

Abstract
Constitutive activation of the NF-κB transcription factor plays a key role in chronic colonic inflammation and colon tumorigenesis. However, the mechanisms by which the tightly regulated NF-κB pathway becomes constitutively activated during colonic pathogenesis remain obscure. Here, we report that PDLIM2, an essential terminator of NF-κB activation, is repressed in various human colorectal cancer cell lines, suggesting one important mechanism for the constitutive activation of NF-κB. Indeed, expression of exogenous PDLIM2 inhibited constitutive NF-κB activation in these colorectal cancer cells. Importantly, the PDLIM2 expression was sufficient to suppress in vitro anchorage-independent growth and in vivo tumor formation of these malignant cells. We have further shown that the PDLIM2 repression involves promoter methylation. Accordingly, treatment of the colorectal tumor cell lines with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-aza-dC) restored PDLIM2 expression and resulted in growth arrest. These studies thus provide new mechanistic insights into colon tumorigenesis by identifying a novel tumor suppressor role for PDLIM2.
Introduction
Colon cancer is the third most common malignancy and the second leading cause of cancer death (1). One major predisposition for colon cancer development is chronic colonic inflammation, particularly in patients with inflammatory bowel diseases (IBD) including Crohn’s disease and ulcerative colitis (2). For example, the cumulative incidence of colitis-associated cancer (CAC) is up to 20–50% in patients with ulcerative colitis, accounting for one sixth of deaths in IBD patients (3). Both mouse and human studies suggest that the NF-κB transcription factor plays a causative role in chronic colonic inflammation and subsequent the pathogeneses of IBD and CAC. An NF-κB defect in mouse intestinal epithelial cells or myeloid cells leads to a significant decrease in CAC (4), constitutively activated NF-κB is detected in gut macrophages and epithelial cells of biopsy specimens as well as in colorectal cancer but not in adjacent normal tissue from IBD patients (5–8). Moreover, inhibition of NF-κB reduces the risk of CAC by 75% to 81% (9, 10).
NF-κB activity is tightly controlled under physiological conditions (11). In response to different stimuli, NF-κB is rapidly activated but usually transiently (12). One essential mechanism for the quick termination of the NF-κB response involves nuclear degradation of its prototypic member p65 (13), which is predominantly mediated by PDLIM2 (14). PDLIM2 is a newestly discovered PDZ-LIM domain-containing protein. It has been suggested that the C-terminal LIM domain of PDLIM2 is required for promoting ubiquitination of nuclear p65, while its N-terminal PDZ domain is involved in shuttling nuclear p65 along the nuclear framework into discrete intranuclear compartments for the proteasome-mediated degradation. Accordingly, PDLIM2 knockout mice are more sensitive to lipopolysaccharide (LPS)-induced shock due to enhanced NF-κB/p65 activation and augmented production of inflammatory cytokines (14).
Currently, the mechanism of constitutive NF-κB activation during intestinal pathogenesis remains largely unknown. Here, we show that PDLIM2 is epigenetically repressed in various human colorectal cancer cell lines. PDLIM2 re-expression inhibited NF-κB constitutive activation, in vitro anchorage-independent growth and in vivo tumor formation of these malignant cells. These studies suggest one important mechanism for the constitutive activation of NF-κB in colon tumorigenesis and a novel tumor suppressor role for PDLIM2 in colorectal cancer.
Materials and Methods
Expression vectors and reagents
pQCXIP-myc-PDLIM2, κb-TATA luciferase reporter constructs and PDLIM2 antibody have been described before (15). The anti-Myc antibody was generated from the 9E10 hybridoma as described (16). Nucleoside analog 5-aza-dC, calcein AM and puromycin were purchased from Sigma.
Cell lines
The human colorectal cancer cell lines HCT116, SNU1040, DLD1, SW480, FET, COLO32 and HT29, the human breast epithelial cell line MCF10A, and the human embryonic kidney (HEK) cell line 293T were obtained from the ATCC and cultured according to their protocols..
Real-time PCR analysis
Total RNA was prepared with TRIZOL reagent and cDNA was generated with SuperScript II reverse transcriptase (Invitrogen), followed by real-time PCR assays as described (17). Primer pairs used were: PDLIM2, forward 5′-GCCCATCATGGTGACTAAGG, reverse 5′-ATGGCCACGATTATGTCTCC; β-actin, forward 5′-ATCAAGATCATTGCTCCTCCT, reverse 5′-GAGAGCGAGGCCAGGATGGA; DNMT1, forward 5′-GGTTCTTCCTCCTGGAGAATGTC, reverse 5′-GGGCCACGCCGTACTG; DNMT3a, forward 5′-GCCTCAATGTTACCCTGGAA; reverse 5′-CAGCAGATGGTGCAGTAGGA; DNMT3b, forward 5′-CCCATTCGAGTCCTGTCATT, reverse 5′-GGTTCCAACAGCAATGGACT.
Cell growth assays
Cells were seeded into 12-well plates at a density of 5000 cells per well, followed by 5-aza-dC (5 μM) or vehicle treatment. The drug-containing medium was replenished each day. Cell density was determined by replacing the medium with 2 μM calcein AM in 1x dissociation solution (Trevigen, Gaithersburg, MD) at the indicated time points. After 1 hour incubation, diesterase activity (relative fluorescence unit, RFU) was measured with a Tecan Infinite 200 Microplate Reader (Durham NC), using an excitation wavelength of 485 nm and emission wavelength of 520 nm (18).
Bisulfite genomic DNA sequencing
Genomic DNA from 5-aza-dC treated or mock-treated cells was isolated using the PureLink Genomic DNA Purification Kit (Invitrogen). Genomic DNA aliquots were there treated with sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research), followed by PCR to amplify the PDLIM2 promoter using Hot-Start Taq enzyme (Qiagen). Primers designed to recognize the bisulfite-modified regions (−1084 to −800) of the PDLIM2 promoter were: forward 5′-AGAGGAGTTTATATATATTTAGG, reverse 5′-TACCTAACAACCCTCTCTCC. The PCR products were then directly for DNA sequencing or subcloned into the SmaI restriction site of pEGFP-N2 (Clontech) for single colony sequencing to determine the methylation status of the CpG dinucleotides with the PDLIM2 promoter.
Luciferase gene reporter assays
The indicated colon cancer cell lines were transfected with a κb-TATA luciferase reporter plasmid in the presence of increasing amount of Myc-PDLIM2. At 40 hrs post-transfection, luciferase activity was measured as described before (15). The expression of the Myc-PDLIM2, p65 and Hsp90 proteins was detected by direct immunoblotting (IB) assays. To reduce background, we used the cytoplasmic extraction for p65 detection.
Retroviral transduction and generation of stable transfectants
HEK293T cells were transfected with pQCXIP-myc-PDLIM2, followed by viral supernatant collection and infection of the indicated colorectal cell lines as described before (19). Stable transfectants were obtained by selection with puromycin.
Colony formation assays
Cell suspension in culture medium containing 0.6% SeaPlaque low melting agarose was added to plates coated with an initial underlay of 1% agarose in culture medium. Colony growth was scored after 21 days.
In vivo tumorigenicity assays
Five-week old female SCID mice (Charles River) were challenged subcutaneously in the hind back with control or modified colorectal cancer cell lines as indicated. The recipient mice were sacrificed for tumor evaluation 14 days post-injection.
Results and Discussion
PDLIM2 expression is repressed in human colon cancer cells
Given the causative role of NF-κB in colon tumorigenesis and the involvement of PDLIM2 in the termination of NF-κB activation, we hypothesized that PDLIM2 may be involved in the pathogenesis of colon cancer. To test the hypothesis, we examined expression levels of PDLIM2 in different colon cancer cell lines, as PDLIM2 expression has recently been found to be repressed by the human T-cell leukemia virus type I (HTLV-I), the etiological agent of adult T-cell leukemia (ATL) (15). Indeed, PDLIM2 protein levels were much lower in all colon cancer cell lines we examined, compared to those observed in MCF10A, a nontumorigenic epithelial cell line (Fig. 1A). In correlation with protein down-regulation, PDLIM2 mRNA levels were also significantly decreased in these colon cancer cell lines (Fig. 1B). These data indicate that PDLIM2 is repressed at both RNA and protein levels in human colon cancer cells.
Figure 1.

PDLIM2 expression is repressed in colorectal cancer cell lines. A, Protein expression of PDLIM2 in the indicated cell lines was analyzed by immunoblotting (IB) using a PDLIM2 specific antibody. B, The relative levels of PDLIM2 mRNAs in the indicated colon cancer cell lines were analyzed by real-time PCR using β-actin mRNA as a control. The data are represented as the mean percent of expression observed in control MCF10A cells ± standard deviation (n = 3).
PDLIM2 repression in colon cancer cells involves DNA methylation
To investigate the mechanism of PDLIM2 repression in colon cancer cells, we examined the potential role of DNA methylation, one major mechanism responsible for repression of tumor suppressor genes in neoplastic cells (17). To do so, we examined expression levels of DNMT1, DNMT3a and DNMT3b, three DNA methyltransferases responsible for DNA methylation. Compared with their expressions in nontumorigenic epithelial cells, all three enzymes were consistently and significantly upregulated in each colon cancer cell line, although to different extents (Fig. 2A).
Figure 2.

PDLIM2 repression in colon cancer cells involves DNA methylation. A, RNA levels of DNMT1, DNMT3a and DNMT3b in the indicated colon cancer cells were analyzed by real-time PCR using β-actin mRNA level as a control and represented as fold induction in mRNA abundance relative to those in MCF10A cells (set as 1). The data presented are the mean ± standard deviation (n = 3). B, The indicated cell lines were treated with the DNMT inhibitor 5-aza-dC (5μM) for 48 h, followed by real-time PCR to determine relative mRNA levels of PDLIM2. Changes in PDLIM2 mRNA abundance following 5-aza-dC treatment are represented as fold induction relative to those observed in an RNA sample from mock-treated cells. C, The indicated colon cancer cell lines were treated with 5 μM 5-aza-dC or vehicle for the indicated time points, followed by cell growth assay. D, The indicated cell lines were treated with 5 μM 5-aza-dC or vehicle for 5 days, followed by the bisulfite genomic DNA sequencing as described in Material and Methods. Each circle represents a CpG site; open circles represent unmethylated CpG dimucleotides whereas filled circles represent methylated CpG sites. The ratios of the filled area in circles represent percentiles of the methylation in the CpG sites. The position of each CpG nucleotide relative to the PDLIM2 transcription initiation site (+1) is indicated at the top.
To establish a connection between DNA methyltransferase up-regulation and PDLIM2 expression, we examined the effect of 5-aza-dC, a highly specific DNA methyltransferase inhibitor, on PDLIM2 expression in these cancer cells. In agreement with our recent finding that HTLV-I-mediated repression of PDLIM2 is mediated by DNA methylation (17), 5-aza-dC treatment efficiently restored PDLIM2 expression in all seven colon cancer cell lines examined (Fig. 2B). These data suggest that like HTLV-I-mediated repression, PDLIM2 repression in colon cancer cells also involves DNA methylation.
In association with recovery of PDLIM2 expression, 5-aza-dC treatment also led to growth inhibition of different colon cells (Fig. 2C, and Supplemental Fig. 1). On the other hand, normal epithelial cells were largely resistant to the 5-aza-dC treatment (Supplemental Fig. 2). These data were consistent with the fact that 5-aza-dC is toxic to cancer cells but not normal cells both in vitro and in vivo (17, 20). Since 5-aza-dC showed an efficient antitumor activity in phase III clinical trials for patients with MDS/AML (20), these studies not only substantiate the PDLIM2 epigenetic repression but also suggest a potential therapeutic strategy for human colon cancer. Clearly, the effect of 5-aza-dC can not only be attributed to the reactivation of PDLIM2. Other yet-identified targets of this anti-tumor drug may also play very important roles in the colon cancer growth inhibition.
To investigate whether the PDLIM2 promoter is methylated in colon cancer cells and whether 5-aza-dC-mediated recovery of PDLIM2 expression involves reverse of the methylation of the PDLIM2 promoter, we performed the bisulfite genomic DNA sequencing. As shown in Fig. 2D, the PDLIM2 promoter was hypermethylated in colon cancer cell lines, in comparison with normal control cell line. As expected, treatment of 5-aza-dC induced a dramatic decrease in methylation of the PDLIM2 promoter. These data strongly suggest that promoter methylation directly controls the expression of PDLIM2.
PDLIM2 re-expression prevents constitutive activation of NF-κB in colon cancer cells
To establish a mechanistic connection between PDLIM2 epigenetic repression and NF-κB in colon tumorigenesis, we performed luciferase gene reporter assays to examine the effect of PDLIM2 restoration on the constitutive activation of NF-κB in human colon cancer cells. As shown in Figs. 3A–3D, PDLIM2 re-expression resulted in a dose-dependent suppression of NF-κB activation in four independent colorectal cancer cell lines, which was associated with decreased p65 expression. Our mechanistic studies further indicated that PDLIM2 shuttled p65 into the nuclear matrix for the proteasomal degradation in these colorectal cancer cells (Supplemental Fig. 3). These data imply that PDLIM2 repression is one important mechanism contributing to the constitutive activation of NF-κB in colon cancer cells.
Figure 3.
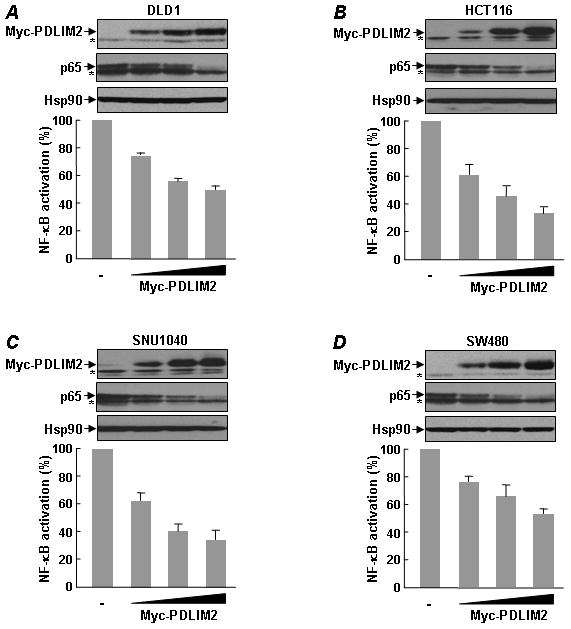
PDLIM2 inhibits NF-κB constitutive activation in colon cancer cells. A–D, The indicated colon cancer cell lines were transfected with κb-TATA driven luciferase reporter in the presence of increasing amounts of Myc-PDLIM2, followed by luciferase assay. The luciferase activities were presented as the percentile of that in cells without Myc-PDLIM2 transfection (denoted as 100). The protein expression levels of Myc-PDLIM2, p65 and Hsp90 were detected by direct immunoblotting assays and the non-specific bands were indicated by the asterisks.
PDLIM2 re-expression suppresses anchorage-independent growth and tumor formation of malignant colon cancer cells
To investigate the significance of PDLIM2 repression in colon tumorigenesis, PDLIM2 was stably expressed in the colorectal cancer cell lines HCT 116 and DLD1 (Supplemental Fig. 4). We then measured soft agar colony forming activity in comparison to the control cells transduced with an empty vector. As expected, the colon cancer cell lines readily formed colonies, indicative of their anchorage-independent growth potential (Figs. 4A & 4B). However, the colon cancer cell lines stably expressing PDLIM2 formed far fewer and much smaller colonies in soft agar. These data suggest that PDLIM2 suppresses the tumorigenicity of colon cancer cells in vitro.
Figure 4.
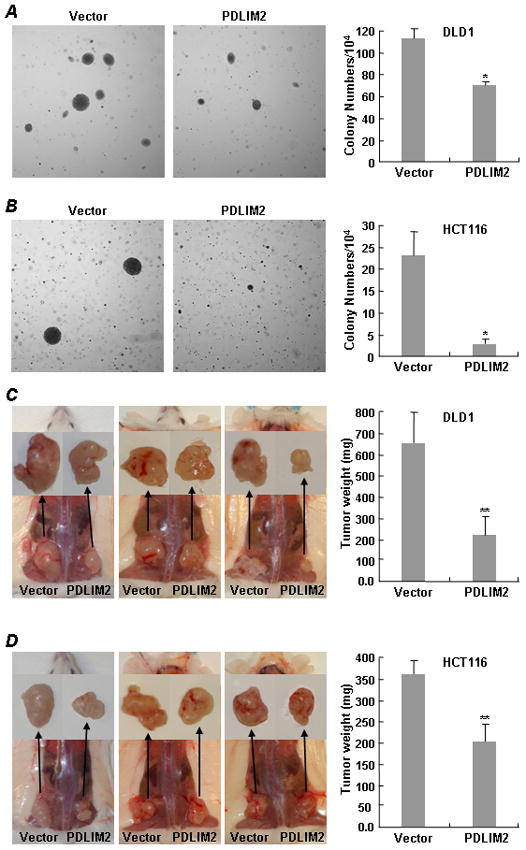
PDLIM2 re-expression suppresses tumorigenicity of colorectal cancer cell lines. A & B, DLD1 and HCT116 colon cancer cells stably expressing Myc-PDLIM2 or an empty vector were plated in soft agar and colony numbers were counted at day 21 after plating. The data presented are the mean ± standard deviation (*p<0.01). C & D, The DLD1 and HCT116 stable cell lines were subcutaneously inoculated into the hind back of the SCID mice. The mice were sacrificed at day 14 after inoculation and tumor weights were measured. The data presented are the mean ± standard deviation (**p<0.05).
To confirm the in vitro studies in vivo, we subcutaneously injected the control and PDLIM2-expressing colon cancer cell lines into opposite flanks of individual SCID mice. As shown in Figs. 4C and 4D, both control and PDLIM2 stable colon cancer cell lines developed tumors at the injection sites in mice. However, the tumors formed by the PDLIM2-expressing cancer cell lines were significantly smaller than those formed by the vector control cell lines. These results indicate that PDLIM2 suppresses the tumorigenicity of colon cancer cells in vivo.
Recent biochemical, pharmacological and genetic studies from both humans and animals strongly support a causative role for NF-κB in intestinal inflammation-associated tumorigenesis (4–10). However, the mechanisms by which NF-κB is constitutively activated during intestinal pathogenesis are largely unclear. Data presented in this study indicate that one important mechanism of NF-κB constitutive activation involves PDLIM2 repression via epigenetic silencing. Importantly, the DNA methyltransferase inhibitor 5-aza-dC not only re-activates PDLIM2 expression in multiple human colorectal cancer cell lines, but also reverts their tumorigenic phenotype. Although this anti-tumor drug also reverses promoter methylation of genes other than PDLIM2, specific re-expression of PDLIM2 alone is able to suppress the tumorigenicity of the malignant colon cancer cells both in vitro and in vivo. These data therefore provide mechanistic insights into colon tumorigenesis and a potential therapeutic strategy for human colon cancer.
Our data also suggest a novel tumor suppression role for PDLIM2. In support of this, our recent studies have shown that PDLIM2 is repressed epigenetically by HTLV-I (17). Similarly, the PDLIM2 repression is one important mechanism of HTLV-I-mediated ATL (15). In addition to colon cancer and ATL, our more recent data suggest that PDLIM2 is repressed in certain other cancer types including breast cancer (21). It is noteworthy that the pathogenesis of these PDLIM2 repression-associated cancers has already been linked to NF-κB, suggesting a common tumor suppressor function of PDLIM2.
Supplementary Material
Acknowledgments
We thank DC Radisky and MJ Bissell for MCF10A cells. This study was supported in part by NIH/NCI grant R01 CA116616, ACS grant RSG-06-066-01-MGO and Hillman Innovative Cancer Research Award to G. Xiao.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Burstein E, Fearon ER. Colitis and cancer: a tale of inflammatory cells and their cytokines. J Clin Invest. 2008;118:464–7. doi: 10.1172/JCI34831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munkholm P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment Pharmacol Ther. 2003;18:1–5. doi: 10.1046/j.1365-2036.18.s2.2.x. [DOI] [PubMed] [Google Scholar]
- 4.Greten FR, Eckmann L, Greten TF, et al. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Rogler G, Brand K, Vogl D, et al. Nuclear factor κB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–69. doi: 10.1016/s0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- 6.Lind DS, Hochwald SN, Malaty J, et al. Nuclear factor-κB is upregulated in colorectal cancer. Surgery. 2001;130:363–9. doi: 10.1067/msy.2001.116672. [DOI] [PubMed] [Google Scholar]
- 7.Yu HG, Yu LL, Yang Y, et al. Increased expression of RelA/nuclear factor-κB protein correlates with colorectal tumorigenesis. Oncology. 2003;65:37–45. doi: 10.1159/000071203. [DOI] [PubMed] [Google Scholar]
- 8.Hardwick JC, van den Brink GR, Offerhaus GJ, van Deventer SJ, Peppelenbosch MP. NF-κB, p38 MAPK and JNK are highly expressed and active in the stroma of human colonic adenomatous polyps. Oncogene. 2001;20:819–27. doi: 10.1038/sj.onc.1204162. [DOI] [PubMed] [Google Scholar]
- 9.Eaden J, Abrams K, Ekbom A, Jackson E, Mayberry J. Colorectal cancer prevention in ulcerative colitis: a case-control study. Aliment Pharmacol Ther. 2000;14:145–53. doi: 10.1046/j.1365-2036.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- 10.Jänne PA, Mayer RJ. Chemoprevention of colorectal cancer. N Engl J Med. 2000;342:1960–8. doi: 10.1056/NEJM200006293422606. [DOI] [PubMed] [Google Scholar]
- 11.Xiao G. Autophagy and NF-κB: fight for fate. Cytokine Growth Factor Rev. 2007;8:233–43. doi: 10.1016/j.cytogfr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao G, Rabson A, Young W, Qing G, Qu Z. Alternative pathways of NF-κB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17:281–93. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. J Exp Med. 2004;200:107–13. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-κB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–91. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 15.Yan P, Fu J, Qu Z, et al. PDLIM2 suppresses HTLV-I Tax-mediated tumorigenesis by targeting Tax into the nuclear matrix for proteasomal degradation. Blood. 2009;113:4370–80. doi: 10.1182/blood-2008-10-185660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qing G, Qu Z, Xiao G. Stabilization of basally translated NF-κB-inducing kinase (NIK) protein functions as a molecular switch of processing of NF-κB2 p100. J Biol Chem. 2005;280:40578–82. doi: 10.1074/jbc.M508776200. [DOI] [PubMed] [Google Scholar]
- 17.Yan P, Qu Z, Ishikawa C, Mori N, Xiao G. Human T-cell leukemia virus type I-mediated repression of PDZ-LIM domain-containing protein 2 involves DNA methylation but independent of the viral oncoprotein Tax. Neoplasia. 2009;11:1036–41. doi: 10.1593/neo.09752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang XM, Terasaki PI, Rankin GW, Jr, et al. A new microcellular cytotoxicity test based on calcein AM release. Hum Immunol. 1993;37:264–70. doi: 10.1016/0198-8859(93)90510-8. [DOI] [PubMed] [Google Scholar]
- 19.Qing G, Qu Z, Xiao G. Proteasome-mediated endoproteolytic processing of the NF-κB2 precursor at the κB promoter. Proc Natl Acad Sci USA. 2007;104:5324–9. doi: 10.1073/pnas.0609914104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Manero G. Demethylating agents in myeloid malignancies. Current Opinion in Oncology. 2008;20:705–10. doi: 10.1097/CCO.0b013e328313699c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qu Z, Fu J, Yan P, Cheng S, Xiao G. Epigenetic repression of PDLIM2: implications for the biology and treatment of breast cancer. J Biol Chem. 2009 doi: 10.1074/jbc.M109.086561. in revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.