

Summary
Imidazole glycerol phosphate synthase (IGPS) is a V-type allosteric enzyme, which is catalytically inactive for glutamine hydrolysis until the allosteric effector, N’-[(5′-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide-ribonucleotide (PRFAR) binds 30 Å away. In the apo state, NMR relaxation dispersion experiments indicate the absence of millisecond (ms) timescale motions. Binding of the PRFAR to form the active ternary complex is endothermic with a large positive entropy change. In addition, there is a protein wide enhancement of conformational motions in the ternary complex, which connect to two active sites. NMR chemical shift changes and acrylamide quenching experiments suggest that little in the way of structural changes accompany these motions. The data indicate that enzyme activation in the ternary complex is primarily due to an enhancement of ms motions that allows formation of a population of enzymatically active conformers.
Introduction
Allostery is a widespread mechanism used by all organisms to regulate enzyme and protein function and adapt to a changing environment(Ackers et al., 1991; Carpenter and Hand, 1986; Liu et al., 1998; Monod et al., 1963; Onuchic et al., 2006), by enabling binding information at one site to be relayed to a distant binding site. The mechanism of information transfer through a densely packed protein is a subject of intense study because it is essential to better understand this phenomenon to advance our knowledge of biological processes as well as aid efforts in drug discovery(Bertrand and Gopalakrishnan, 2007; Raddatz et al., 2007; Ross, 2007; Schweizer et al., 2007; Shi et al., 2006) and protein design(Dueber et al., 2007; Dueber et al., 2003; Ostermeier, 2005). Allostery is classically defined as the regulation of enzyme function by the binding of a molecule at a site other than the active site(Monod et al., 1963). From a thermodynamic perspective, some of the binding energy due to interaction with the allosteric ligand is used to alter either the structure and/or dynamics of the distant active site, thereby resulting in a change in function. Thus the allosteric phenomenon transmits information between binding sites in enzymes, sometimes over significant molecular distances. In many cases binding of an allosteric ligand does not alter the affinity (K-type) of the enzyme for the second ligand but rather alters kcat. Enzymes such as this are referred to as V-type allosteric enzymes. Regardless of whether alterations in affinity or catalysis are the end result, characterization of allostery necessitates focus on the ternary complex and the differences with the singly ligated enzyme forms(Fenton, 2008; Reinhart, 2004), because the full effect of allostery is realized in the ternary complex, whether it be structural or dynamical. Recently experimental and computational studies have revealed the essential role that protein motions can play in conveying an allosteric response(Bruschweiler et al., 2009; Cooper and Dryden, 1984; Das et al., 2008; Fuentes et al., 2004; Jarymowycz and Stone, 2008; Kern and Zuiderweg, 2003; Kimmel and Reinhart, 2000; Koide et al., 1992; Petit et al., 2009; Popovych et al., 2006; Popovych et al., 2009; Yan et al., 2004). These studies have shown that a significant dynamic or entropic component can be involved in the relay of binding information. To date, these studies have largely centered on K-type allosteric proteins; whereas, the role of protein motions in V-type allostery has been largely uncharacterized despite its widespread occurrence in nature.
Here we work to fill this void with a detailed biophysical characterization of the heterodimeric, allosteric enzyme imidazole glycerol phosphate synthase (IGPS). IGPS catalyzes two reactions in two spatially separated active sites located on two non-covalently associated enzymes(Chaudhuri et al., 2001; Douangamath et al., 2002; Omi et al., 2002). The first reaction is the hydrolysis of the substrate glutamine to form glutamate and NH3 by the protein HisH (23kDa)(Klem et al., 2001; Klem and Davisson, 1993). The second reaction involves the coupling of the NH3 produced by HisH with the non-standard nucleotide, N’-[(5′-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide-ribonucleotide (PRFAR) to produce imidazole glycerol phosphate (IGP) and 5-aminoimidazole-4-carboxamide ribotide (AICAR)(Klem et al., 2001; Klem and Davisson, 1993). This cyclization reaction is catalyzed by the protein HisF (28 kDa), which associates with HisH with nanomolar affinity. The product IGP enters the histidine biosynthetic pathway, while AICAR proceeds to the purine biosynthetic pathway. In addition to its biological importance, IGPS exhibits V-type allosterism and provides a model system for addressing the underlying mechanisms that govern this important biological phenomenon. In the absence of PRFAR, the rate of Gln hydrolysis is negligible (~10−3 s−1); however, binding of PRFAR in HisF triggers a 5300-fold increase in the rate of Gln hydrolysis(Myers et al., 2003), nearly 30 Å away. This yields the interesting scenario where PRFAR acts both as an effector for the glutaminase reaction and as a substrate for the cyclization reaction.
The HisH subunit is a type I glutamine amidotransferase(Zalkin and Smith, 1998). As such, the active site contains a conserved catalytic triad formed by C84, H178 and E180. Production of NH3 from Gln begins by thioester bond formation between the Gln substrate and C84 of HisH, with stabilization of the resulting oxyanion tetrahedral intermediate by the backbone HN of V51, followed by hydrolysis to release NH3 and glutamate(Chittur et al., 2001). It is believed that the NH3 travels via a tunnel to the HisF active site(Amaro et al., 2007; Chaudhuri et al., 2001; Douangamath et al., 2002; Krahn et al., 1997; Rudolph and Stubbe, 1995; Thoden et al., 1997). To facilitate the necessary transfer of NH3 to the HisF active site, the sequential HisH and HisF reactions are tightly coupled, though spatially separated. The HisH active site is within 10 Å of the HisF/HisH protein interface. The ammonia molecule enters the interface between HisF and HisH where it encounters a gate formed by four invariant charged side chains in HisF (R5, E46, K99, and E167) (Amaro et al., 2005). NH3 must pass the gate and then travel an additional 20 Å through the hydrophobic tunnel formed by the β-strands of the HisF (β/α)8 barrel leading to the cyclase active site.
The hydrolysis of glutamine to yield ammonia by HisH and its subsequent reaction with PRFAR catalyzed by HisF are highly synchronized, despite the two active sites being separated by 30 Å and residing on different polypeptide chains. This synchrony ensures that Gln is not wasted by needless hydrolysis in the absence of PRFAR and results in a 1:1 stoichiometry for the HisH:HisF reactions. The kinetic mechanism for the overall reaction is random sequential, thus HisH-bound Gln waits in an inactive site until a signal is received that indicates PRFAR is bound to HisF. That signal must travel over 30Å to the HisH active enabling its catalytic activity. Our work indicates that PRFAR binding to IGPS is entropically dominated in the absence and presence of a Gln analog. Furthermore, ms protein motions are an essential feature of the allosteric process in this enzyme and are propagated between PRFAR and Gln binding sites upon ternary complex formation.
Results
Entropically favored ligand binding to IGPS
Substrate binding to IGPS was monitored by isothermal titration calorimetry (ITC) at 303 K. Gln binding to the HisH active site is exothermic (−1.9 ± 0.3 kcal/mol) but also possesses a small favorable entropy value of 2.7 kcal/mol at this temperature (Fig. 1b) (The nomenclature is used to refer to a specific domain of the HisF/HisH heterodimer, HisH in this instance. All experiments in this work are performed on the enzymatically active HisF-HisH 52kDa heterodimer enzyme). In contrast, PRFAR binding to HisF is endothermic (ΔH = 6.3 ± 0.04 kcal/mol) and entropically driven (TΔS = 14.5 kcal/mol). To assess PRFAR binding in the formation of the ternary complex, the Gln analog acivicin was utilized to avoid enzyme turnover during the titration. Acivicin binds at the HisH active site covalently labeling the active site nucleophile C84 and can be considered a suicide inhibitor. In addition, the rate of IGPS inactivation by acivicin is accelerated 50-fold in the presence of PRFAR, supporting its role as a valid Gln mimetic (Fig. 1c)(Chittur et al., 2001). Binding of PRFAR to acivicin-labeled IGPS is also entropically favored with TΔS = 17.8 kcal/mol and ΔH = (10.4 ± 0.06 kcal/mol) (Fig. 1b). The HisH-acivicin complex binds PRFAR (4.5 ± 0.23 μM) with nearly identical affinity and thermodynamic parameters as the free enzyme (1.2 ± 0.09 μM), confirming previous claims that IGPS operates via a random sequential kinetic mechanism and that IGPS is not a K-type allosteric system(Myers et al., 2003). These experiments further demonstrate that formation of the ternary IGPS complex is entropically driven.
Figure 1. Ligand interaction with IGPS.
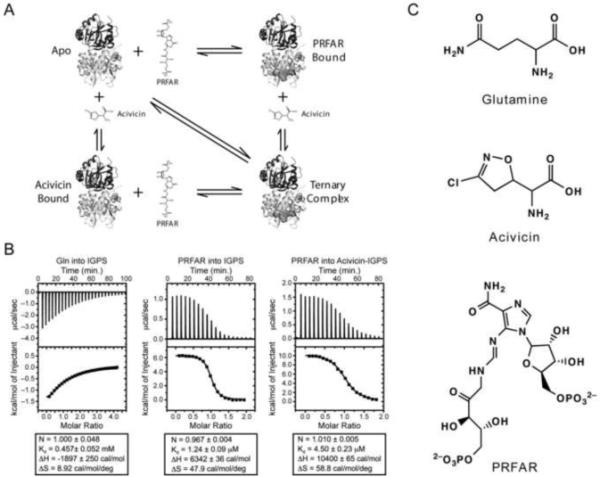
A) Thermodynamic cycle of ligand binding to IGPS. The glutamine analog, acivicin, and PRFAR are shown as sticks when unbound and spheres when bound. B) ITC profiles of ligand binding to IGPS. Thermodynamic parameters are shown below each titration C) Structures of glutamine, acivicin and PRFAR.
Ligand activation of ms motions in HisF
Having characterized the thermodynamics of IGPS ternary complex formation we investigated the motions in IGPS by solution state NMR relaxation measurements. To simplify spectral complexity HisF and HisH were expressed individually and combined during purification to allow selective isotopic enrichment of each protein in the heterodimeric complex. 91% of the backbone amide(Lipchock and Loria, 2009), Ala Cβ, Ileδ, Leuδ and Valγ (ILV) positions have been assigned in the apo, acivicin, PRFAR, and ternary (PRFAR/acivicin bound) complexes of the HisF enzyme in IGPS. Molecular motions in IGPS were characterized by NMR single quantum (SQ) and multiple quantum (MQ) Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion experiments. The expressions for SQ and MQ relaxation dispersion and the details of the fitting procedure are provided in Supplemental Information.
In the apo form, evidence for conformational exchange motion in HisF-IGPS is absent for all backbone amide and Ala Cβ positions using SQ and MQ dispersion experiments, respectively (Supplemental Figure 1). MQ relaxation dispersion experiments that probe the methyl groups of the ILV resonances show upward curving relaxation dispersion profiles for 17 out of 116 resonances. These resonances have dispersion amplitudes ranging from 2-4 s−1. Four representative curves are shown in Figure 2a. All 17 of these resonances (I6δ1, L10δ1, V12γ1, V17γ1, I44δ1, L50δ2, L65δ1, L65δ2, I73δ1, V79γ1, V79γ2, V100γ1, L153δ2, L169δ1 L169δ2, L222δ1, L232δ1) are shown mapped onto the protein structure in Figure 2b. These residues roughly divide the protein in half vertically, with nearly all the residues located in the half with HisF loop 1. Additionally, a nearly unbroken network connects loop 1 with the HisF/HisH interface via residues in β-sheet 1 (I6, L10, V12) and I44. The best model to fit these data is one in which each residue moves independently as opposed to a global model (p-value = 0.04; Supplemental Table 1). The exchange rate constants, kex for this conformational exchange process experienced by these residues ranges from 110 – 440 s−1. The curves obtained from the individual fits are shown plotted with the relaxation data in Figure 2a.
Figure 2. NMR characterization of μs-ms motions in apo ILV 13CH3 methyl labeled HisF.

A) Four representative 13C1H MQ dispersion curves out of the 17 with positive relaxation dispersion amplitudes. B) Structural mapping of ILV dispersion onto IGPS. Residues exhibiting dispersion are shown in light orange spheres. Individual atoms with dispersion are highlighted with bright orange spheres. See also Supplemental Table 1 and Supplemental Figure 1.
Binding of the Gln-analog acivicin to the HisH active site causes minimal perturbation in dispersion amplitudes and in the 13C1H chemical shifts (Supplemental Figure 2) of ILV methyl labeled HisF. Similar to the apo form, 17 resonances out of 116 show dispersion, of which nearly all are equivalent to the apo form. Four representative curves are shown in Figure 3a. As before, these residues generally split HisF vertically, with the loop 1 region showing evidence for millisecond protein motions (Figure 3b). Additional resonances relative to the apo form primarily serve to lengthen the network connecting loop 1 and the interface with the addition of residues V18, L50 and L241. The individual fits for these residues (p-value = 2.3×10−5; Supplemental Table 2) yields kex values ranging from 100 – 490 s−1. These values are very similar to those measured for the apo form, suggesting that motions are relatively unperturbed in HisF upon modification with acivicin in HisH. Furthermore, modification with acivicin caused chemical shift perturbations on the order of parts per billion (Δδ < 0.050 ppm) for HisF, suggesting that relatively few structural changes occur in HisF when ligand is bound in HisH (Supplemental Figure 2).
Figure 3. NMR characterization of μs-ms motions in acivicin bound ILV 13CH3 methyl labeled HisF.

A) Four representative 13C1H MQ dispersion curves out of the 17 with positive relaxation dispersion amplitudes. B) Structural mapping of ILV dispersion onto IGPS. Acivicin is shown bound to HisH in green sticks. Residues exhibiting dispersion are shown in light green spheres. Individual atoms with dispersion are highlighted with bright green spheres. See also Supplemental Table 2.
Unlike the binding of acivicin, addition of PRFAR to the apo form causes significant changes in HisF. The most striking of which is that 63 amide resonances become broadened beyond detection, indicative of intermediate exchange (Supplemental Figure 3). Broadening forms an unbroken network from the PRFAR binding site to the HisF/HisH protein interface and divides the protein roughly in half. It is of note that the broadened half contains the region that exhibits ILV methyl MQ dispersion for the apo and acivicin bound states. Additionally, composite chemical shift changes as large as 0.34 ppm were measured for the amide protons. Many of these changes are localized to the PRFAR binding site and reflect perturbations due to ligand contacts; however, several (R5, I42, G121, E159, T195, F210, R230, I232, D233 and R249) span the region to the protein interface at distances greater than 10 Å from the ligand binding site. To investigate changes in dynamics, 15N SQ and 13C1H MQ relaxation dispersion experiments were completed on PRFAR bound 15N HisF and 13CH3 ILV HisF. Sixty-eight residues were identified to undergo conformational exchange motions in the binary PRFAR form. Four representative curves are shown in Figure 4a. Dispersion amplitudes for the PRFAR bound form are as large as 20 s−1 and are significantly greater than those in the apo or acivicin bound forms. Furthermore, motion is not limited to one half of the protein; although, residues that exhibit dispersion are almost universally located in the central β-sheets, which make up the NH3 tunnel, or point inward from the α-helices (Fig. 4b). Residues exhibiting dispersion and exchange broadening connect the PRFAR binding site and the protein interface, providing clear pathways for the relay of allosteric signals. The kex values from the fit (p-value = 8.9 × 10−15; Supplemental Table 3) range between 100 – 3600 s−1. These values are significantly different from the apo and acivicin bound enzyme.
Figure 4. NMR characterization of μs-ms motions in PRFAR bound 15N and ILV 13CH3 methyl labeled HisF.

A) Four representative 13C1H MQ dispersion curves out of the 68 (includes 15N SQ and 13C1H MQ) with positive relaxation dispersion amplitudes. B) Structural mapping of ILV dispersion onto IGPS. PRFAR is shown bound to HisF in blue sticks. Residues exhibiting dispersion are shown in light blue spheres. Individual atoms with dispersion are highlighted with bright blue spheres. Residues undergoing exchange broadening are shown in black spheres. See also Supplemental Table 3.
Formation of the ternary complex results in a reduction in the number of mobile residues relative to the PRFAR bound enzyme, but the ternary complex still exhibits more flexible residues, on the millisecond timescale, than the apo and acivicin bound states. 13C1H MQ relaxation dispersion experiments were performed for PRFAR bound ILV residues in HisF that had been modified at the HisH active site with acivicin. Thirty-eight residues were identified to have positive dispersion amplitudes. Four representative curves are shown in Figure 5a. As before, these residues form an unbroken network connecting the PRFAR binding site and the HisF/HisH protein interface (Figure 5b). Notably, global data analysis (p-value = 0.24; Supplemental Table 4) yielded kex = 225 ± 30 s−1. The apparent suitability of a single kex to describe all the relaxation dispersion data suggests concerted motion in the ternary complex. Of these 38 resonances, 22 also exhibit dispersion in the PRFAR bound form and 6 have dispersion in all four states. Figure 6 contains overlays of four dispersion curves to highlight the differences between ligand bound states. Leucine 153 is an example of a residue that exhibits dispersion in all four ligand bound forms each with different exchange parameters. Valine 126 has equal dispersion for the PRFAR bound and ternary forms, while V56 has greater dispersion amplitude for the PRFAR bound form relative to the ternary complex. L94 exhibits dispersion in the binary PRFAR form only. This subset of residues demonstrates how finely tuned millisecond motions in IGPS are and how sensitive these residues are to ligand occupancy and ligand structure. A Venn-like diagram visually catalogs the differences and similarities in flexible residues for each IGPS-ligand complex (Fig. 7a). Residues with dispersion in the PRFAR bound and/or ternary states are mapped onto the structure in Figure 7b. The central β-sheet core predominately exhibits dispersion in both states; however, residues with dispersion only in the ternary state are more localized to loop 1 and the PRFAR binding site, while residues showing dispersion in only the PRFAR bound form cluster in the half of the protein opposite loop 1.
Figure 5. NMR characterization of μs-ms motions in ternary ILV 13CH3 methyl labeled HisF.

A) Four representative 13C1H MQ dispersion curves out of the 38 with positive relaxation dispersion amplitudes. B) Structural mapping of ILV dispersion onto IGPS. PRFAR is shown bound to HisF in blue sticks, while acivicin is shown bound to HisH in green sticks. Residues exhibiting dispersion are shown in pink spheres. Individual atoms with dispersion are highlighted with red spheres. See also Supplemental Table 4.
Figure 6. Comparison of dispersion profiles for the apo, acivicin bound, PRFAR bound and ternary states of ILV 13CH3 methyl labeled HisF.

Dispersion overlays highlighting differences in dispersion amplitudes for the four states. Apo data are shown as squares, acivicin bound data are as triangles, PRFAR bound (circles) and ternary complex data as diamonds.
Figure 7. Summary of ms motions.

A) Venn-type diagram illustrating the relation between flexible residues from ILV relaxation dispersion experiments and the enzyme complex in which they occur for apo (black), acivicin (green), PRFAR (blue), and ternary (red). Residues common to all four are shown in a larger font size. B) Residues exhibiting ILV methyl 13C1H MQ dispersion in the PRFAR bound and ternary states are shown mapped onto IGPS. Residues with dispersion in only the PRFAR bound state are shown in blue spheres, while those with dispersion in only the ternary complex are shown in red. Residues with dispersion in both states are shown in magenta.
Picosecond-nanosecond motions are not ligand dependent
To address whether changes in fast time scale (ps – ns) motions accompany the observed activation of ms motions, 1H-1H dipolar cross-correlated relaxation rate constants, η (Tugarinov et al., 2007) were measured for isolated ILV methyl groups in otherwise perdeuterated IGPS in the apo, acivicin, PRFAR, and ternary enzymes (Supplemental Figure 4). The average η values for each enzyme form are equal within the standard deviations (59.1 ± 22.7 s−1, 58.7 ± 20.5 s−1, 61.5 ± 21.4 s−1, and 53.3 ± 18.4 s−1, respectively), indicating that overall fast timescale motions change little in HisF upon formation of the binary and ternary complexes. Analysis of individual residues confirms this trend; however, in a few cases the ternary complex value is altered relative to the apo, acivicin bound and PRFAR bound forms and some of the sites of altered fast motions are also those that experience changes in ms motions (Supplemental Figure 4B). The dominant effect of PRFAR binding to form the binary or ternary complex is changes in ms motions and allostery in IGPS does not appear to depend heavily on motions in the faster time regime, whether this extends to other amino acid side chains or the protein backbone is not currently known.
Synergistic effects detected from chemical shift changes
Non-additive effects in IGPS due to ligand binding can be detected by comparing ligand-induced chemical shift changes (relative to the apo form) for binary and ternary complexes as:
| (1) |
in which ΔδX are the difference in chemical shift between apo IGPS and X-bound IGPS. Sites at which ΔΔδ ≠ 0 are those that experience the synergistic effects of ternary complex formation. In IGPS there are 10 such residues with ΔΔδ values > 1.5 σ from the mean value (Fig. 8) These residues (V12, V48, L50, V126, L153, I168, L222, V226, L234, and L237) span the distance between the glutaminase and cyclase active sites and suggest a pathway by which the HisH active site is coupled to the HisF PRFAR binding site. All but four (I168, L237, V226, and V234) of the residues that exhibit synergistic chemical shift changes also show positive relaxation dispersion amplitudes. The correlation between synergistic chemical shifts and sites of ms motions in the ternary complex further suggests the importance of these residues in allosteric coupling of the two active sites. The four noted above that show no dispersion are located near sites of ms motions and thus synergistic chemical shift analysis may prove useful for additional detection of important residues in allosteric systems.
Figure 8. Synergistic chemical shift changes due to ligand binding.

The 13C ΔΔδ chemical shift differences between the two singly ligated forms of IGPS and the ternary complex is shown versus residue for the Ile, Leu, and Val positions as calculated with equation (1). Residues with a non-additive ΔΔδ value ≥ 1.5 σ from the mean as indicated by the horizontal black lines are mapped onto the IGPS structure in magenta spheres. Acivicin and PRFAR are shown in green and blue sticks, respectively. Not shown are synergistic 1H chemical shift changes, which show similar behavior. See also Supplemental Figures 2 and 3.
Transmission of the PRFAR signal to the oxyanion hole in HisH
Hydrolysis of Gln at the HisH active site requires the presence of PRFAR bound nearly 30 Å away. In addition, stabilization of the tetrahedral Gln-HisH reaction intermediate requires formation of an oxyanion hole much like in serine proteases and in other Gln amidohydrolases such as TrpG/E (Spraggon et al., 2001) and carbamoyl phosphate synthase (Thoden et al., 1998). However in apo IGPS, the HisH residue that would perform this function, V51, is not in optimal position for providing stabilization to the negatively charged tetrahedral intermediate. Rather, the carbonyl oxygen of G50 is oriented toward the site occupied by the negatively charged oxygen atom of the tetrahedral intermediate, resulting in a conformation that would disfavor formation of the oxyanion intermediate (Fig. 9a). Stabilization of this inactive conformation is facilitated by a hydrogen bond between the HN of V51 and C’ oxygen of P10. A 1H,15N NMR spectrum of apo HisH indicates a sharp resonance for G50, suggesting the absence of ms time scale motions. Titration with PRFAR, however, reveals that G50 becomes broadened beyond detection, indicative of conformational exchange motions on the μs-ms timescale (Fig. 9b). Therefore, PRFAR not only induces large-scale millisecond motions in HisF, but also spans across the HisF/HisH protein interface into the active site of HisH. We propose that these motions allow the peptide bond of G50 to rotate and present the stabilizing amide proton of V51, generating a HisH active site primed for catalysis.
Figure 9. Motions at the HisH active site.

HisH is shown in blue cartoon ribbon. The loop comprising the oxyanion hole is highlighted in cyan. The catalytic Cys84 is shown in sticks with the nucleophilic sulfur atom in yellow. The carbonyl oxygen of Gly50 is shown as a red sphere, while the amide proton of Val51 is represented as a gray sphere to emphasize the geometry of the inactive glutamine amidotransferase. Activation of HisH is predicted to require flipping of the amide bond to present the amide proton of Val51 to allow for stabilization of the oxyanion intermediate. B) Resonance broadening of Gly50 in 15N HisH-IGPS upon titration with PRFAR. All spectra were collected under identical conditions with an equal number of scans and are shown contoured equally. Gly50 goes from an intense, well-resolved resonance in the apo form (red) to broadened beyond detection at 0.87 mM PRFAR (purple) (99.8% saturated), with intermediate titration points of 0.038 mM (orange), 0.150 mM (yellow), 0.220 mM (green), and 0.290 mM (blue).
The HisF/HisH protein interface is unaltered by ligand binding
Comparison of the apo crystal structures of IGPS from T. maritima and S. cerevisiae suggested a less solvent accessible HisH/HisF interface with a 15° reduction in the angle between HisF and HisH in S. cerevisiae relative to that in T. maritima(Chaudhuri et al., 2001). This observation led to a model in which PRFAR binding triggers a hinging mechanism at the interface, allowing inter-protein contacts necessary for the stimulation of glutamine hydrolysis. Previous studies utilized tryptophan fluorescence to study the HisF/HisH complex formation, as the lone tryptophan (W123) in HisH exhibits a shift in fluorescence maximum in the presence of HisF suggesting this tryptophan is sensitive to the environment at the protein interface and furthermore that the single buried tryptophan in HisF is not (Beismann-Driemeyer and Sterner, 2001). We monitored acrylamide quenching of tryptophan fluorescence to study changes in the protein interface upon ligand binding. Stern-Volmer plots for each ligand bound state are shown in Figure 10. The Stern-Volmer quenching constants for each are identical (Apo: 1.04 ± 0.01 M−1, Acivicin: 1.03 ± 0.01 M−1, PRFAR: 0.98 ± 0.02 M−1, Ternary: 0.96 ± 0.01 M−1), suggesting little change in the acrylamide accessibility at the interface due to ligand binding.
Figure 10. Acrylamide accessibility assay.
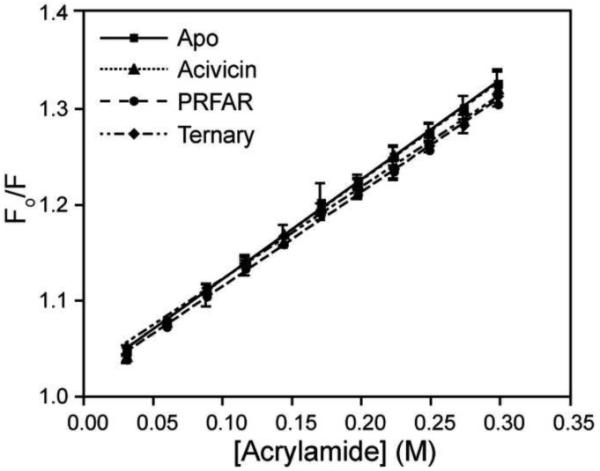
Comparison of Stern-Volmer tryptophan fluorescence quenching with acrylamide for apo (squares), acivicin bound (triangles), PRFAR bound (circles) and ternary (diamonds) IGPS. For each state, 10 μM IGPS was utilized in 50 mM HEPES pH 8.0 at room temperature.
Discussion
The ability to propagate binding information over significant molecular distances to regulate function at a distant site is an impressive biological feat. Allostery in IGPS conveys PRFAR binding information over 30 Å through the HisF protein matrix, across a non-covalent protein interface, and finally to the HisH active site to activate catalysis. In contrast to what is often observed, ligand binding in IGPS increases molecular motions rather than promotes protein rigidification. Furthermore, in IGPS this information transfer appears to utilize little in the way of structural changes. Binding of acivicin results in maximal composite 1H13C chemical shift changes that are < 0.05ppm (Supplemental Fig. 2). Likewise, 1H13C chemical shift changes upon PRFAR binding are also < 0.05 ppm with few exceptions. Composite amide chemical shift changes are similarly of small magnitude upon interaction with PRFAR, with residues not located at the PRFAR binding site, experiencing shifts < 0.1 ppm (Supplemental Fig. 3). Comparison of 1H13C chemical shift changes between the apo and ternary complexes again shows that very small changes (< 0.07 ppm) result upon ligand binding (Supplemental Fig. 2). Combined with the identical Stern-Volmer acrylamide quenching for the apo, both binary, and ternary complexes, these data argue for little change in conformation due to ligand binding (Fig. 10). Residues in loop 1, which have been suggested to change conformation, do not experience chemical shift changes upon ligand binding; however, many become exchange broadened beyond detection (Supplemental Fig. 3) indicating a change in the timescale of molecular motion. The MQ dispersion experiments reveal ms motions in loop 1 for the acivicin (V18Cγ2), PRFAR bound (V18 Cγ1) and ternary (V17 Cγ2 and V18 Cγ1) states (Fig. 7a) despite very little change in chemical shift. Interestingly, mutation of the adjacent residue, K19, has been shown to disrupt the stoichiometry of Gln hydrolysis to IGP and AICAR production(Myers et al., 2003), suggesting that ms motion in the hinge position of loop 1 is important for IGPS function. The endothermic ligand binding determined from the ITC experiments also argues against any large ligand-induced structural changes. However, it is possible that a component of the +ΔH from ligand binding results in weakening the salt bridges that form the gate at the entrance of the NH3 tunnel.
The products of the reaction, IGP and AICAR can activate glutamine hydrolysis, albeit at levels 50 and 170-fold less than PRFAR(Myers et al., 2003). This suggests that the full chemical functionality of PRFAR is essential for complete activation of catalysis but that the imidazole portion may contribute more toward allosteric activation. This portion of PRFAR is closest to β-sheet 1, which has been identified as containing multiple flexible residues and forming part of the HisH/HisF interface. The extended chain of PRFAR is also near several of the residues with non-additive chemical shift changes suggesting importance in the coupling of HisF and HisH active sites. It is noteworthy that the residues that are flexible only in the ternary complex (Fig. 7b red oval) are closest to the IGP portion of PRFAR.
Further insight into the nature of allostery in IGPS can be obtained from analysis of the synergistic shift changes upon ternary complex formation. Despite the small magnitude of the shift changes, 10 residues experience clear non-additivity in their chemical shifts upon forming the ternary complex (Fig. 8). Of the ten residues that show synergistic chemical shifts changes all are highly conserved. V12 is located near D11, which is important for PRFAR binding (Chaudhuri et al., 2003). V48, L50, V126, I168, and L222 form part of the NH3 tunnel; I168, and V48 are adjacent to NH3 gate residues, K99 and E46, respectively. V126 is also near the functionally important residue Q123, which is involved in maintaining the 1:1 reaction stoichiometry (Myers et al., 2003). V234 and L237 are located on α-8, which packs against the C-terminal region, which comprises part of the HisF/HisH interface. L153 and V226 pack against one another and against I168, which as noted is part of the NH3 tunnel. Thus, analysis of the non-additive chemical shifts has identified conserved residues located at or near functionally essential sites. By definition, this analysis identifies sites unique in the ternary complex. As emphasized in earlier work, focus on the ternary complex is essential for a full understanding of allostery (Fenton, 2008; Reinhart, 2004).
Characterization of motions by MQ relaxation dispersion experiments indicates significant enhancement in the number of residues undergoing a millisecond motion conformational exchange process. In addition, the motional parameters in the ternary complex are distinct from those in the apo, acivicin, or PRFAR bound forms. The timescale of these molecular motions are ~45-fold faster than kcat and therefore these motions would not present a bottleneck to overall catalytic turnover of IGPS. Of the 34 resonances showing relaxation dispersion in the ternary complex, there are eight whose ms motions occur only in the ternary complex: V12, V17, V48, L63, I116, V140, V190 and L196. Only L63, I116 and V140 are not highly or completely conserved residues based on a comparison within the IGPS family of enzymes in bacteria, archaea, and lower eukaryotes. V12 and V48 are also identified in the synergistic chemical shift analysis. V17 is in loop 1. L50, which is near V48 forms part of the NH3 tunnel. It is not clear what role V190 plays in IGPS function, though it is near (7Å) the bound PRFAR. As shown in Figure 7b, residues showing dispersion in the ternary complex tend to ‘fill the gaps’ between PRFAR induced flexible sites thereby completing the pathway of ms motions from the PRFAR active site to the HisH active site. The completion of the network of connecting residues in the ternary complex may also be responsible for the apparent switch of the observed ms motions to a concerted process in the ternary complex.
When PRFAR binds to HisF the induced ms motions are relayed to the HisH active site, evidenced by the exchange broadening of HisH active site residue G50 (Fig 9). We suggest that in the absence of PRFAR the HisH active site is rigid with an improperly formed oxyanion hole. Millisecond motions of G50 that are induced by PRFAR create a population of active HisH molecules that are now able to catalyze the hydrolysis of glutamine. This scenario was suggested for yeast IGPS based on x-ray crystal structures(Chaudhuri et al., 2003). In that case B-factors and electron density in the apo enzyme suggested conformational variability at the Gly-Val oxyanion hole region. In the apo T. maritima structure(Douangamath et al., 2002) the B-factors for G50 in HisH are not anomalously high, consistent with our NMR data on the apo enzyme. Many of the flexible residues in the ternary complex are located on α-helix-2 (V56 and L65) and the subsequent loop 4 including I73 and I75. This helix-loop motif spans the PRFAR site to HisH with D74 of the loop making van der Waals contact with E180 of the catalytic triad in HisH. It is therefore proposed that motions along this helix can be propagated to the loop and ultimately provide the motion necessary to free the oxyanion hole from its inactive state. It makes physiological sense to maintain HisH in an inactive state in the absence of PRFAR to prevent wasteful hydrolysis glutamine in the absence of the second substrate for this reaction. Thus, in the absence of PRFAR the enzyme does not populate a small percentage of active HisH enzyme suggesting it does not follow a ‘pre-existing equilibrium’ allosteric model. This may not be true for all V-type allosteric enzymes but likely holds for systems that need to remain inactive until all substrates are present.
The observation that IGPS may operate by a mechanism with structural changes too small for easy detection by NMR should not be surprising(Yu and Koshland, 2001). In general a smaller magnitude change should require less energy to achieve the active conformation than a change of larger amplitude. A significant number of conserved residues in IGPS that experience ms motions in the ternary complex are also functionally important despite not being at the HisH or HisF active site, suggesting that the requirement for motional propagation in the absence of significant conformational changes is under selective pressure. The apparent switch from residue independent motion to a global motional process in the ternary complex highlights the importance of concerted motions in the allosteric process.
Experimental Procedures
Cloning, Expression and Purification
E. coli codon optimized T. maritima genes for HisH and HisF were purchased from Genscript and cloned into pET43.1b using the following NdeI and HindIII and NdeI and XhoI restriction sites for HisF and HisH, respectively. The clone for HisH included a C-terminal histidine tag for Ni/NTA affinity column purification. 15N labeled samples were prepared by growing E. coli BL21(DE3) cells in M9 minimal media with 15NH4Cl as the nitrogen source. Expression was induced with 1.0 mM IPTG at an OD600 = 0.9-1.0 and cells were harvested after 8 hours. δ-13CH3 isoleucine/leucine, and γ-13CH3 valine labeling was achieved by adding 60 mg 2-ketobutyric acid-4-13C (Isotec) and 100 mg 2-keto-3-(methyl-D3)-butyric acid-4-13C (Isotec) per liter of M9 media when OD600 = 0.9-1.0 (Goto et al., 1999). Protons bound to carbon-3 were exchanged for deuterons prior to expression by dissolving 2-ketobutyric acid in 30 mL D2O and 2-keto-3-methyl-butyric acid in 20 mL D2O, followed by overnight incubation with 1 KOH pellet at 37°. Deuterium exchange was monitored by 1H NMR followed by adjustment to 7.0. After addition of the keto-acids to the growth media, cells were grown for one hour, induced with IPTG, and harvested 6 hours later. For resonance assignment of the Ile, Leu and Val methyls, an identical growth as above except keto-acids with 13C labeling in positions 1-4 were utilized and 14NH4Cl (1 g/L) and U-13C-glucose (2 g/L).
For HisF(HisH) purification, cells were harvested by centrifugation and resuspended in 5 mL 100 mM potassium phosphate, 100 mM Tris-HCL pH 9.0 and 1.0 M NaCl per gram of cells and co-lysed by sonication with 1.25-1.50-fold volume excess deuterated HisH(HisF). Cell debris was removed by centrifugation and the supernatant was incubated with Ni/NTA resin. The Ni/NTA column was washed with 100 mM potassium phosphate pH 7.5, 1.0 M NaCl, and 10 mM imidazole. IGPS was eluted in 10 mM CAPS, 10 mM Tris-Base pH 9.5 and 0.25 M imidazole and twice dialyzed against 4.0 L of 10 mM HEPES pH 7.3, 10 mM KCl, 1.0 mM EDTA and concentrated to 550 μl.
NMR Assignment
All NMR data were collected at 30°C on Varian 600 and 800 MHz spectrometers. Spectra were processed using NMRPipe(Delaglio et al., 1995) and analyzed with Sparky(Kneller and Kuntz, 1993). Backbone resonances for perdeuterated 15N HisF bound to unlabeled HisH were assigned by comparison with assigned HisF spectra. Ambiguities were resolved by collecting TROSY-based HN(CA)CB and HNCO experiments(Pervushin et al., 1997) on a HisF sample complexed with unlabeled HisH. Isoleucine, leucine and valine methyls were assigned by HmCmCγCβCα, HmCm(CγCβCα)CO and Cm(CγCβCα)NH experiments(Tugarinov and Kay, 2003).
Synthesis of PRFAR
PRFAR was synthesized enzymatically as previously published using HisA, HisIE, HisG and PRPP synthetase(Davisson et al., 1994; Klem and Davisson, 1993; Myers et al., 2003). Plasmids for these enzymes were obtained from Rebecca Linger (University of Charleston).
NMR Titration of PRFAR
Freshly synthesized PRFAR was dissolved into NMR buffer (10 mM HEPES pH 7.3, 10 mM KCl, 0.5 mM EDTA, 1.0 mM DTT) from a frozen stock and adjusted to pH 7.3. For apo 13CH3 ILV-labeled enzyme, spectra were collected at 0.19, 0.37, 0.56, 0.74, 0.92, 1.81, 2.66 and 3.47 mM PRFAR ([IGPS] = 0.89 mM). For acivicin bound 13CH3 ILV-labeled HisF, spectra were collected at 0.0 and 4.6 mM PRFAR ([IGPS] = 0.72 mM). Saturation was noted by monitoring the shift of NMR resonances upon increasing [PRFAR] and from known ITC-determined Kd values. The pH was adjusted after each addition with 1 M HCl or 1 M KOH as necessary.
Modification of HisH with Acivicin, a glutamine analog
IGPS (HisH-C84) was covalently modified with acivicin by dissolving 1.5 molar equivalents of acivicin into IGPS. The reaction was continued at room temperature to completion as determined by 5,5′-dithiobis(2-nitrobenzoate) (DTNB) analysis.
Isothermal Titration Calorimetry (ITC)
ITC measurements were completed on IGPS using a MicroCalorimeter iTC200 system. IGPS was dialyzed twice in 4.0 L of 50 mM HEPES pH 7.6 with 10 mM KCl. Buffer from the second dialysis step was used for protein dilutions and preparation of ligand solutions. The IGPS concentration was measured in triplicate by UV absorbance (ε280 = 29005 cm−1M−1). For glutamine binding, 20 mM glutamine was titrated into 1.0 mM IGPS. For PRFAR binding, 1.14 mM PRFAR (ε300 = 6069 cm−1M−1) was titrated into 0.15 mM IGPS and 0.15 mM IGPS that was stoichiometrically (1:1) labeled with acivicin. For PRFAR titrations, 19 × 2.0 μL injections were completed into 200 μL of IGPS after an initial 0.2 μL injection. The injection spacing was 250 seconds with a stirring speed was 1000 RPM. For glutamine titrations, 26 × 1.5 μL injections were completed into 200 μL of IGPS after an initial 0.2 μL injection. The injection spacing was 225 seconds and the stirring speed was 1000 RPM.
13CH3 TROSY Multiple Quantum (MQ) CPMG Dynamics
13CH3 MQ CPMG dispersion experiments were completed using published sequences(Korzhnev et al., 2004). Interleaved two-dimensional spectra were collected with CPMG delays, τcp, of 0.0 (2x), 0.4167, 0.50, 0.625, 0.7682 (2x), 1.0, 1.4286, 2.0, 2.5 (2x), 3.333, 5.0 and 10.0 ms during a 40 ms constant relaxation period(Mulder et al., 2001). For all 13CH3 ILV HisF-IGPS experiments, the 1H spectral width was set to 7000 Hz centered at 4.70 ppm. The 13C carrier was at 19.5 ppm with a spectral width of 3200 Hz. The recycle delay was set to 2.0 seconds. Apo, PRFAR-bound and ternary 13CH3 ILV HisF-IGPS spectra were collected with 120 t1 increments. The acivicin-bound enzyme was collected with 135 t1 increments.
13CH3 Single Quantum (SQ) CPMG Dispersion experiments
13CH3 SQ CPMG dispersion experiments was completed using published pulse sequences(Skrynnikov et al., 2001). Interleaved two-dimensional spectra were acquired with CPMG delays, τcp, of 0.0 (2x), 0.4167, 0.50, 0.625, 0.8333 (2x), 1.0714, 1.5, 1.875 (2x), 2.5, 3.75 and 7.5 ms during a 30 ms constant relaxation period(Mulder et al., 2001). The 1H spectral width was set to 7000 Hz and the 1H carrier frequency at 0.75 ppm. The 13C carrier was set to 19.5 ppm with a spectral width of 3200 Hz. The recycle delay was set to 2.0 seconds and 120 increments in t1 were used.
Fluorescence
Tryptophan fluorescence emission scans were monitored from 300-400 nm following excitation at 295 nm with a 1 nm step-size and 1 second averaging time. 1.0 mL of 10 μM IGPS in 50 mM HEPES, pH 8.0, was utilized in each experiment. Data were collected at room temperature (~20°C). PRFAR binary and ternary states utilized 50 μM PRFAR. IGPS was modified with acivicin and confirmed complete by DTNB analysis. Fluorescence quenching was achieved with 10 μL additions of 3.0 M acrylamide in 50 mM HEPES, pH 8.0, for a final acrylamide concentration of 0.30 M.
Highlights.
apo IGPS displays few millisecond motions
allosteric ligand binding is entropically dominated
protein-wide millisecond motions are enhanced upon allosteric ligand binding
the flexible residues form a continuous network between active sites
Supplementary Material
Acknowledgements
This work was supported by NIH grant (R01-GM070823). JML acknowledges a NIH biophysical training grant for support (5T32GM008283). The authors declare no conflict of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackers GK, Doyle ML, Myers D, Daugherty MA. Molecular code for cooperativity in hemoglobin. Science. 1991;255:54–63. doi: 10.1126/science.1553532. [DOI] [PubMed] [Google Scholar]
- Amaro RE, Myers RS, Davisson VJ, Luthey-Schulten ZA. Structural elements in IGP synthase exclude water to optimize ammonia transfer. Biophys J. 2005;89:475–487. doi: 10.1529/biophysj.104.058651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaro RE, Sethi A, Myers RS, Davisson VJ, Luthey-Schulten ZA. A network of conserved interactions regulates the allosteric signal in a glutamine amidotransferase. Biochemistry. 2007;46:2156–2173. doi: 10.1021/bi061708e. [DOI] [PubMed] [Google Scholar]
- Beismann-Driemeyer S, Sterner R. Imidazole glycerol phosphate synthase from Thermotoga maritima - Quaternary structure, steady-state kinetics, and reaction mechanism of the bienzyme complex. Journal of Biological Chemistry. 2001;276:20387–20396. doi: 10.1074/jbc.M102012200. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Gopalakrishnan M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;74:1155–1163. doi: 10.1016/j.bcp.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Bruschweiler S, Schanda P, Kloiber K, Brutscher B, Kontaxis G, Konrat R, Tollinger M. Direct Observation of the Dynamic Process Underlying Allosteric Signal Transmission. J Am Chem Soc. 2009;131:3063–3068. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]
- Carpenter JF, Hand SC. Comparison of pH-dependent allostery and dissociation for phosphofructokinases from Artemia embryos and rabbit muscle: nature of the enzymes acylated with diethylpyrocarbonate. Arch Biochem Biophys. 1986;248:1–9. doi: 10.1016/0003-9861(86)90394-2. [DOI] [PubMed] [Google Scholar]
- Chaudhuri BN, Lange SC, Myers RS, Chittur SV, Davisson VJ, Smith JL. Crystal structure of imidazole glycerol phosphate synthase: a tunnel through a (beta/alpha)8 barrel joins two active sites. Structure. 2001;9:987–997. [PubMed] [Google Scholar]
- Chaudhuri BN, Lange SC, Myers RS, Davisson VJ, Smith JL. Toward understanding the mechanism of the complex cyclization reaction catalyzed by imidazole glycerolphosphate synthase: crystal structures of a ternary complex and the free enzyme. Biochemistry. 2003;42:7003–7012. doi: 10.1021/bi034320h. [DOI] [PubMed] [Google Scholar]
- Chittur SV, Klem TJ, Shafer CM, Davisson VJ. Mechanism for acivicin inactivation of triad glutamine amidotransferases. Biochemistry. 2001;40:876–887. doi: 10.1021/bi0014047. [DOI] [PubMed] [Google Scholar]
- Cooper A, Dryden DTF. Allostery without conformational change. Eur. Biophys. J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- Das R, Mazhab-Jafari MT, Chowdhury S, SilDas S, Selvaratnam R, Melacini G. Entropy-driven cAMP-dependent allosteric control of inhibitory interactions in exchange proteins directly activated by cAMP. J Biol Chem. 2008;283:19691–19703. doi: 10.1074/jbc.M802164200. [DOI] [PubMed] [Google Scholar]
- Davisson VJ, Deras IL, Hamilton SE, Moore LL. A Plasmid-Based Approach for the Synthesis of a Histidine Biosynthetic Intermediate. Journal of Organic Chemistry. 1994;59:137–143. [Google Scholar]
- Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Douangamath A, Walker M, Beismann-Driemeyer S, Vega-Fernandez MC, Sterner R, Wilmanns M. Structural evidence for ammonia tunneling across the (beta alpha)(8) barrel of the imidazole glycerol phosphate synthase bienzyme complex. Structure. 2002;10:185–193. doi: 10.1016/s0969-2126(02)00702-5. [DOI] [PubMed] [Google Scholar]
- Dueber JE, Mirsky EA, Lim WA. Engineering synthetic signaling proteins with ultrasensitive input/output control. Nat Biotechnol. 2007;25:660–662. doi: 10.1038/nbt1308. [DOI] [PubMed] [Google Scholar]
- Dueber JE, Yeh BJ, Chak K, Lim WA. Reprogramming control of an allosteric signaling switch through modular recombination. Science. 2003;301:1904–1908. doi: 10.1126/science.1085945. [DOI] [PubMed] [Google Scholar]
- Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–425. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes EJ, Der CJ, Lee AL. Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. J. Mol. Biol. 2004;335:1105–1115. doi: 10.1016/j.jmb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Goto NK, Gardner KH, Mueller GA, Willis RC, Kay LE. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J Biomol NMR. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- Jarymowycz VA, Stone MJ. Remote changes in the dynamics of the phosphotyrosine-binding domain of insulin receptor substrate-1 induced by phosphopeptide binding. Biochemistry. 2008;47:13371–13382. doi: 10.1021/bi801096b. [DOI] [PubMed] [Google Scholar]
- Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Kimmel JL, Reinhart GD. Reevaluation of the accepted allosteric mechanism of phosphofructokinase from Bacillus stearothermophilus. Proc Natl Acad Sci U S A. 2000;97:3844–3849. doi: 10.1073/pnas.050588097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klem TJ, Chen Y, Davisson VJ. Subunit interactions and glutamine utilization by Escherichia coli imidazole glycerol phosphate synthase. J Bacteriol. 2001;183:989–996. doi: 10.1128/JB.182.3.989-996.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klem TJ, Davisson VJ. Imidazole Glycerol Phosphate Synthase - the Glutamine Amidotransferase in Histidine Biosynthesis. Biochemistry. 1993;32:5177–5186. doi: 10.1021/bi00070a029. [DOI] [PubMed] [Google Scholar]
- Kneller DG, Kuntz ID. Ucsf Sparky - an Nmr Display, Annotation and Assignment Tool. Journal of Cellular Biochemistry. 1993:254–254. [Google Scholar]
- Koide S, Yokoyama S, Matsuzawa H, Miyazawa T, Ohta T. Conformational equilibrium of an enzyme catalytic site in the allosteric transition. Biochemistry. 1992;31:5362–5368. doi: 10.1021/bi00138a017. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Kloiber K, Kay LE. Multiple-quantum relaxation dispersion NMR spectroscopy probing millisecond time-scale dynamics in proteins: theory and application. J. Am. Chem. Soc. 2004;126:7320–7329. doi: 10.1021/ja049968b. [DOI] [PubMed] [Google Scholar]
- Krahn JM, Kim JH, Burns MR, Parry RJ, Zalkin H, Smith JL. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry. 1997;36:11061–11068. doi: 10.1021/bi9714114. [DOI] [PubMed] [Google Scholar]
- Lipchock J, Loria JP. Millisecond dynamics in the allosteric enzyme imidazole glycerol phosphate synthase (IGPS) from Thermotoga maritima. J. Biomol. NMR. 2009;45:73–84. doi: 10.1007/s10858-009-9337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wales ME, Wild JR. Temperature effects on the allosteric responses of native and chimeric aspartate transcarbamoylases. Journal Of Molecular Biology. 1998;282:891–901. doi: 10.1006/jmbi.1998.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Changeux JP, Jacob F. Allosteric proteins and cellular control systems. Journal Of Molecular Biology. 1963;6:306–329. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- Mulder FA, Skrynnikov NR, Hon B, Dahlquist FW, Kay LE. Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15)N relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 2001;123:967–975. doi: 10.1021/ja003447g. [DOI] [PubMed] [Google Scholar]
- Myers RS, Jensen JR, Deras IL, Smith JL, Davisson VJ. Substrate-induced changes in the ammonia channel for imidazole glycerol phosphate synthase. Biochemistry. 2003;42:7013–7022. doi: 10.1021/bi034314l. [DOI] [PubMed] [Google Scholar]
- Omi R, Mizuguchi H, Goto M, Miyahara I, Hayashi H, Kagamiyama H, Hirotsu K. Structure of imidazole glycerol phosphate synthase from Thermus thermophilus HB8: open-closed conformational change and ammonia tunneling. J Biochem. 2002;132:759–765. doi: 10.1093/oxfordjournals.jbchem.a003284. [DOI] [PubMed] [Google Scholar]
- Onuchic JN, Kobayashi C, Miyashita O, Jennings P, Baldridge KK. Exploring biomolecular machines: energy landscape control of biological reactions. Philos Trans R Soc Lond B Biol Sci. 2006;361:1439–1443. doi: 10.1098/rstb.2006.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier M. Engineering allosteric protein switches by domain insertion. Protein Eng Des Sel. 2005;18:359–364. doi: 10.1093/protein/gzi048. [DOI] [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden dynamic allostery in a PDZ domain. Proc Natl Acad Sci U S A. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat Struct Mol Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovych N, Tzeng SR, Tonelli M, Ebright RH, Kalodimos CG. Structural basis for cAMP-mediated allosteric control of the catabolite activator protein. Proc Natl Acad Sci U S A. 2009;106:6927–6932. doi: 10.1073/pnas.0900595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raddatz R, Schaffhauser H, Marino MJ. Allosteric approaches to the targeting of G-protein-coupled receptors for novel drug discovery: a critical assessment. Biochem Pharmacol. 2007;74:383–391. doi: 10.1016/j.bcp.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Reinhart GD. Quantitative analysis and interpretation of allosteric behavior. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- Ross RA. Allosterism and cannabinoid CB(1) receptors: the shape of things to come. Trends Pharmacol Sci. 2007;28:567–572. doi: 10.1016/j.tips.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Rudolph J, Stubbe J. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry. 1995;34:2241–2250. doi: 10.1021/bi00007a019. [DOI] [PubMed] [Google Scholar]
- Schweizer A, Roschitzki-Voser H, Amstutz P, Briand C, Gulotti-Georgieva M, Prenosil E, Binz HK, Capitani G, Baici A, Pluckthun A, Grutter MG. Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure. 2007;15:625–636. doi: 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Shi Z, Resing KA, Ahn NG. Networks for the allosteric control of protein kinases. Curr Opin Struct Biol. 2006;16:686–692. doi: 10.1016/j.sbi.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Skrynnikov NR, Mulder FA, Hon B, Dahlquist FW, Kay LE. Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: application to methionine residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 2001;123:4556–4566. doi: 10.1021/ja004179p. [DOI] [PubMed] [Google Scholar]
- Spraggon G, Kim C, Nguyen-Huu X, Yee MC, Yanofsky C, Mills SE. The structures of anthranilate synthase of Serratia marcescens crystallized in the presence of (i) its substrates, chorismate and glutamine, and a product, glutamate, and (ii) its end-product inhibitor, L-tryptophan. Proc Natl Acad Sci U S A. 2001;98:6021–6026. doi: 10.1073/pnas.111150298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden JB, Holden HM, Wesenberg G, Raushel FM, Rayment I. Structure of carbamoyl phosphate synthetase: a journey of 96 A from substrate to product. Biochemistry. 1997;36:6305–6316. doi: 10.1021/bi970503q. [DOI] [PubMed] [Google Scholar]
- Thoden JB, Miran SG, Phillips JC, Howard AJ, Raushel FM, Holden HM. Carbamoyl phosphate synthetase: caught in the act of glutamine hydrolysis. Biochemistry. 1998;37:8825–8831. doi: 10.1021/bi9807761. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Kay LE. Side chain assignments of Ile delta 1 methyl groups in high molecular weight proteins: an application to a 46 ns tumbling molecule. J Am Chem Soc. 2003;125:5701–5706. doi: 10.1021/ja021452+. [DOI] [PubMed] [Google Scholar]
- Tugarinov V, Sprangers R, Kay LE. Probing side-chain dynamics in the proteasome by relaxation violated coherence transfer NMR spectroscopy. J. Am. Chem. Soc. 2007;129:1743–1750. doi: 10.1021/ja067827z. [DOI] [PubMed] [Google Scholar]
- Yan J, Liu Y, Lukasik SM, Speck NA, Bushweller JH. CBFbeta allosterically regulates the Runx1 Runt domain via a dynamic conformational equilibrium. Nat Struct Mol Biol. 2004;11:901–906. doi: 10.1038/nsmb819. [DOI] [PubMed] [Google Scholar]
- Yu EW, Koshland DE., Jr. Propagating conformational changes over long (and short) distances in proteins. Proc Natl Acad Sci U S A. 2001;98:9517–9520. doi: 10.1073/pnas.161239298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalkin H, Smith JL. Enzymes utilizing glutamine as an amide donor. Adv Enzymol Relat Areas Mol Biol. 1998;72:87–144. doi: 10.1002/9780470123188.ch4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.