

Abstract
In this and a previous issue of Molecular Cell, Oda et al., Abbas et al., and Centore et al. determined that the H4K20 histone methyltransferase PR-Set7/Set8 is post-translationally regulated by the PCNA-dependent CRL4Cdt2 ubiquitin ligase.
Post-translational modification of histones is a critically important mechanism to regulate gene expression and cell cycle progression. The histone methyltransferase PR-Set7/Set8 specifically mono-methylates histone H4 on lysine 20 (H4K20) to maintain silent chromatin and effect chromatin compaction as a pre-requisite for mitotic chromosome separation. PR-Set7 is an essential gene, as deletion results in early embryonic lethality in Drosophila and mice (reviewed in Yang and Mizzen, 2009; Oda et al., 2009). More specifically, PR-Set7 activity regulates cell cycle progression, as knockdown of PR-Set7 in cell lines results in G2 arrest, DNA damage, and chromatin decondensation. Like most cell cycle regulators, PR-Set7 itself is controlled in a cell cycle-dependent manner, with mRNA, protein, and activity levels that are low in G1 and S phase, increase during G2/M, and peak at mitosis. However, the regulatory mechanisms that govern PR-Set7 abundance and activity remained largely elusive. In this and a previous issue of Molecular Cell, Oda et al., Abbas et al., and Centore et al. describe the PCNA-dependent ubiquitination and destruction of PR-Set7 by cullin-RING ligase 4 (CRL4Cdt2) during G1 and/or S phase of the cell cycle and in response to DNA damage (Abbas et al., 2010; Centore et al. 2010; Oda et al., 2010).
The CRL4Cdt2 ubiquitin ligase was previously found to target the DNA replication licensing factor Cdt1 for degradation in S phase and following DNA damage (reviewed in Arias and Walter, 2007). Importantly, only chromatin-bound PCNA recognized the specialized PCNA-interaction protein (PIP) box of Cdt1, and recruited CRL4Cdt2 to ubiquitinate Cdt1 (Havens and Walter, 2009). The presence of the PIP box motif in PR-Set7 led all three groups to identify PR-Set7 as a specific target of the PCNA-dependent CRL4Cdt2 ubiquitin ligase in response to UV-induced DNA damage. Oda et al. further reported that PCNA-bound PR-Set7 recruits 53BP1 to sites of DNA damage, thus initiating the DNA damage response. PCNA subsequently mediates degradation of PR-Set7, which may serve to limit the duration of PR-Set7 residence that would otherwise interfere with the repair of damaged DNA.
The three groups also provide compelling evidence for PCNA-CRL4Cdt2 in the proteolytic removal of PR-Set7 during the cell cycle. However, differences arose regarding the exact timing of PR-Set7 degradation: Oda et al. determined through FACS and immunofluorescent imaging that YFP-tagged PR-Set7 is degraded during early G1 in U2OS cells, and is undetectable throughout S phase, while Abbas et al. and Centore et al. found that PR-Set7 is degraded specifically in S phase using synchronized U2OS cells and Western blotting to detect epitope-tagged PR-Set7. All groups observed that PR-Set7 protein levels recover in late G2, with peak levels observed in mitosis. The findings from Abbas et al. and Centore et al. are consistent with the mode of PCNA-dependent CRLCdt2 action that occurs on chromatin during S phase (Havens and Walter, 2009). Given that PCNA is loaded onto primed DNA by replication factor C from the onset of DNA replication in S phase and is undetectable by immunostaining in G1 (Moldovan et al., 2007; Prasanth et al., 2004), the G1-specific targeting of PR-Set7 observed in Oda et al. suggests a chromatin-independent function of PCNA in coupling PR-Set7 to CRL4Cdt2. However, low levels of PCNA associated with DNA in G1 may be sufficient to target PR-Set7 to CRL4Cdt2 and thus chromatin-dependent degradation prior to S phase cannot be excluded. Moreover, two other ubiquitin ligases, SCFSkp2 and anaphase-promoting complex (APC), were also implicated in PR-Set7 ubiquitination and degradation (reviewed in Yang and Mizzen, 2009). Oda et al. extended their findings to show that chromatin-free PR-Set7 is preferentially degraded by SCFSkp2 in addition to the CRL4Cdt2 pathway for PR-Set7 ubiquitination. However, Abbas et al. found that silencing of the CUL1 subunit of SCFSkp2 does not alter PR-Set7 degradation, arguing against a major role for the SCF ubiquitin ligase in controlling PR-Set7 levels. A role for APC-Cdh1 in PR-Set7 degradation was not determined, but APC presumably functions as a G1-specific E3 ligase for PR-Set7 since it is inactive in S phase.
To delineate the role of PR-Set7 degradation in cell cycle progression, both Abbas et al. and Centore et al. ectopically expressed the PCNA binding-deficient PR-Set7 mutant that is resistant to ubiquitination by CRL4Cdt2. Centore et al. reported G2 arrest, chromatin compaction and H4K20me1 accumulation, while Abbas et al. observed a G2 delay with enlarged nuclei, which is indicative of DNA re-replication, and chromatin relaxation due to the suppression of histone gene expression. The divergent phenotypes can potentially be reconciled by the different methods and durations of degradation-resistant PR-Set7 expression: Centore et al. employed a tetracycline-inducible system and assessed the phenotypes within hours, while Abbas et al. utilized retroviral expression vectors and examined the consequences at much later timepoints. Additionally, the degradation-resistant PR-Set7 mutant acted on non-histone targets (e.g. p53) that contributed to the alteration of the transcriptional profile and may account for the spectrum of cellular changes described by Abbas et al. during the prolonged course of enforced expression.
The three groups clearly delineated the biochemical mechanism underlying the post-translational regulation of PR-Set7 by the PCNA-dependent CRL4Cdt2 ubiquitin ligase. Precise regulation of PR-Set7 activity is critically important to limit the adverse effects of H4K20 monomethylation on cell cycle progression, including premature chromatin condensation and DNA re-replication. The latter outcome was independently described by Abbas et al. and Tardat et al., who recently showed that H4K20 monomethylation by PR-Set7 facilitates loading of the pre-replication complex at the replication origins, and that the degradation-resistant PR-Set7 mutant causes re-replication (Tardet et al., 2010). Future studies should address the interplay between the CRL4Cdt2 and SCFSkp2 (or APCCdh1) ubiquitin ligases that specify PR-Set7 levels at different cell cycle stages, the temporal control of CRL4Cdt2-dependent PR-Set7 turnover, and the developmental or pathological conditions associated with perturbation of the intricate proteolytic control of PR-Set7 abundance.
Figure 1.
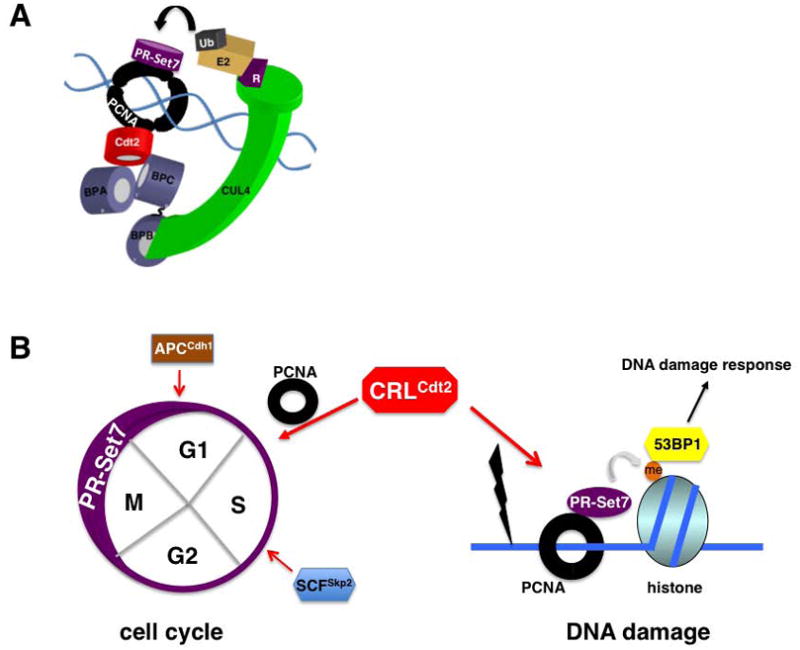
PR-Set7 is targeted for ubiquitination by the PCNA-dependent CRL4Cdt2 ubiquitin ligase.
A. Schematic representation of CRLCdt2-dependent ubiquitination of PR-Set7 orchestrated by chromatin-bound PCNA.
B. Post-translational regulation of PR-Set7 stability during the cell cycle and in response to DNA damage. Multiple ubiquitin ligases have been indicated in regulating PR-Set7 levels to limit H4K20 monomethylation and allow for orderly cell cycle progression. PR-Set7 is degraded by PCNA-dependent CRLCdt2 in response to DNA damage following H4K20 monomethylation and recruitment of 53BP1.
We thank Jianxuan Zhang for assistance with preparation of this figure.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. Mol Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias EE, Walter JC. Genes Dev. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. Mol Cell. 2010;40:22–33. doi: 10.1016/j.molcel.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens CG, Walter JC. Mol Cell. 2009;35:93–104. doi: 10.1016/j.molcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, Pfander B, Jentsch S. Cell. 2007;129:665–79. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM, Torres-Padilla ME, Heard E, Reinberg D. Mol Cell Biol. 2009;29:2278–95. doi: 10.1128/MCB.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. Mol Cell. 2010;40(this issue):pgs. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth SG, Méndez J, Prasanth KV, Stillman B. Philos Trans R Soc Lond B Biol Sci. 2004;359:7–16. doi: 10.1098/rstb.2003.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. Nat Cell Biol. 2010;12 doi: 10.1038/ncb2113. [DOI] [PubMed] [Google Scholar]
- Yang H, Mizzen CA. Biochem Cell Biol. 2009;87:151–61. doi: 10.1139/O08-131. [DOI] [PubMed] [Google Scholar]