

Abstract
The expression of forms of synaptic plasticity, such as the phenomenon of long-term potentiation, requires the activity-dependent regulation of synaptic proteins and synapse composition. Here we show that ARMS (ankyrin repeat-rich membrane spanning protein)/Kidins220, a transmembrane scaffold molecule and BDNF TrkB substrate, is significantly reduced in hippocampal neurons after potassium chloride depolarization. The activity-dependent proteolysis of ARMS/Kidins220 was found to occur through calpain, a calcium-activated protease. Moreover, hippocampal long-term potentiation in ARMS/Kidins220+/− mice was enhanced, and inhibition of calpain in these mice reversed these effects. These results provide an explanation for a role for the ARMS/Kidins220 protein in synaptic plasticity events and suggest that the levels of ARMS/Kidins220 can be regulated by neuronal activity and calpain action to influence synaptic function.
Keywords: Calcium, Calpain, Neurotrophin, Protein Degradation, Synapses, Long-term Potentiation, Neuronal Depolarization, Scaffold Protein, Synaptic Plasticity
Introduction
Activity-dependent changes in protein function and composition are known to accompany changes in synaptic efficacy. Such processes are frequently mediated by calcium signaling and have been well studied in the synaptic plasticity process of long-term potentiation (LTP).4 Activity-induced synaptic changes can occur through up-regulation of proteins via novel gene transcription (1). Synaptic proteins can also undergo regulated turnover or degradation through proteasome-mediated (2) or calpain-mediated (3, 4) proteolysis.
The cysteine protease calpain is ideal for executing activity-dependent proteolysis of proteins because its enzymatic activity is induced by increases in intracellular calcium levels (5). Calpain activation leads to cleavage of synaptic targets such as the postsynaptic density (PSD) scaffold molecules glutamate receptor-interacting protein (GRIP) and PSD-95 (6, 7), as well as cytoskeletal molecules such as spectrin (8), which allows the remodeling of the synapse that accompanies synaptic plasticity. Calpain can also alter glutamate receptor function by targeting receptor subunits (9–16).
One molecule that may play a role in synaptic plasticity is ARMS (ankyrin repeat-rich membrane spanning protein) or Kidins220 (kinase D-interacting substrate of 220 kDa). Initially identified as an interactor of Trk and p75 neurotrophin receptors (17), as well as a protein kinase D substrate (18), ARMS/Kidins220 is a transmembrane scaffold protein that is highly expressed in the nervous system. Strikingly, it is phosphorylated by Trk and Eph receptors, but not by other growth factor tyrosine kinase receptors such as EGF (17). Therefore, ARMS/Kidins220 represents a receptor tyrosine kinase substrate that lends signaling specificity to certain neuronal pathways (17, 19). In the central nervous system, the TrkB receptor phosphorylates ARMS/Kidins220 after brain-derived neurotrophic factor (BDNF) binding. Because BDNF and TrkB are highly responsive to neuronal activation, we hypothesized that downstream signal transducers such as ARMS/Kidins220 might also be involved in activity-dependent events as a mechanism for facilitating synaptic plasticity.
Here we show that the calcium-activated protease calpain is responsible for degrading ARMS/Kidins220 after neuronal activation. In vivo, genetic reduction of ARMS/Kidins220 levels leads to enhanced LTP, and this effect of ARMS/Kidins220 on LTP is dependent on calpain. These data indicate that ARMS/Kidins220 levels can be regulated by neuronal activity through calpain proteolysis, which has consequences for hippocampal synaptic plasticity.
EXPERIMENTAL PROCEDURES
Materials
The following pharmacological reagents were obtained from Sigma-Aldrich: epoxomicin (1 μm), lactacystin (10 μm), chloroquine (50 μm), MDL28170 (20 μm), and EGTA (2.5 mm). N-Acetyl-Leu-Leu-Met (25 μm) was obtained from Calbiochem. The calpain inhibitor BDA-410 (100 nm) was kindly provided by the Mitsubishi Tanabe Pharma Corp.
The following antibodies were used: anti-ARMS/Kidins220 C-terminal rabbit polyclonal (1:4000) (17), anti-actin mouse monoclonal (1:1000; A4700, Sigma), anti-spectrin mouse monoclonal (1:5000; MAB1622, Chemicon), anti-GluA1 (formerly GluR1) C-terminal rabbit polyclonal (1:1000, AB1502, Chemicon), and anti-GluA2 (formerly GluR2) rabbit polyclonal (1:1000; Chemicon).
Production of the ARMS/Kidins220 N-terminal Antibody
The coding sequence of amino acids 1–402 of ARMS/Kidins220 was inserted into a M15 pQE31 vector (Qiagen) to generate a His6-tagged version. Following transformation and growth of Escherichia coli to log phase, protein expression was induced with 0.4 mm isopropyl β-d-1-thiogalactopyranoside. The cells were grown for an additional 3.5 h at 25 °C, pelleted, and then resuspended in 25 mm Tris-HCl (pH 7.8), 300 mm KCl, 2 mm β-mercaptoethanol, and protease inhibitors. The cells were lysed using a French press. After centrifugation, the pellet was resuspended in 6 m urea, PBS, and 0.1% Triton X-100, and after removal of cell debris, the supernatant was incubated with nickel-nitrilotriacetic acid-agarose beads (Qiagen) at 25 °C for 45 min. Subsequently, the beads were washed with 100 mm Tris-HCl (pH 8), 6 m urea, and 20 mm imidazole, and the His-tagged protein was eluted with 500 mm imidazole. The protein was dialyzed in 20 mm HEPES-NaOH (pH 7.4), 0.1 m urea, 5% glycerol, and 150 mm NaCl and stored at −20 °C.
The purified His-tagged N-terminal fragment (amino acids 1–402) of ARMS/Kidins220 was used for immunization of two rabbits by BioGenes GmbH following standard procedures. Affinity-purified antibodies were isolated using antigen-specific CNBr-Sepharose columns and tested for immunoreactivity. The antibody was used in Western blots at a dilution of 1:200.
Primary Hippocampal Cultures
Primary hippocampal neurons were dissected from day 18–19 Sprague-Dawley rat embryos in Ca2+- and Mg2+-free Hanks' balanced salt solution (Invitrogen) supplemented with 0.37% glucose, digested with 0.05% trypsin, mechanically dissociated with fire-polished Pasteur pipettes, and plated in poly-d-lysine-coated 12-well plates at 250,000 neurons per well. Cells were grown in Neurobasal medium (Invitrogen) supplemented with B27 supplement (Invitrogen), 0.37% glucose, 0.5 mm glutamine (Invitrogen), and 1.2 μg/ml 5-fluoro-2-deoxyuridine. Fresh medium was added to the cells every 3–4 days.
Cells were stimulated at 14 to 21 days in vitro with 50 mm KCl or 200 μm glutamate. For KCl treatments, cells were collected at time points after the addition of 50 mm KCl to the cell medium. For glutamate treatments, 200 μm glutamate was added to the medium for 1 min, and then the medium was completely replaced with conditioned medium without glutamate. Cells were collected at time points after the removal of glutamate. When pharmacological inhibitors were used, they were applied to the cells 30 min prior to stimulation and were maintained throughout the stimulation.
Western Blots
Cells were collected using 80 μl of 1× SDS buffer per well of each 12-well plate and boiled for 5 min. A 12-μl sample was separated on SDS-polyacrylamide gels, transferred to PVDF membranes (Millipore), and incubated with primary and HRP-conjugated secondary antibodies. Immunoreactive proteins were visualized by ECL detection (Amersham Biosciences) and film autoradiography. Band intensities were quantified using ImageJ software, and statistical analysis was performed using GraphPad Prism software. All results were obtained from at least three independent experiments.
Electrophysiology in ARMS/Kidins220+/− Mice
ARMS/Kidins220+/− mice have been described previously (20). Hippocampi were dissected from 3–6-month-old male ARMS/Kidins220+/− mice and wild-type littermates and cut into 400-mm transverse slices with a tissue chopper (Electron Microscopy Sciences). Slices were incubated in an interface chamber at 29 °C for 90 min before recording, where they were subfused with artificial cerebrospinal fluid consisting of 124 mm NaCl, 4.4 mm KCl, 1.0 mm Na2HPO4, 25 mm NaHCO3, 2.0 or 2.5 mm CaCl2, 2.0 or 1.3 mm MgSO4, and 10 mm glucose and bubbled with 95% O2 and 5% CO2. A bipolar tungsten-stimulating electrode and a glass micropipette (5–10 megohms, filled with artificial cerebrospinal fluid) recording electrode were placed in the stratum radiatum in the CA1 region, and extracellular field potentials were recorded. Basal synaptic transmission was assayed by plotting the stimulus voltages (V) against the slopes of the field excitatory postsynaptic potentials to generate input-output relations. For LTP experiments, baseline stimulation was delivered every minute for 15 min at an intensity that evoked a response of ∼35% of the maximum evoked response. LTP was induced using a θ-burst stimulation of four pulses at 100 Hz, with bursts repeated at 5 Hz, and each tetanus including three 10-burst trains separated by 15 s. In experiments using the calpain inhibitor BDA-410, slices were perfused with the inhibitor (100 nm) 20 min before LTP induction.
RESULTS
LTP Is Enhanced in ARMS/Kidins220+/− Mice
We previously reported a line of mice heterozygous for the ARMS/Kidins220 gene that display a 30–40% reduction of ARMS/Kidins220 protein compared with wild-type animals (20). Homozygous mutants die at an embryonic age. To examine the effect of ARMS/Kidins220 reduction on synaptic plasticity, acute hippocampal slices were taken from 3–6-month-old ARMS/Kidins220+/− mice, and electrophysiology measurements were performed. LTP at the Schaffer collateral-CA1 synapse was induced using a θ-burst stimulus, and the slopes of the field excitatory postsynaptic potentials were recorded. Strikingly, in ARMS/Kidins220+/− mice, LTP was enhanced compared with wild-type animals (Fig. 1A; F(1,17) = 5.084, p < 0.05, two-way analysis of variance). ARMS/Kidins220+/− mice also exhibited increased synaptic transmission at lower stimulus intensities (Fig. 1B). These data indicate that levels of ARMS/Kidins220 in vivo regulate synaptic transmission and synaptic plasticity.
FIGURE 1.

Decreased levels of ARMS/Kidins220 in vivo lead to enhanced LTP and increased basal synaptic transmission at the Schaffer collateral-CA1 synapse. A, ARMS/Kidins220+/− mice display enhanced LTP. LTP was induced by a θ-burst stimulus (arrow) in acute hippocampal slices from 3–6-month-old ARMS/Kidins220+/− mice and wild-type littermates. Each point represents the average of three successive events (WT, black circles, n = 8; ARMS/Kidins220+/−, white circles, n = 11; data are represented as the means ± S.E.). B, input-output curve shows increased basal synaptic transmission at lower stimulus intensities in ARMS/Kidins220+/−mice. Input-output curve of field excitatory postsynaptic potential slope (fEPSP) (V s−1) versus stimulus (V) at Schaffer collateral-CA1 synapses in adult hippocampal slices from ARMS/Kidins220+/− and wild-type mice (WT, black circles, n = 11; ARMS/Kidins220+/−, white circles, n = 15; *, p < 0.05, t test; data are represented as the means ± S.E.).
ARMS/Kidins220 Is Degraded after Neuronal Activation by KCl Depolarization
It was previously shown that ARMS/Kidins220 protein levels respond to long-term changes in neuronal activity (21). When the sodium channel blocker tetrodotoxin was used to inhibit hippocampal cultures over a course of 48 h, ARMS/Kidins220 protein levels increased, and when the γ-aminobutyric acid receptor inhibitor bicuculline was used to increase activity in cultures, ARMS/Kidins220 protein levels decreased. Because such treatments have been shown to induce homeostatic changes in neurons (22), these data suggest that ARMS/Kidins220 acts as an activity sensor with a possible role in synaptic plasticity.
To study the regulation of ARMS/Kidins220 due to acute neuronal activation, we depolarized rat hippocampal cultures with 50 mm KCl, a stimulus that is widely used to study activity-dependent neuronal changes (23–25). Cultures were treated with KCl, and lysates were collected at different times and probed by Western blot for ARMS/Kidins220 protein using antibodies directed against the C terminus (17) and the N terminus.
Neuronal activation caused a reduction in ARMS/Kidins220 levels to ∼50% of basal levels within 3 h as a result of protein degradation (Fig. 2, A and B). The N-terminal antibody not only showed degradation of the full-length 220-kDa ARMS/Kidins220 protein, but also detected smaller sized degradation products at ∼140, 120, and 85 kDa. These were present at low levels in untreated cultures and accumulated with prolonged neuronal activation (Fig. 2A, arrows a, b, and c). Addition of the calcium chelator EGTA to the culture medium prevented KCl-induced degradation of ARMS/Kidins220, indicating that extracellular calcium influx after membrane depolarization is required (Fig. 3, A and B). Taken together, these data indicate that KCl-induced neuronal activation causes calcium-mediated degradation of full-length ARMS/Kidins220 to smaller sized fragments.
FIGURE 2.
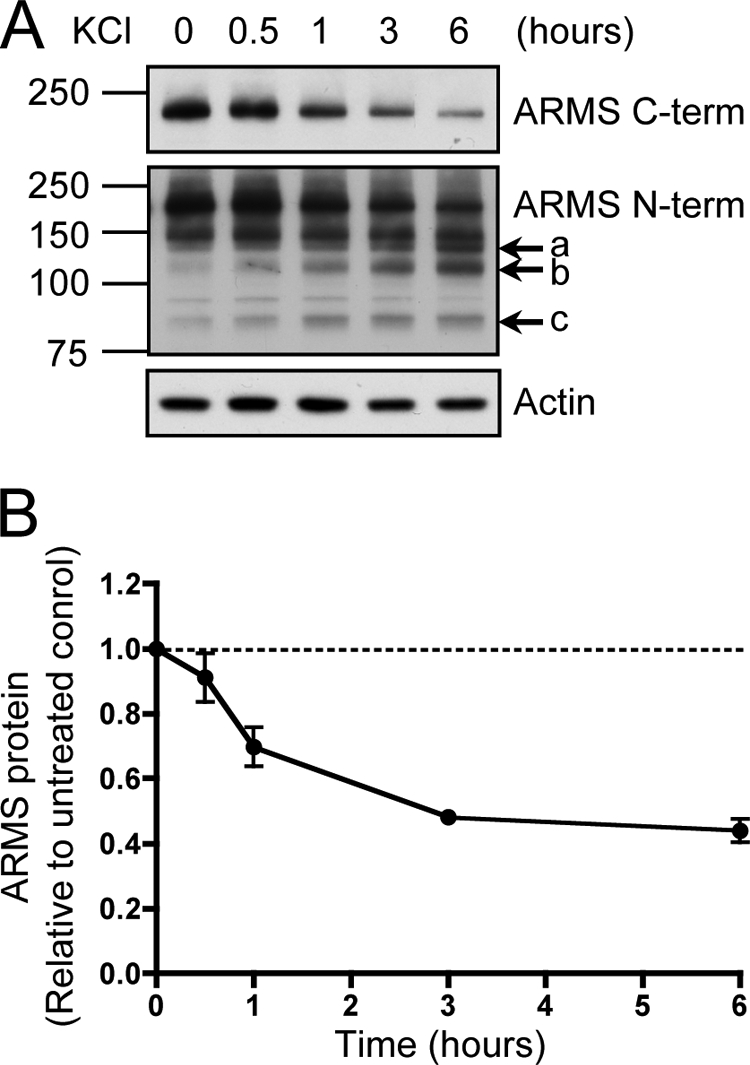
ARMS/Kidins220 is degraded upon KCl activation of hippocampal neurons. A, ARMS/Kidins220 is degraded upon neuronal activation by KCl. Hippocampal cultures (14–21 days in vitro) were depolarized with 50 mm KCl for the indicated times. Cell lysates were immunoblotted for ARMS/Kidins220 using antibodies against the C or N terminus and for actin as a loading control. In response to neuronal activation, 220-kDa full-length ARMS/Kidins220 as detected by both C- and N-terminal antibodies was degraded, leading to the accumulation of smaller sized fragments as detected by the N-terminal antibody (arrows), estimated to be 140 (a), 120 (b), and 85 (c) kDa. 95- and 150-kDa nonspecific bands were detected by the N-terminal antibody. B, quantification of ARMS/Kidins220 protein levels in hippocampal neurons after KCl depolarization, as detected with the C-terminal antibody, normalized to actin levels, and plotted relative to untreated control (n ≥ three independent experiments; data are represented as the means ± S.E.).
FIGURE 3.
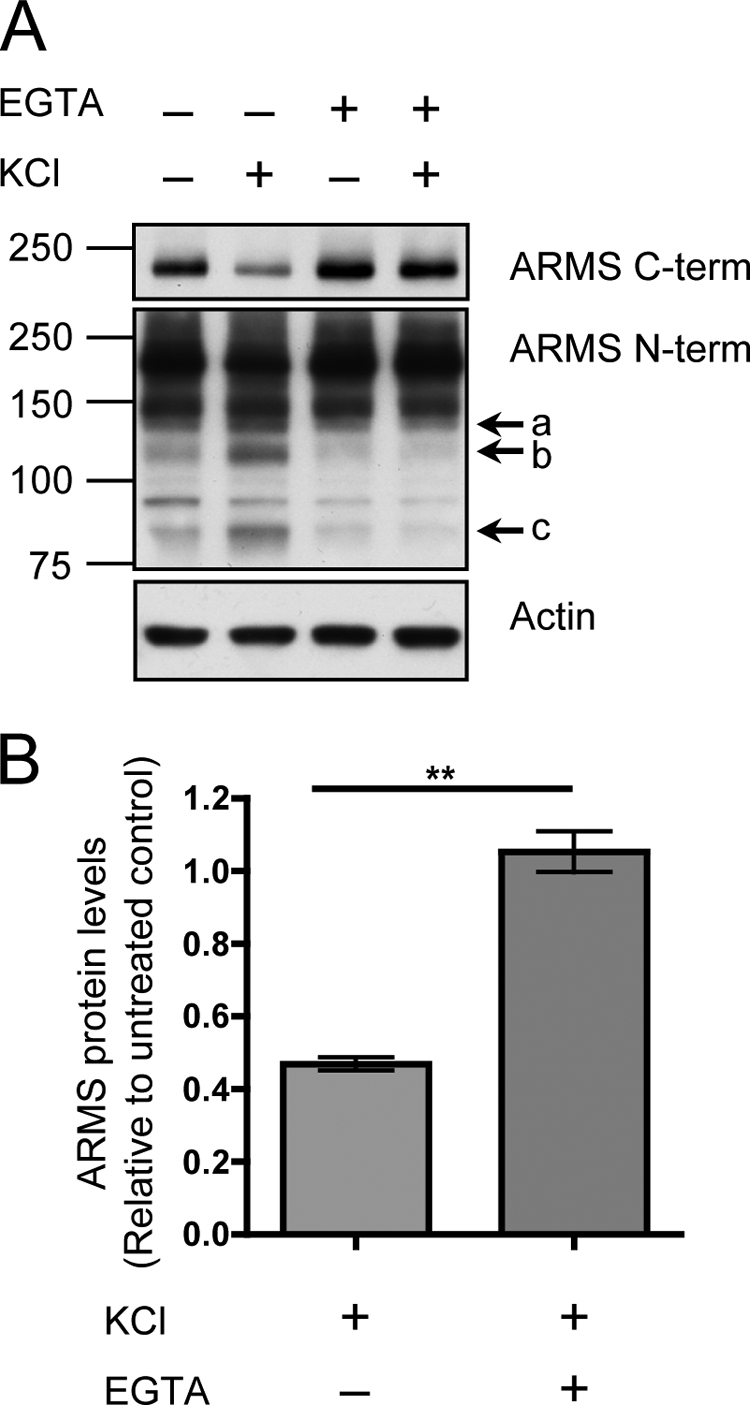
ARMS/Kidins220 degradation induced by KCl depolarization requires extracellular calcium. A, hippocampal cultures were depolarized with 50 mm KCl for 3 h in the presence of the calcium chelator EGTA, and ARMS/Kidins220 protein levels were assessed by Western blot. EGTA fully rescued the degradation of full-length ARMS/Kidins220 and the appearance of N-terminal degradation fragments (arrows a, b, and c). Actin is shown as a loading control. 95- and 150-kDa nonspecific bands were detected by the N-terminal antibody. B, quantification of ARMS/Kidins220 protein levels in A, as detected with the C-terminal antibody, normalized to actin levels, and plotted relative to untreated control (n ≥ three independent experiments; **, p < 0.01, t test; data are represented as the means ± S.E.).
Glutamate-Induced Neuronal Activation Causes ARMS/Kidins220 Degradation
In addition to KCl, glutamate, the most prominent excitatory neurotransmitter in the nervous system, is commonly used to activate cultured neurons to induce physiological synaptic activity (26). To demonstrate that activity-induced ARMS/Kidins220 degradation is not restricted to KCl depolarization, we assayed ARMS/Kidins220 protein levels after glutamate treatment of hippocampal cultures. Similar to the effect of KCl, full-length ARMS/Kidins220 was degraded when neurons were activated by glutamate (Fig. 4A). Additionally, the same N-terminal degradation products also were detected (Fig. 4A, arrows a, b, and c). The degradation of ARMS/Kidins220 and the accumulation of the smaller fragments were prevented by the addition of EGTA (Fig. 4B). These results indicate that ARMS/Kidins220 degradation can result from multiple forms of neuronal activation, including that induced by glutamate.
FIGURE 4.
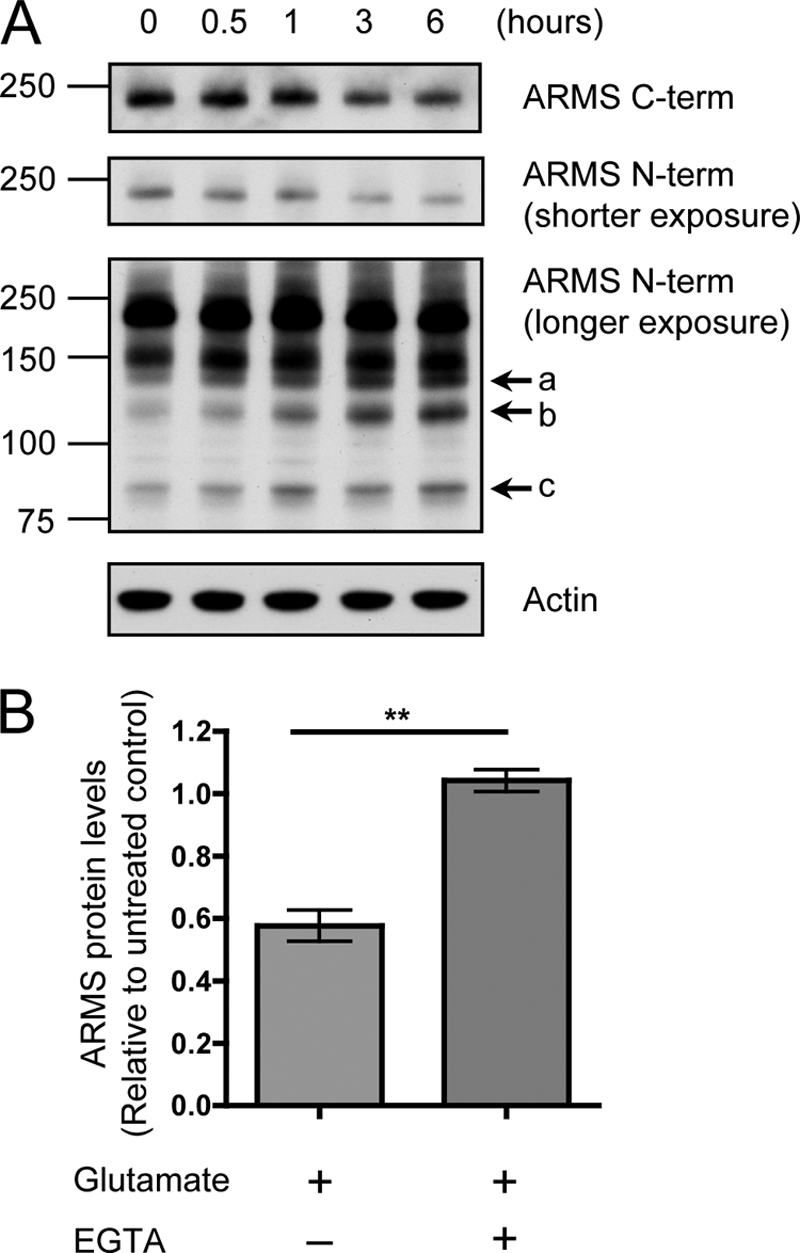
Glutamate activation of hippocampal neurons induces ARMS/Kidins220 degradation through a calcium-mediated process. A, ARMS/Kidins220 is degraded upon glutamate treatment of hippocampal neurons. Hippocampal cultures were stimulated with 200 μm glutamate for 1 min, and cell lysates were collected at the indicated times after stimulation and immunoblotted for ARMS/Kidins220. As was the case after KCl depolarization, full-length ARMS/Kidins220 was degraded, leading to the accumulation of similar smaller sized N-terminal fragments (arrows a, b, and c). Actin is shown as a loading control. A 150-kDa nonspecific band was detected by the N-terminal antibody. B, glutamate-mediated ARMS/Kidins220 degradation requires extracellular calcium. Hippocampal cultures were stimulated with 200 μm glutamate for 1 min in the presence of EGTA, and cell lysates were collected 3 h after stimulation. ARMS/Kidins220 protein levels, as detected by the C-terminal antibody, were assessed by Western blot and quantified. ARMS/Kidins220 levels were normalized to actin levels and plotted relative to untreated control (n ≥ three independent experiments; **, p < 0.01, t test; data are represented as the means ± S.E.).
ARMS/Kidins220 Degradation Is Independent of Proteasomes or Lysosomes
Activity-dependent turnover of proteins at the PSD is a mechanism for protein composition regulation at the highly dynamic synapse (27). Proteasomal activity is implicated in the turnover of PSD proteins (2) and can be regulated by BDNF (28). We first hypothesized that the degradation of ARMS/Kidins220 after neuronal activity was due to the action of proteasomes. Inhibitors of proteasomes were used to determine whether they could prevent ARMS/Kidins220 degradation. Incubation of hippocampal cultures with the proteasomal inhibitor epoxomicin or lactacystin during KCl stimulation did not attenuate ARMS/Kidins220 degradation, indicating that proteasomal digestion was not the primary mechanism of degradation (Fig. 5, A and C).
FIGURE 5.

Activity-dependent degradation of ARMS/Kidins220 protein is not mediated by proteasomes and lysosomes. A, inhibition of proteasomes does not rescue activity-induced degradation of ARMS/Kidins220. Hippocampal cultures were depolarized with 50 mm KCl for 3 h in the presence of the proteasomal inhibitor epoxomicin or lactacystin, and ARMS/Kidins220 protein levels were assessed by Western blot. Actin is shown as a loading control. B, inhibition of lysosomes does not rescue activity-induced degradation of ARMS/Kidins220. Experiments were performed as in A using the lysosomal inhibitor chloroquine. Actin is shown as a loading control. C, quantification of ARMS/Kidins220 protein levels in A and B normalized to actin levels and plotted relative to untreated control (n ≥ three independent experiments; data are represented as the means ± S.E.).
Another degradation pathway is through lysosomes, where hydrolases digest proteins in acidic compartments after internalization from the cell membrane. Because ARMS/Kidins220 is a transmembrane protein, we investigated whether blocking lysosomal action using chloroquine, which deacidifies lysosomes and inactivates lysosomal proteases, could block ARMS/Kidins220 degradation. Incubation of hippocampal cultures with chloroquine during KCl stimulation had no significant effect on ARMS/Kidins220 degradation, suggesting that the degradation did not occur through the lysosomal pathway (Fig. 5, B and C).
ARMS/Kidins220 Degradation Is Due to Calpain Proteolysis
Alternatively, activity-induced proteolysis of ARMS/Kidins220 could occur through the action of proteases that are activated by calcium. One potential candidate is calpain. The two major isoforms of calpain, μ- and m-calpain, are highly expressed in neurons and respond to micro- and millimolar concentrations of intracellular calcium, respectively, in part through the binding of calcium directly to domains in the molecule (5). Calpain acts at neutral pH in the cytosol and at the cell membrane to cleave a wide range of targets. Furthermore, calpain activity has been implicated in synaptic functions and neurodegenerative diseases (3, 4).
To investigate whether calpain degrades ARMS/Kidins220, hippocampal cultures were incubated with the calpain inhibitor MDL28170 or N-acetyl-Leu-Leu-Met during KCl stimulation. Calpain inhibitors rescued degradation of full-length ARMS/Kidins220, as well as the accumulation of degradation products (Fig. 6, A and B). Calpain inhibitors also rescued the ARMS/Kidins220 degradation induced by glutamate treatment (Fig. 6C).
FIGURE 6.

ARMS/Kidins220 is degraded by calpain upon neuronal activation. A, inhibition of the protease calpain rescues KCl-induced degradation of ARMS/Kidins220. Hippocampal cultures were depolarized with 50 mm KCl for 3 h in the presence of the calpain inhibitor MDL28170 or N-acetyl-Leu-Leu-Met (ALLM), and ARMS/Kidins220 protein levels were assessed by Western blot. Both of these inhibitors significantly reduced the degradation of ARMS/Kidins220 and prevented the appearance of N-terminal degradation fragments (arrows a, b, and c). The 145- and 150-kDa breakdown products of spectrin mark calpain activity, and actin is shown as a loading control. 95- and 150-kDa nonspecific bands were detected by the N-terminal antibody. B, quantification of ARMS/Kidins220 protein levels in A, as detected by the C-terminal antibody, normalized to actin levels, and plotted relative to untreated control (n ≥ three independent experiments; **, p < 0.01, t test; data are represented as the means ± S.E.). C, hippocampal cultures were stimulated with 200 μm glutamate for 1 min in the presence of the calpain inhibitor MDL28170 or N-acetyl-Leu-Leu-Met, and cell lysates were collected at the indicated times after stimulation. ARMS/Kidins220 protein levels, as detected by the C-terminal antibody, were assessed by Western blot and quantified. ARMS/Kidins220 levels were normalized to actin levels and plotted relative to untreated control (n ≥ three independent experiments; *, p < 0.05, t test; data are represented as the means ± S.E.). D, hippocampal cultures were depolarized with 50 mm KCl for the indicated times in the presence or absence of the calpain inhibitor MDL28170, and the indicated proteins were probed by Western blot. The degradation of full-length ARMS/Kidins220, as well as the appearance of N-terminal degradation fragments (arrows a, b, and c), was correlated with the degradation of GluA1, and the degradation of both proteins was prevented by calpain inhibition. In contrast, GluA2 was not degraded.
To demonstrate that the degradation of ARMS/Kidins220 is not due to nonspecific proteolysis of membrane proteins, we probed for the AMPA receptor subunits GluA1 and GluA2, two synaptic membrane proteins. After KCl depolarization, GluA1 was degraded in a calpain-dependent manner, which is consistent with previous studies (Fig. 6D) (9, 16, 29). However, GluA2 remained intact, indicating that the activity of calpain was directed at specific targets. Taken together, these data indicate that ARMS/Kidins220 is a specific target of calpain, which degrades the scaffold molecule in an activity-dependent manner.
Inhibition of Calpain Activity Rescues LTP Enhancement in ARMS/Kidins220+/− Mice
A possible explanation for the enhancement of LTP in ARMS/Kidins220+/− mice is that a further reduction of the protein levels in these animals by calpain cleavage unmasks a role for ARMS/Kidins220 in regulating the amounts of synaptic potentiation. To demonstrate a link between the enhancement of LTP observed in ARMS/Kidins220+/− mice and calpain-dependent ARMS/Kidins220 proteolysis, we studied the effect of calpain inhibition in the mice. Acute hippocampal slices were taken from 3–6-month-old ARMS/Kidins220+/− mice, and LTP was induced in the presence and absence of BDA-410, a compound that has been shown to be a potent and selective calpain inhibitor (30, 31). In a previous study, calpain inhibition by BDA-410 had no effect on the expression of LTP or on basal transmission in wild-type mice (32). Our experiments confirmed that this inhibitor also did not affect LTP in our wild-type animals (Fig. 7; wild-type mice treated with vehicle (black circles) versus BDA-410 (black triangles); F(1,13) = 0.14, p > 0.05, two-way analysis of variance). However, the inhibitor did rescue the enhancement of LTP in ARMS/Kidins220+/− mice (Fig. 7; ARMS/Kidins220+/− mice treated with vehicle (white circles) versus BDA-410 (white triangles); F(1,12) = 4.907, p < 0.05, two-way analysis of variance). These data indicate that the effects of ARMS/Kidins220 expression on LTP can be mediated by calpain.
FIGURE 7.

Inhibition of calpain prevents enhancement of LTP in ARMS/Kidins220+/− mice. Acute hippocampal slices from 3–7-month-old ARMS/Kidins220+/− mice and wild-type littermates were incubated with the calpain inhibitor BDA-410 before and during LTP induction by a θ-burst stimulus (arrow). Calpain blockade did not affect LTP expression in wild-type animals, whereas it prevented enhancement of LTP in ARMS/Kidins220+/− mice. Each point represents the average of three successive events (WT: vehicle, black circles, n = 7; BDA-410, black triangles, n = 8; ARMS/Kidins220+/−: vehicle, white circles, n = 7; BDA-410, white triangles, n = 9; data are represented as the means ± S.E.). fEPSP, field excitatory postsynaptic potential.
DISCUSSION
Activity-dependent regulation of synaptic proteins is a critical process responsible for many observations of synaptic plasticity. Here we demonstrated that decreased levels of the ARMS/Kidins220 scaffold protein in vivo led to an enhancement of LTP. In cultured neurons, ARMS/Kidins220 was down-regulated in an activity-dependent manner, and the decrease in ARMS/Kidins220 levels was regulated by calpain. Inhibition of calpain activity in ARMS/Kidins220+/− mice rescued the changes in LTP. Taken together, our data suggest that the activity-dependent regulation of ARMS/Kidins220 by calpain mediates its effects on synaptic plasticity.
It has been proposed that regulated physiological activation of calpain is critical for synaptic plasticity and memory formation, whereas the pathological hyperactivation of calpain leads to neurodegenerative processes (3, 4). Normal activation leads to key signaling processes, whereas abnormal activation leads to dysregulated degradation and neurotoxicity. López-Menéndez et al. (33) have shown that ARMS/Kidins220 plays a key role during NMDA-mediated excitotoxicity that is mediated by calpain. Here we found that calpain cleavage of ARMS/Kidins220 is involved in its ability to influence LTP. We previously showed that alterations in ARMS/Kidins220 levels in culture led to changes in synaptic charge, suggesting that ARMS/Kidins220 regulates synaptic activity (21). Recent studies in hippocampal neurons indicate that the level of ARMS/Kidins220 protein expression can influence both excitatory (34) and inhibitory neurotransmission (35). Intriguingly, lowered ARMS/Kidins220 protein levels in vivo caused an enhancement of LTP.
We used the highly selective BDA-410 calpain inhibitor to assess whether calpain activity is linked to the changes in LTP observed in heterozygous mice expressing reduced levels of ARMS/Kidins220. Consistent with previous findings, the BDA-410 inhibitor did not have any effect upon the generation of LTP or of basal synaptic transmission properties in wild-type mice (32). However, treatment of hippocampal slices from ARMS/Kidins220+/− mice resulted in a reversal of the enhancement of LTP.
These results imply that calpain activity is related specifically to the generation of LTP in mice with a reduced level of ARMS/Kidins220. Steady-state levels of ARMS/Kidins220 in wild-type animals may place a constitutive restraint upon potentiation, perhaps through the inhibition of a rate-limiting step. Decreases in the ARMS/Kidins220 protein in ARMS/Kidins220+/− mice would prime or facilitate the ability to generate LTP at Schaffer collateral-CA1 synapses. The specific action of the BDA-410 calpain inhibitor upon the ARMS/Kidins220+/− mice suggests that calpain cleavage of ARMS/Kidins220 or its many associated proteins may be responsible for changing hippocampal plasticity.
Calpain has also been shown to regulate MAPK signaling involved in memory processes through cleavage of the suprachiasmatic nucleus circadian oscillatory protein (SCOP), an inhibitor of MAPK (36). Interestingly, BDNF was implicated in the induction of calpain to regulate SCOP. Notably, the calpain activation can also be regulated by BDNF through MAPK phosphorylation events (37). These studies provide evidence that calpain is directly involved in synaptic plasticity through the regulation of several different mechanisms. The calpain cleavage of glutamate receptor subunits is regulated by phosphorylation (29). The kinases Fyn and Ca2+/calmodulin-dependent protein kinase II, among others, have been shown to regulate the sensitivity of targets to calpain cleavage (38, 39). Because ARMS/Kidins220 is a molecule with many potential phosphorylation sites, its cleavage may be regulated by the phosphorylation state of the protein.
It is likely that different physiological functions are carried out by ARMS/Kidins220 because it has multifunctional interactions with many different signaling proteins, such as Trio, septins, Trk receptors, CrkL, and AMPA and NMDA receptor subunits (33, 34, 40–42). As a scaffold protein at postsynaptic sites, ARMS/Kidins220 can potentially participate in protein-protein interactions in dendritic spine compartments. Conspicuously, the C terminus of ARMS/Kidins220 contains a PDZ domain-binding motif, which has the potential to interact with PDZ domain-containing proteins in the PSD to exert its scaffolding functions. As a substrate of neurotrophin and ephrin receptors, as well as calpain, regulation of ARMS/Kidins220 levels may serve as an important convergence point for transmitting signals to mediate synaptic plasticity.
Acknowledgments
We thank Katrin Deinhardt and Vladimir Camarena for critical reading of the manuscript; Karishma Dagar and Archana Vasudevan for technical support; and members of the Chao, Hempstead, and Lee laboratories for helpful discussions. We also thank Hiroshi Kinoshita for the BDA-410 inhibitor and Giampietro Schiavo for advice.
This work was supported, in whole or in part, by National Institutes of Health Grants NS21072 and HD23315 (to M. V. C.) and NS049442 (to O. A.). This work was also supported by the Medical Scientist Training Program (to S. H. W.), and a Marie Curie International Reintegration Grant within the 7th European Community Framework Programme (to J. C. A.).
- LTP
- long-term potentiation
- PSD
- postsynaptic density.
REFERENCES
- 1.Flavell S. W., Greenberg M. E. (2008) Annu. Rev. Neurosci. 31, 563–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehlers M. D. (2003) Nat. Neurosci. 6, 231–242 [DOI] [PubMed] [Google Scholar]
- 3.Liu J., Liu M. C., Wang K. K. (2008) Sci. Signal. 1, re1. [DOI] [PubMed] [Google Scholar]
- 4.Wu H. Y., Lynch D. R. (2006) Mol. Neurobiol. 33, 215–236 [DOI] [PubMed] [Google Scholar]
- 5.Goll D. E., Thompson V. F., Li H., Wei W., Cong J. (2003) Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 6.Lu X., Rong Y., Baudry M. (2000) Neurosci. Lett. 286, 149–153 [DOI] [PubMed] [Google Scholar]
- 7.Lu X., Wyszynski M., Sheng M., Baudry M. (2001) J. Neurochem. 77, 1553–1560 [DOI] [PubMed] [Google Scholar]
- 8.Siman R., Baudry M., Lynch G. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 3572–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bi X., Chen J., Dang S., Wenthold R. J., Tocco G., Baudry M. (1997) J. Neurochem. 68, 1484–1494 [DOI] [PubMed] [Google Scholar]
- 10.Bi X., Rong Y., Chen J., Dang S., Wang Z., Baudry M. (1998) Brain Res. 790, 245–253 [DOI] [PubMed] [Google Scholar]
- 11.Guttmann R. P., Baker D. L., Seifert K. M., Cohen A. S., Coulter D. A., Lynch D. R. (2001) J. Neurochem. 78, 1083–1093 [DOI] [PubMed] [Google Scholar]
- 12.Guttmann R. P., Sokol S., Baker D. L., Simpkins K. L., Dong Y., Lynch D. R. (2002) J. Pharmacol. Exp. Ther. 302, 1023–1030 [DOI] [PubMed] [Google Scholar]
- 13.Lu X., Rong Y., Bi R., Baudry M. (2000) Brain Res. 863, 143–150 [DOI] [PubMed] [Google Scholar]
- 14.Simpkins K. L., Guttmann R. P., Dong Y., Chen Z., Sokol S., Neumar R. W., Lynch D. R. (2003) J. Neurosci. 23, 11322–11331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H. Y., Yuen E. Y., Lu Y. F., Matsushita M., Matsui H., Yan Z., Tomizawa K. (2005) J. Biol. Chem. 280, 21588–21593 [DOI] [PubMed] [Google Scholar]
- 16.Yuen E. Y., Gu Z., Yan Z. (2007) J. Physiol. 580, 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong H., Boulter J., Weber J. L., Lai C., Chao M. V. (2001) J. Neurosci. 21, 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iglesias T., Cabrera-Poch N., Mitchell M. P., Naven T. J., Rozengurt E., Schiavo G. (2000) J. Biol. Chem. 275, 40048–40056 [DOI] [PubMed] [Google Scholar]
- 19.Chao M. V. (1992) Cell 68, 995–997 [DOI] [PubMed] [Google Scholar]
- 20.Wu S. H., Arévalo J. C., Sarti F., Tessarollo L., Gan W. B., Chao M. V. (2009) Dev. Neurobiol. 69, 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cortés R. Y., Arévalo J. C., Magby J. P., Chao M. V., Plummer M. R. (2007) Dev. Neurobiol. 67, 1687–1698 [DOI] [PubMed] [Google Scholar]
- 22.Turrigiano G. G., Leslie K. R., Desai N. S., Rutherford L. C., Nelson S. B. (1998) Nature 391, 892–896 [DOI] [PubMed] [Google Scholar]
- 23.Bartel D. P., Sheng M., Lau L. F., Greenberg M. E. (1989) Genes Dev. 3, 304–313 [DOI] [PubMed] [Google Scholar]
- 24.Rosen L. B., Ginty D. D., Weber M. J., Greenberg M. E. (1994) Neuron 12, 1207–1221 [DOI] [PubMed] [Google Scholar]
- 25.Sheng M., McFadden G., Greenberg M. E. (1990) Neuron 4, 571–582 [DOI] [PubMed] [Google Scholar]
- 26.Malgaroli A., Tsien R. W. (1992) Nature 357, 134–139 [DOI] [PubMed] [Google Scholar]
- 27.Yi J. J., Ehlers M. D. (2005) Neuron 47, 629–632 [DOI] [PubMed] [Google Scholar]
- 28.Jia J. M., Chen Q., Zhou Y., Miao S., Zheng J., Zhang C., Xiong Z. Q. (2008) J. Biol. Chem. 283, 21242–21250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bi R., Bi X., Baudry M. (1998) Brain Res. 797, 154–158 [DOI] [PubMed] [Google Scholar]
- 30.Battaglia F., Trinchese F., Liu S., Walter S., Nixon R. A., Arancio O. (2003) J. Mol. Neurosci. 20, 357–362 [DOI] [PubMed] [Google Scholar]
- 31.Li X., Chen H., Jeong J. J., Chishti A. H. (2007) Mol. Biochem. Parasitol. 155, 26–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trinchese F., Fa' M., Liu S., Zhang H., Hidalgo A., Schmidt S. D., Yamaguchi H., Yoshii N., Mathews P. M., Nixon R. A., Arancio O. (2008) J. Clin. Invest. 118, 2796–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.López-Menéndez C., Gascón S., Sobrado M., Vidaurre O. G., Higuero A. M., Rodríguez-Peña A., Iglesias T., Díaz-Guerra M. (2009) J. Cell Sci. 122, 3554–3565 [DOI] [PubMed] [Google Scholar]
- 34.Arévalo J. C., Wu S. H., Takahashi T., Zhang H., Yu T., Yano H., Milner T. A., Tessarollo L., Ninan I., Arancio O., Chao M. V. (2010) Mol. Cell. Neurosci. 45, 92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutachan J. J., Chao M. V., Ninan I. (2010) J. Neurosci. Res. 88, 3447–3456 [DOI] [PubMed] [Google Scholar]
- 36.Shimizu K., Phan T., Mansuy I. M., Storm D. R. (2007) Cell 128, 1219–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zadran S., Jourdi H., Rostamiani K., Qin Q., Bi X., Baudry M. (2010) J. Neurosci. 30, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H. Y., Hsu F. C., Gleichman A. J., Baconguis I., Coulter D. A., Lynch D. R. (2007) J. Biol. Chem. 282, 20075–20087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuen E. Y., Liu W., Yan Z. (2007) J. Biol. Chem. 282, 16434–16440 [DOI] [PubMed] [Google Scholar]
- 40.Neubrand V. E., Thomas C., Schmidt S., Debant A., Schiavo G. (2010) J. Cell Sci. 123, 2111–2123 [DOI] [PubMed] [Google Scholar]
- 41.Park H. J., Park H. W., Lee S. J., Arevalo J. C., Park Y. S., Lee S. P., Paik K. S., Chao M. V., Chang M. S. (2010) Mol. Cell 30, 143–148 [DOI] [PubMed] [Google Scholar]
- 42.Arévalo J. C., Yano H., Teng K. K., Chao M. V. (2004) EMBO J. 23, 2358–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]