

The efficient synthesis of highly substituted cyclopentanols is an important task given the prevalence of this class of compounds in nature. Nitrocyclopentanols are of particular value due to the rich chemistry associated with the nitro group[1] and their potential use as aminocyclopentitol progenitors.[2] Aminocyclopentitols have generated considerable attention because of their significant biological activity and synthetic challenges presented by their often dense functionality and contiguous chiral centers. As such, the development of a flexible synthesis of functionalized nitrocyclopentanols would be a welcome addition to the synthetic toolbox. Herein we report the three-component coupling of silyl glyoxylates, CH2=CHMgBr, and nitroalkenes that selectively affords (Z)-silyl enol ether products through a unique vinylogous Michael cascade. The resulting functionality enables the immediate implementation of a second-stage Henry cyclization for the expeditious, diastereoselective synthesis of functionalized nitrocyclopentanols.
Silyl glyoxylates are conjunctive reagents for the union of complementary nucleophilic and electrophilic partners linked at a protected glycolic acid junction.[3] The use of these reagents in coupling reactions with alkide and hydride nucleophiles[4] and carbonyl secondary electrophiles has been documented. We endeavoured to expand the utility of silyl glyoxylate chemistry to include Michael acceptors as the secondary electrophile. Nitroalkenes were chosen by virtue of their highly electrophilic character and the synthetic versatility of the nitro functionality. The proposed transformation outlined in Scheme 1 involves the addition of vinyl Grignard to the silyl glyoxylate 1 to reveal, after [1,2]-Brook rearrangement,[5] the (Z)-metallodienolate 2.[4a,c] This intermediate could act as a transient secondary nucleophile capable of engaging the nitroalkene 4 in a vinylogous Michael addition to provide enolsilane 6. As such, the combination of 1 and CH2=CHMgBr would function as the synthetic equivalent of the unusual α-keto ester homoenolate synthon, perhaps by way of the C-metalated tautomer 3 of the (Z)-metallodienolate (Scheme 2).[6]
Scheme 1.

Divergent silyl glyoxylate reactivity.
Scheme 2.

Proposed access to the α-ketoester homoenolate synthon.
An open question at the outset of this inquiry was whether α/γ selectivity would exhibit electrophile dependence. Previous silyl glyoxylate-based couplings predominantly provided α-selectivity from metallodienolate intermediates.[4a,c] Moreover, the vinylogous Michael reaction of metallodienolates is infrequently deployed due to an innate kinetic preference for α-addition: evaluation of frontier-orbital densities and HOMO coefficients establish the α-carbon as the more nucleophilic site.[7] While some exceptions exist,[8] successful vinylogous Michael reactions frequently require considerable prefunctionalization to control selectivity and are almost exclusively performed with butenolide derivatives[9] and α,α-dicyanoolefins.[10] A second challenge lies in the management of the relevant rate constants. Stepwise reagent introduction was viewed as untenable in light of the known nucleophile-initiated oligomerization of 1 in the absence of a secondary electrophile.[4a,c] The one-pot coupling would be efficient only if CH2=CHMgBr exhibited high chemoselectivity for 1 over 4 and the derived intermediate 3 manifested the opposite preference.
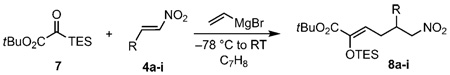
Initial experiments with tert-butyl dimethylsilyl glyoxylate, β-nitrostyrene, and vinyl Grignard at −78°C provided the desired vinylogous Michael reactivity albeit in an unsatisfactory 43% yield for the (Z)-silyl enol ether product with silyl glyoxylate oligomers as the major byproducts. The (Z)-geometry was determined by NOESY analysis of enolsilane and confirmed by NOESY analysis of the derived silyloxyepoxide. This observed (Z)-selectivity is consistent with the Kuwajima-type intermediate 3;[6] a Diels–Alder pathway through the s-cis conformer of 2 would have given the (E)-enolsilane. This result also demonstrates that vinyl Grignard does act as a discriminating nucleophile with a strong preference for the silyl glyoxylate over the nitroalkene. The predominance of the vinylogous reactivity in this reaction is likely due to steric hindrance congesting the α-addition pathway in conjunction with γ-preference of the (Z)-metallodienolate based on the hard/soft character of the electrophile.[7] Such an analysis provides a plausible explanation for divergence from previous silyl glyoxylate coupling reactions that have thus far been limited to hard electrophiles.
Optimization of the reaction conditions required 50% excess of silyl glyoxylate and vinyl Grignard to compensate for the partial oligomerization of the silyl glyoxylate. A temperature of −78°C or lower was required since the [1,2]-Brook rearrangement and subsequent reactivity leads to decomposition at higher temperatures. The silyl group was also varied to determine its effect on the reaction. The triethylsilyl (TES) and tert-butyldimethylsilyl (TBS) groups were found to provide similar yields, with the bulkier triisopropylsilyl (TIPS) group providing a diminished yield (54%) and the trimethylsilyl (TMS) group performing the poorest (30%). The triethylsilyl glyoxylate was chosen for the substrate scope over the tert-butyldimethylsilyl glyoxylate due to its more facile deprotection and functionalization. The optimized conditions for the coupling involved dropwise addition of vinyl Grignard to a solution of the silyl glyoxylate and the nitroalkene at −78°C followed by warming to room temperature to afford the desired product 8a in a 72% yield based on β-nitrostyrene.[11]
An examination of various alkyl, aryl, and heteroaryl nitroalkenes was conducted with the results summarized in Table 1. The yields ranged from 36–72% with aryl and heteroaryl nitroalkenes outperforming the alkyl substrates. The efficiency of the reaction for the alkyl examples tended to decrease with the increasing steric hindrance from α-branching on the R group.
Table 1.
Substrate scope for three-component coupling.
 | ||
|---|---|---|
| Product[a] | R | Yield [%] |
| 8a | Ph | 72[b] |
| 8b | 4-Cl-C6H4 | 70[b] |
| 8c | 2-thienyl | 65[b] |
| 8d | 2-furyl | 64[b] |
| 8e | (E)-CH=CH-C6H5 | 66[b] |
| 8 f | H | 57[c] |
| 8g | n-pentyl | 58[b] |
| 8h | iPr | 53[c] |
| 8i | tBu | 36[c] |
Reagents: Silyl glyoxylate (1.5 equiv), nitroalkene (1 equiv), CH2= CHMgBr (1.5 equiv), [4]0=0.1m in C7H8;
Yield of isolated product.
Yields were determined by 1H NMR spectroscopy of crude product compared to an internal standard of mesitylene.
The silyl enol ethers 8a–i are assembled in the correct oxidation state to enable a deprotection/Henry cyclization cascade to furnish nitrocyclopentanols with three contiguous stereocenters. The intramolecular Henry reaction is an infrequently utilized transformation due to the difficulty in synthesizing the requisite bifunctional nitrocarbonyl precursors.[12] Our next set of experiments evaluated optimal conditions to reveal the latent nitroketone functionality thereby enabling in situ Henry cyclization. Various deprotection methods were screened for the desired cascade using the substrate 8a. Fluoride-based deprotections accomplished the desired transformation; however, the diastereomeric ratios were inconsistent, and mixtures of the acyclic α-ketoester and the desired nitrocyclopentanol 9a were isolated in varying ratios. Substituting the fluoride deprotections for a basic methanol deprotection at −5°C provided the desired adduct 9a in 78% yield with a greater than 20:1 diastereo-selection.[13]
We then examined the scope of the Henry cyclization for the enolsilanes 8a–i (Table 2). Yields ranged from 58–94% with alkyl, aryl, and heteroaryl substituents all being well tolerated. The diastereomeric ratio was controlled by the identity of the side chain. The branched substrates containing aryl, heteroaryl, iPr, and tBu groups provided the highest diastereoselectivies (≥20:1) while the products with unbranched R groups such as styryl (9e), hydrogen (9 f), and n-pentyl (9g) were obtained with diminished diastereoselection.
Table 2.
Substrate scope for deprotection/diastereoselective Henry cyclization cascade.
 | ||||
|---|---|---|---|---|
| Product[a] | R | t [h] | Yield [%] | d.r. |
| 9a | Ph | 3 | 78 | >20:1 |
| 9b | 4-Cl-C6H5 | 7 | 69 | 20:1 |
| 9c | 2-thienyl | 3 | 94 | 20:1 |
| 9d | 2-furyl | 3 | 54 | 20:1 |
| 9e | (E)-CH=CH-C6H4 | 12 | 64 | 3:1 |
| 9 f | H | 12 | 70 | 1.5:1 |
| 9g | n-pentyl | 12 | 58 | 5:1 |
| 9h | iPr | 16 | 56 | >20:1 |
| 9i | tBu | 16 | 59 | >20:1 |
Reagents: Silyl enol ether 8a–i (1 equiv), NaOH (1.2 equiv), [8]0= 0.01m in (1:1) MeOH:CH2Cl2.
Stereostructures were determined through NOESY experiments and X-ray crystallography.
This intramolecular Henry cascade provides a complementary method to that recently reported by Zhong et al. in their synthesis of nitrocyclopentanols through an organocatalytic Michael/Henry domino reaction.[14] Where the Zhong work provides a tertiary benzylic alcohol, the present chemistry installs a tertiary glycolic acid moiety, the reduced form of which is present in numerous cyclopentanol natural products including trehazolin[15] and pactamycin.[16] This method also addresses one of the few limitations of their published methodology, which is the requirement of aryl and heteroaryl nitroalkenes. Since the three-component coupling reaction can employ a wide variety of nitroalkenes, it arrives at nitrocyclopentanols with reasonable flexibility in the side chain identity. Moreover, utilization of the nucleophilicity of the silyl enol ether moiety prior to cyclization could enable the synthesis of tetrasubstituted cyclopentanols.
Lastly, we sought to develop an asymmetric variant of this three-component coupling. We examined silyl glyoximides, a new class of reagents recently synthesized by Hsung and co-workers in conjunction with ynamide oxidation studies.[17] Exposing silyl glyoximide 10 to the three-component coupling conditions with vinyl Grignard and β-nitrostyrene gratifyingly provided the desired product 11 in 75% yield and 20:1 diastereoselection (Scheme 3). The absolute configuration of the product was determined by conversion to the known nitroaldehyde by ozonolysis.[18] Studies are underway to determine the potential of silyl glyoximides as a new class of conjunctive reagents and to understand this remarkable long-range stereochemical transmission.[19]
Scheme 3.

Diastereoselective silyl glyoximide coupling.
In summary, a sequential vinylation/[1,2]-Brook rearrangement/vinylogous Michael reaction incorporating silyl glyoxylates, vinyl Grignard, and nitroalkenes has been developed that provides (Z)-silyl enol ether products. This marks the first use of a Michael acceptor as the secondary electrophile in silyl glyoxylate-based cascades and is unique due to the vinylogous reactivity of the (Z)-metallodienolate that diverges from previous silyl glyoxylate couplings. The silyl enol ether products were utilized in a merged deprotection/Henry cyclization sequence that furnishes nitrocyclopentanols with three contiguous stereocenters in two steps from readily available starting materials. We further demonstrated that electrophile facial selectivity is possible in the title coupling through the application of a chiral auxiliary.
Experimental Section
The silyl glyoxylate (123 mg, 0.5 mmol, 1.5 equiv) and β-nitrostyrene (50 mg, 0.33 mmol, 1.0 equiv) were added to an oven-dried vial. The vial was then purged with N2 and toluene (3.3 mL) was added. The resulting solution was cooled to −78°C using an acetone and dry ice bath. Vinylmagnesium bromide (0.5 mL, 0.5 mmol, 1.5 equiv) was added dropwise to the solution. Once the addition was complete the reaction was allowed to warm to room temperature, diluted with ethyl acetate (5 mL), and quenched with saturated ammonium chloride (5 mL). The resulting mixture was stirred for 10 min. The layers were separated, and the aqueous layer was extracted with ethyl acetate (3 × 5 mL). The organic extracts were combined, washed with brine (5 mL), dried with magnesiumsulfate, and concentrated in vacuo. The crude mixture was purified by flash chromatography eluting with 2.5% EtOAc/hexanes to yield 102 mg (72%) of the desired product 8a as a yellow oil. Additional details and full characterization data are presented in the Supporting Information.
Supplementary Material
Footnotes
This project was supported by Award Number R01 GM084927 from the National Institute of General Medical Sciences. Additional support from Novartis and Amgen is gratefully acknowledged. Glyoximide 10 was generously provided by Prof. Richard Hsung and Dr. Ziyad Al-Rashid. X-ray crystallography was performed by Dr. Peter White.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201003470.
References
- 1.Ono N. The Nitro Group in Organic Synthesis. Weinheim: Wiley-VCH; 2001. [Google Scholar]
- 2.Review of aminocyclopentitol syntheses and biological activity: Berecibar A, Grandjean C, Siriwardena A. Chem. Rev. 1999;99:779–844. doi: 10.1021/cr980033l. Flatt P, Mahmud T. Nat. Prod. Rep. 2007;24:358–392. doi: 10.1039/b603816f.
- 3.Nicewicz DA, Brétéché G, Johnson JS. Org. Synth. 2008;85:278–286. [Google Scholar]
- 4.a) Nicewicz DA, Johnson JS. J. Am. Chem. Soc. 2005;127:6170–6171. doi: 10.1021/ja043884l. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Linghu X, Satterfield AD, Johnson JS. J. Am. Chem. Soc. 2006;128:9302–9303. doi: 10.1021/ja062637. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nicewicz DA, Satterfield AD, Schmitt DC, Johnson JS. J. Am. Chem. Soc. 2008;130:17281–17283. doi: 10.1021/ja808347q. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Greszler SN, Johnson JS. Org. Lett. 2009;11:827–830. doi: 10.1021/ol802828d. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Greszler SN, Johnson JS. Angew. Chem. 2009;121:3743–3745. doi: 10.1002/anie.200900215. Angew. Chem. Int. Ed.2009, 48, 3689 – 3691; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Schmitt DC, Johnson JS. Org. Lett. 2010;12:944–947. doi: 10.1021/ol9029353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brook AG. Acc. Chem. Res. 1974;7:77–84. [Google Scholar]
- 6.a) Kuwajima I, Kato M. J. Chem. Soc. Chem. Commun. 1979:708–709. [Google Scholar]; b) Reich HJ, Olson RE, Clark MC. J. Am. Chem. Soc. 1980;102:1423–1424. [Google Scholar]; c) Kato M, Mori A, Oshino H, Enda J, Kobayashi K, Kuwajima I. J. Am. Chem. Soc. 1984;106:1773–1778. [Google Scholar]; d) Enda J, Kuwajima I. J. Am. Chem. Soc. 1985;107:5495–5501. [Google Scholar]; e) Ahlbrecht H, Beyer U. Synthesis. 1999:365–390. [Google Scholar]
- 7.a) Rassu G, Appendino G, Casiraghi G, Zanardi F. Chem. Rev. 2000;100:1929–1972. doi: 10.1021/cr990247i. [DOI] [PubMed] [Google Scholar]; b) Denmark SE, Heemstra JR, Jr, Beutner GL. Angew. Chem. 2005;117:4760–4777. doi: 10.1002/anie.200462338. Angew. Chem. Int. Ed.2005, 44, 4682 – 4698. [DOI] [PubMed] [Google Scholar]
- 8.For examples of direct vinylogous Michael reactions: Hitce J, Trost BM. J. Am Chem. Soc. 2009;131:4572–4573. doi: 10.1021/ja809723u. Wang J, Qi C, Ge Z, Cheng T, Li R. Chem. Commun. 2010;46:2124–2126. doi: 10.1039/b923925a.
- 9. Brown SP, Goodman NC, MacMillan DWC. J. Am Chem. Soc. 2003;125:1192–1194. doi: 10.1021/ja029095q. and references therein.
- 10. Lui T-Y, Cui H-L, Long J, Li B-J, Wu Y, Ding L-S, Chen Y-C. J. Am. Chem. Soc. 2007;129:1878–1879. doi: 10.1021/ja068703p. and references therein.
- 11.Certain substrates required alterations in procedure. See the Supporting Information for details.
- 12.Luzzio FA. Tetrahedron. 2001;57:915–945. [Google Scholar]
- 13.The Supporting Information contains more detailed information regarding the optimization of the deprotection/Henry cyclization and a discussion of the stereochemical assignment based on X-ray crystallography and NMR analysis. CCDC 788056 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 14.Tan B, Chua PJ, Zeng X, Lu M, Zhong G. Org. Lett. 2008;10:3489–3492. doi: 10.1021/ol801273x. [DOI] [PubMed] [Google Scholar]
- 15.a) Murao S, Sakai T, Gibo H, Nakayama T, Shin T. Agric. Biol. Chem. 1991;55:895. [Google Scholar]; b) Ohkuma Y, Kinoshita T, Enokita R. J. Antibiot. 1991;44:1165. doi: 10.7164/antibiotics.44.1165. [DOI] [PubMed] [Google Scholar]
- 16.Wiley PF, Jahnke HK, MacKellar F, Kelly RB, Argoudelis AD. J. Org. Chem. 1970;35:1420–1425. doi: 10.1021/jo00830a035. [DOI] [PubMed] [Google Scholar]
- 17.Al-Rashid ZF, Johnson WL, Hsung RP, Wei Y, Yao P-Y, Liu R, Zhao K. J. Org. Chem. 2008;73:8780–8784. doi: 10.1021/jo8015067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotoh H, Ishikawa H, Hayashi Y. Org. Lett. 2007;9:5307–5309. doi: 10.1021/ol702545z. [DOI] [PubMed] [Google Scholar]
- 19. Nakamura T, Shirokawa S, Hosokawa S, Nakazaki A, Kobayashi S. Org. Lett. 2006;8:677–679. doi: 10.1021/ol052871p. Shirokawa S, Kamiyama M, Nakamura T, Okada M, Nakazaki A, Hosokawa S, Kobayashi S. J. Am. Chem. Soc. 2004;126:13604–13605. doi: 10.1021/ja0465855. and references therein.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.