs. S4 and S5)].
Fig. 4.

RVM improves the performance of APV within anota. (A). Translational activity data (indicated as translation on each y-axis) and cytosolic mRNA data (indicated as transcription on each x-axis) from the top two genes (ranked by p-value) that were identified among the top 5% differentially translated (ranked by p-value) by anota RVM but not by the log-ratio approach are shown. The two sample categories are indicated by squares and triangles. The same scale as in Fig. 2 is used. Effects calculated by anota RVM and by the log-ratio approach are indicated together with their corresponding p-values. The differences in means between the sample classes for the translational activity data only and the cytosolic mRNA data only are indicated as delta translation and delta transcription, respectively. Lines represent the regression lines estimated by anota for the two sample categories. (B). Volcano plots for anota and anota RVM. The - log 10 p-value is plotted as a function of the effect. (C). A comparison of statistical power between anota and anota RVM. Shown is the cumulative distribution function (CDF) of all p-values from anota and anota RVM. CDFs for both the raw p-values and p-values adjusted for multiple testing as described in the text are shown.
We searched for enrichment of genes with shared biological functions among genes identified by anota RVM and the log-ratio approach to assess whether the former enabled new biological insights. Genes identified as differentially translated by anota RVM shared functions to a larger extent than genes identified as translationally regulated by the log-ratio approach in all three datasets shown in Fig. 1F (Figs. S6–S8). This suggests that anota RVM is associated with fewer biological false positive and false negative findings inasmuch as a completely random set of genes would not enable identification of any enriched biological functions. Thus, anota identifies differential translation that is more biologically coherent compared to log ratios and thereby offers greater promise for biological discoveries.
Conclusions
Regulation of translation is important for many biological processes and dysregulation is present in diseases such as cancer and fibrosis. Despite this, genome-wide studies of differential translation have been rare. Downstream of the technical challenges, data analysis has in the past relied on simple intuitive approaches that we show are ineffective for analysis of differential translation and have led to many false inferences that may have hampered the enthusiasm for genome wide studies of translational control. Our approach to analysis of translational activity provides an advancement that is necessary to understand how mRNA specific translation is regulated on a translatome wide scale and shows promise for generating reproducible and more biologically sound findings.
We show that our approach for analysis of differential translation is versatile and can be used both on count data from sequencing as well as microarray data. Moreover, our approach is not limited to studies of translational control but could be used in, for example, ribonucleoprotein immunoprecipitation–microarray (RIP-chip) studies where the observed differential level of immunoprecipitated mRNA will be partially dependent on the total mRNA amount. In general, anota can be used profitably with any datasets containing paired-controls.
Defining which genes are under translational control only represents the first step toward mechanistic understanding of translational regulation and its underlying organization. Efforts are currently being made toward identifying tools and approaches that will be necessary to discover mechanisms that define the common regulatory patterns as described in the posttranscriptional regulon theory and by the originally proposed informosomes (33–36). We believe that our methodological improvements for analysis of genome wide datasets are in this spirit and will help to understand how the translatome is regulated from a systems perspective. Such characterization will be necessary to understand how dysregulation of translational control leads to human diseases.
Materials and Methods
Sampling of Example Data.
Data shown in Fig. 1 A–D were sampled from normal distributions. First, 5 cytosolic mRNA data points were sampled and then a set of corresponding translational activity data points were sampled using the values of the sampled cytosolic mRNA data as distribution means. A second sample class was generated in the same manner. The different examples in Fig. 1 A–D were generated by changing the means and the standard deviations of the normal distributions within the “rnorm” function in R.
A Survey of the Occurrence of Spurious Correlations in Published Datasets.
We searched both the ArrayExpress (37) and the Gene Expression Omnibus (38) databases for a set of studies of translational activity. Datasets generated using Affymetrix GeneChips were normalized with the robust multiarray average method (39, 40) using updated probe set definitions when available (41, 42). For the cDNA dataset (11), we used the normalized data provided by the authors. For the sequencing dataset (24), we used the count data supplied by the authors, filtered for identifiers originating from the coding regions, and used quantile normalization and a transformation to stabilize the variance. For all studies, we identified paired translational activity and cytosolic mRNA data and calculated correlations between the log 2 difference scores and the cytosolic mRNA data per gene across all samples in each study.
Analysis of Differential Translation Using Three Datasets.
We identified two sample classes from each study to be used for the analysis of differential translation. For the Kitamura et al. dataset (21), we compared “no treatment” to “LPS treatment 4 h.” For the Otulakowski et al. dataset (9), we compared “FD19” to “P1.” The sequencing-based dataset contained only two samples classes (“rich” and “starved”) and only two samples per class (24). Because anota requires at least 3 samples per sample class, we simulated a third sample for each class and type of data (actively translated and cytosolic mRNAs). The simulation was done using the transformed and normalized data and by sampling one new data point from a normal distribution with a mean and a standard deviation derived from the data points of that gene in the empirical data. Correlation between the simulated and empirical data was similar to the correlation between the two empirical data replicates. To simulate the presence of outliers, we also generated a dataset for which 5% of all data points were shifted by adding or subtracting 3 gene-specific standard deviations to/from the mean of the normal distribution from which the data point was sampled. The two simulated datasets generated similar results; we present results from the dataset without outliers. We generated log ratios using data from actively translated mRNAs and cytosolic mRNAs that originated from the same sample. Student’s t test (two-tailed unequal variance) was used to compare the log ratios between the sample categories.
Supplementary Material
Acknowledgments.
We thank Richard Simon and George Wright for permission to incorporate some of their original RVM code into anota. O.L. was initially supported by a fellowship from the Knut and Alice Wallenberg Foundation. Partial support was provided by Le Fonds Québécois de la Recherche sur la Nature et les Technologies (FQRNT) Grant 119258 (R.N.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1006821107/-/DCSupplemental.
References
- 1.Mathews M, Sonenberg N, Hershey JBW. Translational Control in Biology and Medicine. New York: CSHL Press; 2007. p 1. [Google Scholar]
- 2.Polunovsky VA, Bitterman PB. The cap-dependent translation apparatus integrates and amplifies cancer pathways. RNA Biol. 2006;3:10–17. doi: 10.4161/rna.3.1.2718. [DOI] [PubMed] [Google Scholar]
- 3.Petroulakis E, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009;16:439–446. doi: 10.1016/j.ccr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 4.Silvera D, et al. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol. 2009;11:903–908. doi: 10.1038/ncb1900. [DOI] [PubMed] [Google Scholar]
- 5.Larsson O, Nadon R. Gene expression—Time to change point of view? Biotechnol Genet Eng. 2008;25:77–92. doi: 10.5661/bger-25-77. [DOI] [PubMed] [Google Scholar]
- 6.Arava Y, Boas FE, Brown PO, Herschlag D. Dissecting eukaryotic translation and its control by ribosome density mapping. Nucleic Acids Res. 2005;33:2421–2432. doi: 10.1093/nar/gki331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearson K. On a form of spurious correlation which may arise when indices are used in the measurement of organs. P R Soc London. 1896;60:489–498. [Google Scholar]
- 8.Cohen J, Cohen P, West SG, Aiken L. Applied Multiple Regression/Correlation Analysis for the Behavioral Sciences. London: Erlbaum; 2003. [Google Scholar]
- 9.Otulakowski G, Duan W, O’Brodovich H. Global and gene-specific translational regulation in rat lung development. Am J Respir Cell Mol Biol. 2009;40:555–567. doi: 10.1165/rcmb.2008-0284OC. [DOI] [PubMed] [Google Scholar]
- 10.Sampath P, et al. A hierarchical network controls protein translation during murine embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2008;2:448–460. doi: 10.1016/j.stem.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 11.Lu X, de la Pena L, Barker C, Camphausen K, Tofilon PJ. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res. 2006;66:1052–1061. doi: 10.1158/0008-5472.CAN-05-3459. [DOI] [PubMed] [Google Scholar]
- 12.Chen YW, et al. Response of rat muscle to acute resistance exercise defined by transcriptional and translational profiling. J Physiol. 2002;545:27–41. doi: 10.1113/jphysiol.2002.021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajasekhar VK, et al. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell. 2003;12:889–901. doi: 10.1016/s1097-2765(03)00395-2. [DOI] [PubMed] [Google Scholar]
- 14.Iguchi N, Tobias JW, Hecht NB. Expression profiling reveals meiotic male germ cell mRNAs that are translationally up- and down-regulated. Proc Natl Acad Sci USA. 2006;103:7712–7717. doi: 10.1073/pnas.0510999103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spence J, Duggan BM, Eckhardt C, McClelland M, Mercola D. Messenger RNAs under differential translational control in Ki-ras-transformed cells. Mol Cancer Res. 2006;4:47–60. doi: 10.1158/1541-7786.MCR-04-0187. [DOI] [PubMed] [Google Scholar]
- 16.Larsson O, et al. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814–6824. doi: 10.1158/0008-5472.CAN-07-0752. [DOI] [PubMed] [Google Scholar]
- 17.Parent R, Kolippakkam D, Booth G, Beretta L. Mammalian target of rapamycin activation impairs hepatocytic differentiation and targets genes moderating lipid homeostasis and hepatocellular growth. Cancer Res. 2007;67:4337–4345. doi: 10.1158/0008-5472.CAN-06-3640. [DOI] [PubMed] [Google Scholar]
- 18.Genolet R, Araud T, Maillard L, Jaquier-Gubler P, Curran J. An approach to analyze the specific impact of rapamycin on mRNA-ribosome association. BMC Med Genomics. 2008;1:33. doi: 10.1186/1755-8794-1-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsson O, et al. Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PLoS One. 2008;3:e3220. doi: 10.1371/journal.pone.0003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grech G, et al. Igbp1 is part of a positive feedback loop in stem cell factor-dependent, selective mRNA translation initiation inhibiting erythroid differentiation. Blood. 2008;112:2750–2760. doi: 10.1182/blood-2008-01-133140. [DOI] [PubMed] [Google Scholar]
- 21.Kitamura H, et al. Genome-wide identification and characterization of transcripts translationally regulated by bacterial lipopolysaccharide in macrophage-like J774. 1 cells. Physiol Genomics. 2008;33:121–132. doi: 10.1152/physiolgenomics.00095.2007. [DOI] [PubMed] [Google Scholar]
- 22.Parent R, Beretta L. Translational control plays a prominent role in the hepatocytic differentiation of HepaRG liver progenitor cells. Genome Biol. 2008;9:R19. doi: 10.1186/gb-2008-9-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramirez-Valle F, Braunstein S, Zavadil J, Formenti SC, Schneider RJ. eIF4GI links nutrient sensing by mTOR to cell proliferation and inhibition of autophagy. J Cell Biol. 2008;181:293–307. doi: 10.1083/jcb.200710215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YY, et al. Eukaryotic initiation factor 4E binding protein family of proteins: Sentinels at a translational control checkpoint in lung tumor defense. Cancer Res. 2009;69:8455–8462. doi: 10.1158/0008-5472.CAN-09-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ceppi M, et al. Ribosomal protein mRNAs are translationally-regulated during human dendritic cells activation by LPS. Immunome Res. 2009;5:5. doi: 10.1186/1745-7580-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang Do AN, Kimball SR, Cavener DR, Jefferson LS. eIF2alpha kinases GCN2 and PERK modulate transcription and translation of distinct sets of mRNAs in mouse liver. Physiol Genomics. 2009;38:328–341. doi: 10.1152/physiolgenomics.90396.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zid BM, et al. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schleifer SJ, Eckholdt HM, Cohen J, Keller SE. Analysis of partial variance (Apv) as a statistical approach to control day-to-day variation in immune assays. Brain Behav Immun. 1993;7:243–252. doi: 10.1006/brbi.1993.1025. [DOI] [PubMed] [Google Scholar]
- 30.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: From disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 31.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 32.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- 33.Fan D, Bitterman PB, Larsson O. Regulatory element identification in subsets of transcripts: Comparison and integration of current computational methods. RNA. 2009;15:1469–1482. doi: 10.1261/rna.1617009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keene JD. RNA regulons: coordination of post-transcriptional events. Nat Rev Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- 35.Spirin AS. The second Sir Hans Krebs Lecture. Informosomes. Eur J Biochem. 1969;10:20–35. doi: 10.1111/j.1432-1033.1969.tb00651.x. [DOI] [PubMed] [Google Scholar]
- 36.Foat BC, Stormo GD. Discovering structural cis-regulatory elements by modeling the behaviors of mRNAs. Mol Syst Biol. 2009;5:268. doi: 10.1038/msb.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parkinson H, et al. ArrayExpress update—From an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Res. 2009;37:D868–D872. doi: 10.1093/nar/gkn889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrett T, et al. NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–D890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 40.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai M, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sandberg R, Larsson O. Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinformatics. 2007;8:48. doi: 10.1186/1471-2105-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.