

Abstract
Hsp90 is an ATP-dependent molecular chaperone, which facilitates the activation and stabilization of hundreds of client proteins in cooperation with a defined set of cofactors. Many client proteins are protein kinases, which are activated and stabilized by Hsp90 in cooperation with the kinase-specific co-chaperone Cdc37. Other Hsp90 co-chaperones, like the ATPase activator Aha1, also are implicated in kinase activation, and it is not yet clear how Cdc37 is integrated into Hsp90 co-chaperone complexes. Here, we studied the interaction between Cdc37, Hsp90, and other Hsp90 co-chaperones from the nematode Caenorhabditis elegans. Nematode Cdc37 binds with high affinity to Hsp90 and strongly inhibits the ATPase activity. In contrast to the human Hsp90 system, we observed binding of Cdc37 to open and closed Hsp90 conformations, potentially reflecting two different binding modes. Using a novel ultracentrifugation setup, which allows accurate analysis of multifactorial protein complexes, we show that cooperative and competitive interactions exist between other co-chaperones and Cdc37-Hsp90 complexes in the C. elegans system. We observed strong competitive interactions between Cdc37 and the co-chaperones p23 and Sti1, whereas the binding of the phosphatase Pph5 and the ATPase activator Aha1 to Cdc37-Hsp90 complexes is possible. The ternary Aha1-Cdc37-Hsp90 complex is disrupted by the nucleotide-induced closing reaction at the N terminus of Hsp90. This implies a carefully regulated exchange process of cofactors during the chaperoning of kinase clients by Hsp90.
Keywords: ATPases, C. elegans, Chaperone Chaperonin, Protein Chemistry, Protein Kinases, AUC, Cdc37, Hsp90, Co-chaperones, Ternary Complexes
Introduction
The ATP-dependent molecular chaperone Hsp902 is a ubiquitous protein that interacts with a large number of client proteins, conferring activity or stability to them (1–4). The process of client protein activation of this chaperone requires the hydrolysis of ATP (5, 6). The hydrolysis energy is assumed to be transmitted to the client proteins via conformational changes within Hsp90 (7, 8). Hsp90 contains an N-terminal nucleotide-binding domain (NBD),3 a middle domain, and a C-terminal dimerization domain. A cyclical reaction pathway, in which Hsp90 goes from an open to a closed conformation, has been proposed for ATP hydrolysis. During this reaction cycle, the two N-terminal domains approach each other and thereby activate the hydrolysis reaction (9, 10). The interaction of the N-terminal domains is initiated by a lid segment (ATP-lid), which closes over the nucleotide-binding pocket and subsequently exposes the N-terminal interaction surfaces (11–15). This model has been derived from extensive studies of yeast and human Hsp90 proteins and is believed to be conserved for all Hsp90s (14, 16, 17).
Co-chaperone regulation is an important feature of Hsp90 function, and a defined set of co-chaperones has been described for eukaryotic Hsp90 proteins (18). Inhibitory effects on the turnover rate of Hsp90 have been documented for the co-chaperones Sba1/p23 (9, 19, 20), Sti1/Hop (21, 22), and Cdc37 (23–25), whereas Aha1 and its homolog Hch1 are the only known activating cofactors to date (26–28). Interestingly, remarkable differences have been observed between the yeast and human systems with respect to the binding affinity of these cofactors and, more importantly, regarding their inhibitory potential. For example, Sba1/p23 inhibits the ATPase activity of human Hsp90 (29) but not of yeast Hsp90 (19, 30). Sti1/Hop inhibits the ATPase activity of yeast (21) but not of human Hsp90 (29). These differences add to the vastly different ATPase rates of human and yeast Hsp90 proteins, with human Hsp90 hydrolyzing only one ATP molecule every 20 min (29) and yeast Hsp90 being 20 times more active (5). It is generally assumed that each co-chaperone recognizes a specific conformation of Hsp90 and thus influences its conformational cycle in a defined way. Sti1/Hop binds the open conformation of Hsp90 (22), whereas Sba1/p23 recognizes an N-terminally dimerized state and remains bound to Hsp90 during the hydrolysis event (12, 19, 31). Aha1 accelerates the formation of the N-terminally dimerized conformation (28, 32, 33). It is well documented that the cofactors Sba1/p23 and Sti1/Hop regulate the Hsp90-dependent activation process of the steroid hormone receptors by interacting at defined stages of the “chaperone cycle” (34–36).
For Cdc37, this information is mostly lacking. Cdc37 was identified as an essential component in Hsp90-kinase complexes (37–42). Although the human Cdc37-Hsp90 complex can be assembled in vitro, the weak ATP hydrolysis activity of human Hsp90 makes mechanistic studies difficult (23, 25). In the yeast system, however, the interaction between Cdc37 and Hsp90 can hardly be detected due to a binding constant of about 100 μm (23). Therefore, most biochemical data were obtained in systems composed of human Cdc37 and yeast Hsp90. There, human Cdc37 binds and inhibits the ATPase activity of the chaperone (23). Structural studies demonstrated that Cdc37 binds to the ATP-lid within the NBD of Hsp90, suggesting that the ATP lid is then unable to perform the movements involved in ATP turnover (24, 43). In addition, it was shown that the binding of Cdc37 to Hsp90 may also involve the middle domain (25).
Like the binding properties, also the regulation of Cdc37-Hsp90 complexes by nucleotides and other cofactors seems to be different in the yeast and human systems. ATP analogs disrupt Cdc37-Hsp90 complexes in the human system (44) but not in yeast (20, 45). In addition, the TPR domain-containing cofactors PP5, Hop, and FKBP52 block binding of Cdc37 to human Hsp90 (46, 47). In contrast, the yeast PP5 homolog Ppt1 was found to be a cofactor in ternary Ppt1-Cdc37-Hsp90 complexes during the chaperoning of kinase client proteins (48). Furthermore, Cdc37 directly interacts with Sti1 in yeast (45, 49), and ternary Cdc37-Sti1-Hsp90 complexes have been proposed in which one co-chaperone connects the other to the complex (45). Also, gel filtration experiments have shown that the simultaneous binding of Aha1 and Cdc37 to Hsp90 is not possible but have suggested a potential Cdc37-p23-Hsp90 complex (45). In contrast, other studies employing differential CD spectroscopy have demonstrated Aha1 and Cdc37 binding together to Hsp90, whereas p23 interaction is mutually exclusive with Cdc37 binding (20). These studies used mixed systems of human Cdc37 and yeast Hsp90.
These differences between eukaryotic Hsp90 systems and the need to use mixed yeast/human systems in many studies prompted us to investigate the biochemistry of the Hsp90 system in Caenorhabditis elegans. This system allows characterization of the enzymatic activity of Hsp90 and its interaction with endogenous cofactors (50). The sequence homology of the nematode proteins is closer to the mammalian proteins than to the yeast proteins (Table 1). To study the formation of Cdc37-containing protein complexes, we employed a novel fluorescence detection system during analytical ultracentrifugation that allowed us to accurately resolve the ability of different co-chaperones to simultaneously assemble on the Hsp90 scaffold. We show that the formation of Cdc37-Hsp90 complexes is influenced by other co-chaperones and regulated by conformational changes within Hsp90.
TABLE 1.
Proteins of the C. elegans Hsp90 system and the respective homologs
The percentage of sequence identity to the nematode homolog is shown in parentheses and was determined using the SIM alignment tool with default settings. The C. elegans proteins are abbreviated throughout for clarity by omitting the hyphen (utilized name/official name).
| C. elegans | Saccharomyces cerevisiae | Homo sapiens |
|---|---|---|
| Hsp90/DAF-21 | Hsp90 (62.9%) | Hsp90α (75.4%) |
| Cdc37/CDC-37 | Cdc37 (26.4%) | Cdc37 (39.8%) |
| Aha1/C01G10.8 | Aha1 (21.1%) | Aha1 (36.8%) |
| p23/ZC395.10 | Sba1 (35.8%) | p23 (28.2%) |
| Sti1/STI-1 | Sti1 (41.2%) | Hop (56.4%) |
| Pph5/PPH-5 | Ppt1 (39.2%) | PP5 (59.3%) |
EXPERIMENTAL PROCEDURES
Cloning, Protein Expression, and Protein Purification
The C. elegans homolog proteins of Hop/Sti1 (STI-1, R09E12.3) Hsp90 (DAF-21, C47E8.5), the N-terminal TPR domain of nematode Sti1, and Hsp90-ΔMEEVD were generated as described previously (50). Expression constructs for the C. elegans homologs of p23/Sba1 (ZC395.10), Aha1 (C01G10.8), Cdc37 (CDC-37, W08F4.8), Aha1-N (from amino acid 1 to 179), and PP5/Ppt1 (PPH-5, Y39B6A.2) were generated using full-length cDNA containing plasmids as templates (Open Biosystems, Huntsville, AL). PCR products were subcloned into the pET28b expression plasmid (Merck), generating fusions with an N-terminal His6 tag. The F8A, F122A, and F340E mutants of nematode Hsp90 were generated using site-directed mutagenesis protocols. Protein expression was performed in the E. coli strain BL21-CodonPlus(DE3)-RIL (Stratagene, La Jolla, CA) by inducing an exponentially growing culture with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Bacterial cells were harvested and lysed in a Constant Systems TS 0.75 cell disruption instrument (IUL Instruments, Koenigswinter, Germany). The cleared lysate was applied to a HisTrap FF 5-ml column (GE Healthcare). Protein was eluted in a buffer containing 300 mm imidazole and was further purified using a Resource Q 6-ml ion exchange column and a HiLoad 26/60 Superdex 75 prep grade gel filtration column (both from GE Healthcare). Protein purity was generally higher than 95% as judged by SDS-PAGE. The molecular mass was confirmed by MALDI-TOF/TOF mass spectrometry. Purified proteins were dialyzed against 40 mm HEPES/KOH, pH 7.5, 20 mm KCl, 0.2 mm EDTA and stored at −80 °C. If proteins contained cysteine residues, like Pph5, Sti1, Hsp90, and Cdc37, the storage buffer was supplemented with 1 mm DTT. Final protein concentrations were 55 μm for Hsp90, 100 μm for Aha1, 65 μm for p23, 220 μm for Cdc37, 45 μm for Sti1, and 60 μm for Pph5.
Protein Stability Measurements
The stability of the purified proteins was investigated because the physiological growth temperature of C. elegans is lower compared with the mammalian and yeast systems. Thermal unfolding transitions were recorded to determine the unfolding temperature of the C. elegans proteins p23, Aha1, Cdc37, and Pph5. Protein concentrations were 0.2 mg/ml in 10 mm KH2PO4, pH 7.5. Using CD spectroscopy, thermal denaturation transitions were recorded in a J-715 spectrophotometer (Jasco, Gross-Umstadt, Germany) from 10 to 90 °C with a heating rate of 30 °C/h. The wavelength was set to 220 nm. All proteins were stably folded at 30 °C, suggesting that an enzymatic analysis at 25 °C is appropriate for these proteins.
Determination of Oligomerization Properties by Analytical Ultracentrifugation
Analytical ultracentrifugation was used to determine the molecular weights of individual proteins. Samples contained a protein concentration of 0.5 mg/ml. Sedimentation experiments were performed in 350-μl samples at 42,000 rpm using an XL-I analytical ultracentrifuge (Beckman Coulter, Brea, CA) equipped with a UV-visible detection system. UV scans at 280 nm were recorded every 5 min. The sedimentation profiles of the individual proteins were fit to determine the sedimentation coefficient (s20,w), the diffusion coefficient (D20,w), and the corresponding molecular weight of the protein. Data analysis was carried out with the software UltraScan version 9.0 using the modules for “c(s) analysis” and “Finite Element (DUD)” analysis (51).
Furthermore, sedimentation equilibrium experiments were performed for Hsp90 cofactors at concentrations of 0.3 and 1 mg/ml at 4 °C. Protein samples of 250 μl were centrifuged at 15,000 rpm until a sedimentation equilibrium was reached. Data analysis was performed using the Origin 8G software (OriginLab Corp., Northampton, MA).
ATPase Activity Measurements
ATPase assays were performed using an ATP-regenerating system as described previously (5). An ATPase premix was generated using phosphoenolpyruvate, NADH, l-lactate dehydrogenase, and pyruvate kinase (Roche Applied Science) in a low salt buffer composed of 40 mm HEPES/KOH, pH 7.5, 5 mm MgCl2, and 20 mm KCl, unless indicated otherwise. Assays were performed at 25 °C. The Hsp90 concentration was 2.7 μm in reaction mixtures. The Hsp90 ATPase activity was specifically inhibited by using the Hsp90-targeting compound radicicol (Sigma) at concentrations of 40 μm. Background activities were subtracted. In cases where KD values for cofactor-Hsp90 interactions needed to be derived, variable concentrations of co-chaperone were added, and the resulting activities were analyzed as described previously (22).
Fluorescein-labeling of Proteins
Cdc37 was labeled at cysteine residues using Alexa Fluor 488 C5-maleimide (Invitrogen). With this aim, a 3-fold molar excess of dye was added to a Cdc37 sample in 40 mm HEPES/KOH, pH 7.5, 20 mm KCl. The reaction was allowed to proceed for 2 h at room temperature and was stopped by the addition of 1 mm DTT. Protein was separated from free label by size exclusion chromatography on a Superdex 75HR column (GE Healthcare) in 40 mm HEPES/KOH, pH 7.5, 300 mm KCl, 1 mm DTT. The labeling efficiency was determined using UV spectroscopy and the extinction coefficients provided by the manufacturer. The efficiency was 1.2 labels per Cdc37 molecule. The labeled protein is designated *Cdc37.
Fluorescence Detection during Analytical Ultracentrifugation
A novel AUC approach (52, 53) was employed to investigate the binding of Cdc37 to Hsp90. *Cdc37 was subjected to AUC in a Beckman XL-A analytical ultracentrifuge (Beckman Coulter, Brea, CA) equipped with an Aviv AU-FDS fluorescence detection system (Aviv Biomedical Inc., Lakewood, NJ). Sedimentation velocity experiments were performed in standard two-sector centerpieces in a Ti50 rotor at 42,000 rpm. Scans at an excitation wavelength of 488 nm were recorded in 90-s intervals. *Cdc37 showed unchanged sedimentation parameters compared with the unlabeled Cdc37.
Hsp90 and other cofactors were added at the indicated concentrations, and influences on the sedimentation behavior of *Cdc37 were recorded. The concentration of *Cdc37 was 150 nm, unless indicated otherwise. We determined binding constants by a titration procedure. Different concentrations of Hsp90 were added to *Cdc37, and the samples were sedimented at 42,000 rpm. Ultracentrifugation experiments were analyzed by subtracting nearby scans and converting the difference into Δc/Δt plots according to the method described by Stafford (54). The plots generally correlated with those from the SEDVIEW dc/dt program (55). In cases where the temperature deviated from 20 °C, we performed a temperature correction of the sedimentation coefficients. Data in Δc/Δt plots were then fit to bi-Gaussian functions using the ORIGIN 8G software package in order to determine the s20,w values and the corresponding peak amplitude for free *Cdc37 (Peak 1, [Cdc37]) and for complexed *Cdc37 (Peak 2, [Cdc37-Hsp90]). Using several different Hsp90 concentrations, we determined the binding constant of the *Cdc37-Hsp90 interaction by fitting the reduction in Peak1 and the increase in Peak2. The values A1 and A2 in Equations 2 and 3 are proportionality factors, which convert the peak amplitude to the species concentration. Cdc370 and Hsp900 correspond to the total added concentration of Cdc37 and Hsp90.
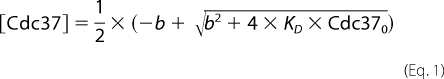 |
with b = Hsp900 + KD − Cdc370.
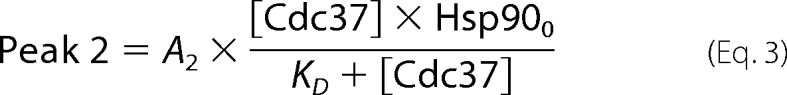 |
More complex binding reactions involving three proteins or competition experiments, including several cofactors, were analyzed qualitatively by comparing Δc/Δt plots of individual sedimentation experiments. It is important to note that the observed peaks in Δc/Δt plots represent reaction boundaries and thus do not give the s20,w values of defined and stable complexes. This is in particular true because the studied protein complexes are dynamic, and the protein concentrations generally are not high enough to ensure full complex formation for *Cdc37. Thus, a potentially reduced s20,w value of the respective ternary protein complex is observed. Quantitative analysis of the species distribution also was performed using the c(s) analysis module of UltraScan (51) because this method provides percentage values for each species detected in the protein sample. Because the fluorescence properties of *Cdc37 did not change during binding events, the species were grouped according to their s20,w values into a “free Cdc37” group and a “complexed Cdc37” group. Generally, deviations were in the range of 2–4 percentage points for the amounts of different species. Also, the standard deviation of the determined s20,w values was ∼0.2 S. Reproducibility of the effects was determined by at least three independent runs for each experiment.
Surface Plasmon Resonance Spectroscopy
The interaction between Aha1 and Hsp90 was recorded by surface plasmon resonance spectroscopy using a Biacore X instrument (GE Healthcare). About 1500 resonance units of Aha1 were coupled at lysine residues to the surface of a CM5 sensor chip using NHS/EDC. The coupling buffer was 40 mm acetate, pH 5. Hsp90-containing protein samples were injected in 40 mm HEPES/KOH, pH 7.5, 80 mm KCl, 5 mm MgCl2. The binding kinetics of Hsp90 was recorded in the absence and presence of 2 mm concentrations of different nucleotides.
RESULTS
C. elegans Cdc37 Inhibits the ATPase Activity of Hsp90
The interaction between Hsp90 and Cdc37 varies strongly between the yeast and human proteins, with the affinity of the yeast complex being about 40-fold weaker (20, 23, 25). Because the activity of human Hsp90 is hardly detectable (14, 29), most studies rely on the interaction between human Cdc37 and yeast Hsp90 in order to analyze the influence on ATP turnover. We recently reported a high ATPase activity of the nematode Hsp90 protein (50), which allows analysis of the regulation of Hsp90 by Cdc37 in the C. elegans system. We purified C. elegans Cdc37 and observed a strong inhibition of the ATPase activity of Hsp90 (Fig. 1A). The turnover rate of the saturated Hsp90-Cdc37 complex was reduced by >85%. The apparent KD for the Hsp90-Cdc37 interaction was found to be 6 ± 2 μm in standard low salt buffer (Fig. 1A). The inhibition was much weaker at high salt concentrations (KD >20 μm; Fig. 1A). Thus, the binding of Cdc37 to Hsp90 in C. elegans appears to be ∼15-fold stronger than the binding of yeast Cdc37 to yeast Hsp90 (KD ∼100 μm) (20). This implies that, due to the higher affinity interaction and the high ATPase activity of nematode Hsp90, the C. elegans system can be used to analyze the mechanism of Cdc37 action on Hsp90.
FIGURE 1.

Cdc37 binds to Hsp90 and inhibits its ATPase activity. A, Hsp90 ATPase activities were measured with increasing concentrations of Cdc37 in standard buffer with low salt (20 mm KCl) (○) or high salt concentration (150 mm KCl) (●). Data were analyzed as outlined under “Experimental Procedures.” B, individual scans of a sedimentation velocity experiment of 600 nm *Cdc37 in standard buffer containing low salt (20 mm KCl) were monitored by fluorescence detection at 42,000 rpm. C, individual scans of a sedimentation velocity experiment of 600 nm *Cdc37 in the presence of 2 μm Hsp90 in standard buffer containing low salt (20 mm KCl) were monitored by fluorescence detection at 42,000 rpm. D, sedimentation velocity experiments of 600 nm *Cdc37 containing 0, 2, 4, 7, and 10 μm Hsp90 were converted into Δc/Δt plots and fit as outlined under “Experimental Procedures.” E, quantitative evaluation of the amplitudes for free and complexed *Cdc37 obtained from the *Cdc37-Hsp90 titration. The amplitudes of free (●) and complexed *Cdc37 (▾) under low salt conditions (20 mm KCl; filled symbols) and high salt conditions (150 mm KCl; open symbols) are plotted against the Hsp90 concentration. Data analysis was performed as described under “Experimental Procedures.” a.u., arbitrary units. Error bars, S.D.
Analytical Ultracentrifugation Allows Quantification of the Hsp90-Cdc37 Complex Formation
In order to analyze the interaction between Cdc37 and Hsp90 directly, Cdc37 was labeled with Alexa Fluor 488 at cysteine residues (*Cdc37) and subjected to sedimentation velocity experiments in an analytical ultracentrifuge (Fig. 1B). We analyzed the sedimentation data for *Cdc37 using the UltraScan software package. The molecular weight derived from the sedimentation coefficient (s20,w = 2.9 ± 0.5 S) and the diffusion coefficient of the protein (D20,w = 5.9 × 10−7 ± 1.1 × 10−7 cm2 liter/mol) reveals that nematode Cdc37 is a monomeric protein (molecular mass ∼41 kDa). The monomeric organization was also confirmed by sedimentation equilibrium ultracentrifugation of non-labeled Cdc37 using a UV detector (data not shown). These data are unexpected, given that human Cdc37 was reported to be a dimer (24), albeit with a very weak dimerization constant of 80 ± 20 μm (25).
*Cdc37 sedimentation was then monitored in the presence of Hsp90. Here, the sedimentation profile showed the formation of a large protein complex (Fig. 1C). Using higher concentrations of Hsp90, we obtained full saturation of *Cdc37. Conversion of the sedimentation data into Δc/Δt plots clearly demonstrates the formation of complexes at 6.6 S (Fig. 1D). By analyzing the amplitudes of free *Cdc37 and complex-bound *Cdc37 for each Hsp90 concentration, we obtained a binding constant of 1.4 ± 0.4 μm at low salt conditions (Fig. 1E). Also, UltraScan c(s) analysis of the sedimentation velocity experiment revealed that, at the highest concentration of Hsp90, more than 90% of *Cdc37 was part of Hsp90 complexes. At high salt conditions (150 mm KCl), significantly higher concentrations of Hsp90 were required to achieve complex formation, and the KD was determined to be 14 ± 4 μm (Fig. 1E). These results confirm that the salt-dependent effects observed in the ATPase assays are due to a weaker binding affinity at high ionic strength (see Fig. 1A). Given the monomeric nature of Cdc37 and the large excess of Hsp90 in the AUC experiments, it can be assumed that the observed protein complexes contain one Cdc37 molecule per Hsp90 dimer.
Cdc37 Binding Is Enhanced by Nucleotide-induced Conformational Changes
Human Cdc37 had been shown to bind to the N-terminal NBD of Hsp90 (24, 25, 43). Two mutations that we generated in the NBD of C. elegans Hsp90 (F8A and F122A) prevented the formation of a *Cdc37-Hsp90 complex (data not shown), confirming that also in C. elegans the main interaction site for binding Cdc37 resides in the NBD of Hsp90, but there may also be additional sites in the full-length protein (25).
Next we used AUC to investigate how the Cdc37-Hsp90 complex is regulated by nucleotides because previous studies reported contradictory results for the yeast and human proteins (20, 44, 45). We performed sedimentation velocity experiments under weak binding conditions (150 mm KCl) in the absence and presence of nucleotides (Fig. 2A). The addition of ADP did not change the sedimentation compared with the nucleotide-free set-up (Fig. 2A). Both conditions resulted in about 14% of the *Cdc37 being part of complexes that sedimented at 6.5 S. However, the addition of the non-hydrolyzable ATP analog AMP-PNP and the slowly hydrolyzing variant ATPγS had strong effects on the sedimentation behavior. First, the ATP analogs increased the affinity between *Cdc37 and Hsp90 slightly, as implicated by a reduction in free *Cdc37 (at s20,w = 2.9 S) and an increase of *Cdc37 as part of oligomeric protein complexes from 14 to 26% (Fig. 2A). Second, the sedimentation of the *Cdc37-Hsp90 complex fraction was faster in the presence of the nucleotide analogs (s20,w = 7.3 S versus s20,w = 6.5 S). Similar effects were also observed at low salt conditions (data not shown). This sedimentation behavior implies that the nucleotides alter the shape of the *Cdc37-containing protein complex. Recently, it was observed that the addition of AMP-PNP or ATPγS increases the sedimentation coefficient of yeast Hsp90 from 5.6 to 6.8 S, reflecting the nucleotide-induced closing of the N-terminal domains in yeast Hsp90 (8). It is therefore reasonable to assume that the nucleotide-induced closing of the N-terminal domains leads to an increase in the affinity of *Cdc37 for Hsp90. Thus, Cdc37 interacts both with the open and with the closed form of Hsp90.
FIGURE 2.

Cdc37 binds open and closed Hsp90 conformations. A, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37 in standard buffer containing high salt (150 mm KCl) in the absence (black) or presence of 4 μm Hsp90 (blue). The influence of nucleotides was detected by the addition of 4 mm AMP-PNP (red) or 4 mm ADP (green) to 150 nm *Cdc37 and 4 μm Hsp90. Data fitting was performed as described under “Experimental Procedures.” B, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37 and 2 μm F340E Hsp90 in standard buffer containing low salt (20 mm KCl) in the absence of nucleotides (blue) or in the presence of 4 mm AMP-PNP (green) or 4 mm ATPγS (red). The schemes represent the proposed conformations of Hsp90, with *Cdc37 depicted in green.
To test this notion, we generated an Hsp90 mutant, which has an altered interface between the NBD and the middle domain and is therefore unable to respond to nucleotide binding. We substituted the phenylalanine at position 340 to glutamate because a corresponding change in yeast Hsp90 at this site had been found to inhibit the closing reaction (20). In the absence of nucleotides, the complex formation between *Cdc37 and F340E Hsp90 was not influenced by the mutation (Fig. 2B). However, both nucleotide-dependent effects were dramatically altered. First, the shift of the complex peak to a higher s20,w value in response to AMP-PNP or ATPγS was no longer observed. Furthermore, both nucleotides weakened rather than strengthened the interaction between *Cdc37 and Hsp90 (Fig. 2B). UltraScan c(s) analysis revealed that the amount of Hsp90-bound *Cdc37 decreased from 65 to 48% in the presence of AMP-PNP and even further to 36% in the presence of ATPγS. These data suggest that the interaction between Cdc37 and Hsp90 is influenced by nucleotide-dependent rearrangements that ultimately lead to an enhanced binding affinity of Cdc37 to a closed conformation of the chaperone.
Sti1 Competes with Cdc37 for Binding to the Open Conformation of Hsp90
It is important to understand how Cdc37 is integrated into the network of Hsp90 co-chaperones, especially because Cdc37 apparently binds to the open and closed conformation of Hsp90. The AUC assay based on *Cdc37 sedimentation is perfectly suited to address the formation of these multimeric protein assemblies under conditions that can be quantitatively evaluated. We initially tested the influence of the cofactor Sti1, an Hsp90 ATPase inhibitor that binds the open conformation of Hsp90 (22), on *Cdc37 complexes with Hsp90. In the absence of Sti1, 57% of *Cdc37 was part of Hsp90-containing complexes at 6.5 S. These complexes completely disappeared when Sti1 was present and only unbound *Cdc37 was detectable (Fig. 3A). Apparently, binding of Sti1 to Hsp90 expels *Cdc37 from Hsp90 complexes. To determine the specificity of this effect, we replaced wild-type Hsp90 with Hsp90-ΔMEEVD because this variant is not capable of binding Sti1 and other TPR-containing cofactors due to a deletion of the TPR-interacting MEEVD motif at the C-terminal end (56, 57). Here, no changes upon the addition of Sti1 could be observed, confirming that the interaction between Sti1 and the MEEVD motif of Hsp90 is critically important for this competition to occur (Fig. 3A).
FIGURE 3.

Influence of the Hsp90 cofactors Sti1, p23, and Pph5 on the Cdc37-Hsp90 complex. A, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37 in standard buffer containing low salt (20 mm KCl) in the absence (black) or in the presence of 2 μm Hsp90 (blue). 4 μm Sti1 was either added to wild-type Hsp90 (red) or to Hsp90-ΔMEEVD (green). B, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37 in standard buffer containing low salt (20 mm KCl) in the presence of 2 μm Hsp90 without nucleotide (blue), with 4 mm ATPγS (green), or with 4 mm ATPγS and 5 μm Cep23 (red). C, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37 in low salt buffer (20 mm KCl) in the absence (black) or in the presence of 2 μm Hsp90 (blue). 4 μm Pph5 was added either to wild-type Hsp90 (red) or to Hsp90-ΔMEEVD (green). The schemes represent the proposed conformations of Hsp90, with *Cdc37 depicted in green and Pph5 in blue.
It is remarkable that Sti1 binding at the C terminus of Hsp90 can inhibit binding of another protein to the N-terminal domain. Accordingly, a second binding interface with Hsp90 had been proposed for Sti1, which potentially resides in the N-terminal domain of Hsp90 (21, 22) and might be recognized by the second TPR domain of Sti1 (50). Indeed, when we deleted the C-terminal TPR domain of Sti1, we did not observe the disruption of *Cdc37-Hsp90 complexes (data not shown). This shows that the competition with Cdc37 binding requires the C-terminal TPR domain of Sti1, which also is required for the inhibition of the Hsp90 ATPase (50). It is important to note that the Sti1 proteins of nematode species lack the TPR1 domain of other eukaryotic species, which had been implicated in a direct interaction with Cdc37 before (49).
p23 Displaces Cdc37 from the Closed Conformation of Hsp90
p23 is an important ATPase regulatory cofactor during later stages of the chaperone cycle. It is thought to exclusively bind to closed Hsp90 conformations (12, 58–60). Thus, the interaction between Cdc37 and p23 could be either synergistic or mutually exclusive. Two previous studies performed on yeast Hsp90 bound to human Cdc37 employed either CD spectroscopy (20) or size exclusion chromatography (45) and had obtained opposing results regarding this question.
If nucleotides were omitted, the plots of *Cdc37-Hsp90 complexes in the presence and absence of p23 were identical, implying that no interaction of p23 occurred in the absence of nucleotides (data not shown). We then tested complex formation in the presence of ATPγS (Fig. 3B). As described before (see Fig. 2A), the addition of ATPγS alone shifted the *Cdc37-Hsp90 complex fraction from 6.6 to 7.5 S, representing the closing of the N-terminal domains. In addition, the affinity of *Cdc37 for Hsp90 increased slightly, with 83% of *Cdc37 being bound to Hsp90 versus 66% in the absence of ATPγS (Fig. 3B). However, in the presence of excess p23, the complex between *Cdc37 and Hsp90 was disrupted, and most *Cdc37 sedimented in a manner indistinguishable from that of the monomeric protein (Fig. 3B). *Cdc37-Hsp90 complexes accounted for less than 4% of the species distribution according to c(s) analysis.
To determine the influence of Hsp90 rearrangements on the competition between p23 and Cdc37, we used the F340E Hsp90 mutant, which is incapable of forming the closed conformation. As expected, the addition of p23 did not lead to any additional displacement of *Cdc37 beyond that already observed for the addition of ATPγS or AMP-PNP (data not shown; for comparison, see Fig. 2B). Therefore, the ability of Hsp90 to form the closed conformation is critically required for the cofactor p23 to compete with *Cdc37 at the N-terminal domains of Hsp90. Thus, it is interesting to note that, besides the early acting cofactor of the steroid hormone receptor assembly pathway, Sti1, also the late acting cofactor p23 strongly competes with the kinase-specific cofactor Cdc37.
Complexes Are Formed between Hsp90, Cdc37, and the TPR-containing Co-chaperone Pph5
We then tested cofactors that participate in kinase activation in vivo. As such, the yeast homolog of protein phosphatase 5, Ppt1, had recently been shown to be part of Cdc37-Hsp90 complexes (48) and to be important for activation of the kinase v-Src (61). In contrast, earlier studies in the human Hsp90 system had found all TPR proteins, including the Ppt1 homolog PP5, in competition with Cdc37 (46). Given the conflicting results, we set out to understand the influence of the nematode homolog Pph5 on Cdc37 binding to Hsp90.
We analyzed the formation of *Cdc37-Hsp90 complexes in the presence and absence of Pph5. Although about 57% of *Cdc37 was integrated in 6.5 S Hsp90-containing complexes in the absence of Pph5, these complexes changed dramatically when Pph5 was added (Fig. 3C). The average complex size increased to 8.3 S, demonstrating that Pph5 bound in addition to Cdc37 to the Hsp90-containing complexes (Fig. 3C). The amount of free *Cdc37 was not altered significantly, with 43% free *Cdc37 versus 46% in the presence of Pph5, implying that the binding affinity of *Cdc37 to Hsp90 was not affected. Thus, the two co-chaperones bind independently to the Hsp90 scaffold. We also wanted to confirm that the interaction of Pph5 with the *Cdc37-Hsp90 complexes is specific and utilizes the Hsp90 scaffold as the linking protein. We therefore investigated this interaction using Hsp90-ΔMEEVD (56, 57). As expected, the formed complexes did not show a shift in the s20,w value in the presence of Pph5, proving that binding of Pph5 to *Cdc37-Hsp90 complexes requires the interaction of the TPR domain with the MEEVD motif (Fig. 3C). Thus, in the nematode system, Pph5 has the capability of forming ternary complexes with Cdc37 and Hsp90. The broad complex peak and the strong shift to higher s20,w values in response to Pph5 binding may imply that these complexes can contain either one or two Pph5 molecules.
Aha1 Forms Ternary Complexes with Cdc37 and Hsp90 in the Absence of Nucleotides
The divergent behavior of the cofactors Sti1, p23, and Pph5 prompted us to investigate the influence of the ATPase activator Aha1 on Cdc37-Hsp90 complexes. Aha1 is involved in kinase maturation in yeast, although its function in this context is not clear (26, 27). We purified C. elegans Aha1 and found it capable of activating the ATPase activity of Hsp90 with an apparent binding constant of 14.6 ± 5.2 μm (Fig. 4A). The addition of a large excess of Cdc37 decreased this stimulatory effect only moderately (Fig. 4A). This is surprising because Cdc37 is an ATPase inhibitor with similar affinity toward Hsp90. It is therefore important to understand whether Aha1 replaces Cdc37 or whether it alters the ATPase activity in the ternary complex. We employed the AUC-based interaction assay to gain insight into the *Cdc37-Hsp90 complex formation in the presence of Aha1 (Fig. 4B). Comparison of the Δc/Δt plots of the sedimentation runs revealed that two events occurred. First, the amount of uncomplexed *Cdc37 increased more than 3-fold in the presence of Aha1, demonstrating that the ability of *Cdc37 to bind to Hsp90 is significantly reduced by Aha1 (Fig. 4B). According to c(s) analysis, only about 20% of the *Cdc37 was observed in larger complexes, whereas 71% had been part of Hsp90 complexes in the absence of Aha1. Also, the properties of the complexed fraction changed in the presence of Aha1; the s20,w value of the complex peak increased from 6.6 to 7.9 S, which clearly shows that this fraction of *Cdc37 now is part of larger complexes, which have to contain *Cdc37, Aha1, and Hsp90. Gel filtration experiments had suggested a competition between Aha1 and Cdc37 before (45), whereas difference circular dichroism studies implied simultaneous binding of Aha1 and Cdc37 to Hsp90 (20). The AUC-based assay shows that in the nematode system, Aha1 binds to Cdc37-Hsp90 complexes, although it reduces the affinity of Cdc37 for Hsp90.
FIGURE 4.

Interaction of Aha1 with Cdc37-Hsp90 complexes. A, stimulation of the ATPase activity of Hsp90 was determined in low salt buffer (20 mm KCl) with increasing amounts of Aha1 either in the absence of Cdc37 (●) or in the presence of 10 μm Cdc37 (○). Data were analyzed as described under “Experimental Procedures.” B, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37, either in the absence (black) or in the presence of 3 μm Hsp90 (blue). 5 μm Aha1 was added to 150 nm *Cdc37 and 3 μm Hsp90, either without the addition of nucleotide (red) or with the addition of 4 mm AMP-PNP (green). Experiments were performed in standard buffer containing low salt (20 mm KCl). The schemes represent the proposed conformations of Hsp90, with *Cdc37 depicted in green and Aha1 in red. C, surface plasmon resonance analysis of the interaction of chip-bound Aha1 with Hsp90. Experiments were performed in standard buffer containing 80 mm KCl. The interaction of Hsp90 with immobilized Aha1 was studied either in the absence of nucleotides (black) or in the presence of 2 mm AMP-PNP (red), 2 mm ATPγS (green), or 2 mm ADP (blue). a.u., arbitrary units. Error bars, S.D.
We further addressed how this complex formation is regulated by nucleotides. We were surprised to see that the addition of AMP-PNP or ATPγS to the same complexes led to full displacement of *Cdc37 by Aha1 (Fig. 4B). The addition of ADP, instead, had no effect (data not shown). The displacement of *Cdc37 is unexpected because *Cdc37 in principle favors the closed conformation of Hsp90 (see Fig. 2A). These results therefore suggest that Aha1 as well binds with higher affinity to Hsp90 in the presence of AMP-PNP or ATPγS. We performed a surface plasmon resonance-based interaction assay to confirm the nucleotide dependence of the Aha1-Hsp90 interaction. We coupled Aha1 to the surface of a CM5 sensor chip and recorded the binding kinetics of Hsp90 in the presence of different nucleotides. Binding and dissociation kinetics in the absence of nucleotides were fast (Fig. 4C). The addition of ADP to the solution did not alter the binding properties, whereas the addition of AMP-PNP and ATPγS led to stronger binding and a drastically reduced dissociation rate (Fig. 4C). Thus, ATP analogs shift the binding properties to favor the binding of Aha1, leading to the full displacement of Cdc37 from Hsp90-Aha1 complexes.
Full Replacement of Cdc37 Requires the C-domain of Aha1 and Conformational Changes within Hsp90
We were intrigued by the mechanistic aspects of the Aha1-Hsp90-Cdc37 interaction. Recent studies demonstrated the importance of the two-domain structure of Aha1 for the stimulation of the ATPase of Hsp90. The N-terminal domain of Aha1 (Aha1-N) was found to contact the middle domain, and Aha1-C was found to contact the NBD of Hsp90 (32, 33).
We purified the N-terminal domain of nematode Aha1 (Aha1-N) and investigated its influence on the ATPase activity of Hsp90. Aha1-N was severely compromised in its ability to stimulate the Hsp90 ATPase activity (data not shown). However, Aha1-N could bind to Hsp90, as shown by competition experiments with full-length Aha1 in which a marked decrease of the stimulated ATPase activity was observed (Fig. 5A). We used the AUC assay to examine whether Aha1-N is capable of displacing *Cdc37 from Hsp90 complexes. Applying the same conditions as before, we again observed a reduction in the amount of bound *Cdc37 in the presence of Aha1-N (Fig. 5B). However, in contrast to full-length Aha1, we did not observe the full displacement of *Cdc37 after the addition of the ATP analog AMP-PNP (Fig. 5B) or ATPγS (data not shown). Instead, we recorded a shift to higher s20,w values and similar amounts (19% versus 17% bound) of *Cdc37 as a part of protein complexes. Therefore, the N-terminal domain of Aha1 alone cannot fully displace Cdc37 from the Hsp90 scaffold. However, its binding to Hsp90 severely reduces complex formation between Cdc37 and Hsp90, although the binding sites of Cdc37 and Aha1-N reside in different domains.
FIGURE 5.

Cdc37 displacement from the Hsp90 scaffold is dependent on the Aha1 C-domain and requires conformational changes within the Hsp90 dimer. A, the competition experiment between the N-terminal domain of Aha1 and full-length Aha1 was performed by adding increasing concentrations of Aha1-N to Hsp90 stimulated with 2 μm Aha1. Assays were performed in standard buffer containing low salt (20 mm KCl). B, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37 in the presence of 2 μm Hsp90 (blue). 5 μm Aha1-N was added to 150 nm *Cdc37 and 2 μm Hsp90 either in the absence (red) or in the presence of 4 mm AMP-PNP (green). Experiments were performed in standard buffer containing low salt (20 mm KCl). C, Δc/Δt plots were generated from sedimentation velocity experiments of 150 nm *Cdc37 in the presence of 2 μm F340E Hsp90 without cofactors (blue), with 5 μm Aha1 (red), or with 5 μm Aha1 and 4 mm AMP-PNP (green). The schemes represent the proposed conformations of Hsp90, with *Cdc37 depicted in green and Aha1 in red. Error bars, S.D.
To address whether nucleotide-induced conformational changes at the N-M interface are required for the observed effects in the presence of Aha1, we again utilized F340E Hsp90. We formed complexes of *Cdc37 and F340E Hsp90, leading to the sedimentation of *Cdc37-containing species with an s20,w value of 6.5 S in the absence of Aha1 (Fig. 5C). UltraScan c(s) analysis determined the amount of *Cdc37 in Hsp90 complexes to be 61% under the conditions used. In the presence of Aha1, we observed an increase in unbound *Cdc37 (from 39 to 81%) concomitant with an increase in the observed sedimentation coefficient of the remaining complex-bound *Cdc37 to 7.9 S (Fig. 5C). This behavior is similar to the results obtained before for wild-type Hsp90. For the F340E Hsp90 mutant, however, the addition of AMP-PNP (Fig. 5C) or ATPγS (data not shown) did not result in the disappearance of the complexed fraction, and its amount decreased only marginally from 19 to 14%. We therefore conclude that the nucleotide-induced closing reaction of Hsp90 and the C-terminal domain of Aha1 are required for the full displacement of Cdc37 from Hsp90 complexes in the presence of AMP-PNP or ATPγS.
DISCUSSION
Interaction of Cdc37-Hsp90 Complexes with Nucleotides and Other Hsp90 Cofactors
Formation of protein complexes between the molecular chaperone Hsp90 and its regulatory cofactors is an important part of its functional cycle. In many cases, these cofactors bind in a conformation-sensitive manner to Hsp90 and contribute to the maturation process of Hsp90 clients at different stages. This concept has been developed mainly for steroid hormone receptors as Hsp90 clients, where the participation of several cofactors, including Sti1/Hop and p23, is required to achieve the active hormone binding conformation (36, 62).
Here we set out to define the potential cooperative and competitive cofactors for the kinase-specific Hsp90 partner protein Cdc37. Our results show that the co-chaperones p23, Sti1, Pph5, and Aha1 differ dramatically in their interaction with Cdc37-Hsp90 complexes. p23 and Sti1, both of which are not expected to participate in Cdc37-dependent kinase maturation, prevent Cdc37 binding to Hsp90. Although Sti1/Hop displaces Cdc37 from apparently open Hsp90 complexes by virtue of its two TPR domains and its second interaction site, p23 displaces Cdc37 only from closed Hsp90 complexes. Thus, Cdc37 competes with important cofactors of the steroid hormone receptor pathway, which represent early and late stages of the steroid hormone maturation cycle. It is reasonable to assume that these cofactors act in different but parallel pathways, by differentially functionalizing Hsp90 molecules in the cell in order to ensure the concomitant processing of steroid hormone receptors and kinases.
We show that the two cofactors Pph5 and Aha1 can form ternary complexes with Cdc37-Hsp90. Like Cdc37, they are suspected to participate in the maturation of protein kinases (45, 48). For both proteins, it had been shown that their knock-out results in markedly reduced kinase levels in yeast (26, 45, 61). Based on our AUC-data, we generated a model describing the conformation-driven exchange of Hsp90 cofactors (Fig. 6). Initially, Cdc37-Hsp90 complexes can adopt open and closed conformations but barely hydrolyze ATP due to the inhibitory action of Cdc37 on Hsp90 (Fig. 6). Pph5 binds independently of Cdc37 at the C-terminal end of Hsp90 without an influence on the ATPase activity (data not shown) or on the conformational changes within Hsp90. In contrast, Aha1 has a strong influence on the affinity of Cdc37 for Hsp90. The function of Aha1 may be to trigger the hydrolysis event of the ATPase cycle, which is inhibited by Cdc37. Our data show that this is achieved by a two-step procedure. The action of the N-terminal domain of Aha1 reduces the affinity of Cdc37 to Hsp90 and leads to a ternary complex containing Hsp90, Cdc37, and Aha1 (Fig. 6). The effect of Aha1-N on the affinity of Cdc37 for Hsp90 indicates that the two cofactors do not interact independently with different domains of Hsp90 but potentially recognize partially overlapping binding sites. Aha1 then facilitates the closing reaction by virtue of its C-terminal domain in the presence of ATP analogs. This action leads to the full displacement of Cdc37 from the Hsp90 complex, closing of the N-terminal domains of Hsp90, and activation of the ATP hydrolysis reaction (Fig. 6). Recent studies had demonstrated that the C-terminal domain of Aha1 is required for the stimulation of the ATPase activity (32, 33). We observe that deletion of the C-terminal domain of Aha1 or failure to perform the closing reaction in an inactive Hsp90 mutant also prevents the full displacement of Cdc37 from Hsp90 complexes.
FIGURE 6.

Model for the proposed interaction between Cdc37, Hsp90, and Aha1, including the influence of nucleotide-induced conformational changes. Starting from Cdc37-Hsp90 complexes, ATP binding results in an open-closed equilibrium. The hydrolysis is inhibited by Cdc37. Aha1 binds at the middle domain of Hsp90 by virtue of its N-terminal domain. After nucleotide-induced conformational changes have taken place, the hydrolysis-competent conformation of Hsp90 is achieved, and Cdc37 is fully released from the complex by the action of the Aha1 C-terminal domain. The interaction of Aha1 with the middle and N-terminal domains of Hsp90 is based on recent work from Retzlaff et al. (32) and Koulov et al. (33).
Based on these data, one might assume that kinase activation not only requires the formation of stable Hsp90-Cdc37-kinase complexes but also requires the disruption of these complexes by Aha1, which acts to replace Cdc37 and stimulates the turnover of Hsp90. It appears that the cofactors Cdc37, Aha1, and Pph5 interact in a successive manner with the Hsp90 scaffold to activate protein kinases.
Conservation of the Cdc37-Hsp90 Interaction in Yeast, Nematodes, and Humans
We had performed our study using the nematode Hsp90 system. This is currently the only system where the ATPase activity of Hsp90 is significant and the binding of Cdc37 occurs with reasonable affinity. It is now important to evaluate whether the observed cooperative and competitive effects can be generalized for other Hsp90 systems because previous studies using a heterologous system of yeast Hsp90 and human Cdc37 delivered significantly different results (20, 45). Indeed the degree of conservation between the Hsp90 proteins and their cofactors may argue for a conserved mechanism. In addition, all Hsp90 proteins studied to date share general aspects of the mechanism of nucleotide-induced transient N-terminal dimerization (14, 16, 17, 63), but certainly the basic ATP turnover rates differ between 1 ATP/min in the yeast Hsp90 and 1 ATP/20 min in the human Hsp90s (5, 29). Several cofactors were found to react differently in the yeast and human Hsp90 systems before. As such, Hop/Sti1, p23/Sba1, and Cdc37 inhibit ATP turnover by Hsp90 to different degrees and with vastly different binding affinities when the yeast and human Hsp90 systems are compared (19, 21, 23, 29, 30). Here we find that nematode Cdc37 can bind to open and closed forms of Hsp90. This contradicts data for the human system, where AMP-PNP disrupted Cdc37-Hsp90 complexes in immunoprecipitation experiments (44). Certainly, the human Hsp90 protein is special among eukaryotic Hsp90 proteins due to its extremely low ATP-hydrolyzing activity. It might be that the difficulty in forming the closed, hydrolysis-active conformation is responsible for this effect. Thus, the binding of cofactors might be differently regulated because differences in basic Hsp90 activities can be correlated with a coevoluted activity of the cofactors. This adaptation to the intrinsic ATPase rate of the respective Hsp90 protein may lead to some of the observed differences in the human, nematode, and yeast Hsp90 system. Furthermore, it implies that the analysis of mixed systems, composed of yeast Hsp90 and human cofactors, may lead to unphysiological results. In particular, using the human Cdc37 protein, which binds with 40-fold higher affinity than the corresponding yeast protein, may mask parts of the regulatory interactions between cofactors observed in our study.
The studies, which addressed the potential formation of ternary complexes with Cdc37-Hsp90 before, do not agree in many aspects (20, 23, 45, 46). Indeed different technologies have been applied, which may detect different types of interactions with different sensitivities. Whereas analytical ultracentrifugation can provide a holistic analysis of the complexes, resulting in quantitative information on affinity and size, other methods, like CD spectroscopy (20, 23), size exclusion chromatography of protein complexes (45), or co-immunoprecipitations (46) may detect only subsets of these effects. Therefore, it may well be that the observed interaction patterns turn out to be conserved throughout the eukaryotic Hsp90 systems.
The results presented here together with those of a previous study on the Sti1-Hsp90 interaction in C. elegans (50) provide a basic picture of the traits of this Hsp90 system and the interplay between activating and inhibitory cofactors. It will be interesting to see in the long run how these proteins and their interactions are utilized by the specialized cells of the nematode in order to chaperone the large set of Hsp90 client proteins in a specific manner.
Acknowledgments
We thank Borries Demeler for providing the analysis software UltraScan. We also thank Veronika Haslbeck, Julia Eckl, and Katharina Papsdorf for excellent practical assistance and Christoph Kaiser and Johannes Buchner for critical reading of the manuscript.
This work was supported by Deutsche Forschungsgemeinschaft Grant RI1873/1-1 (to K. R.) and a grant from the Fonds der Chemischen Industrie (to K. R.).
For clarity, the protein designations Hsp90 (heat-shock protein 90), p23 (Hsp90 co-chaperone p23), Aha1 (activator of heat shock protein 90 ATPase), Cdc37 (cell division cycle 37 protein), Sti1 (stress-inducible protein 1), and Pph5 (protein phosphatase 5) are presented independent of species.
- NBD
- nucleotide-binding domain
- AUC
- analytical ultracentrifugation
- TPR
- tetratricopeptide repeat
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate
- ATPγS
- adenosine 5′-O-(thiotriphosphate).
REFERENCES
- 1.Picard D. (2002) Cell Mol. Life Sci. 59, 1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wandinger S. K., Richter K., Buchner J. (2008) J. Biol. Chem. 283, 18473–18477 [DOI] [PubMed] [Google Scholar]
- 3.Pratt W. B., Toft D. O. (2003) Exp. Biol. Med. 228, 111–133 [DOI] [PubMed] [Google Scholar]
- 4.Pearl L. H., Prodromou C. (2006) Annu. Rev. Biochem. 75, 271–294 [DOI] [PubMed] [Google Scholar]
- 5.Panaretou B., Prodromou C., Roe S. M., O'Brien R., Ladbury J. E., Piper P. W., Pearl L. H. (1998) EMBO J. 17, 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obermann W. M., Sondermann H., Russo A. A., Pavletich N. P., Hartl F. U. (1998) J. Cell Biol. 143, 901–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grenert J. P., Sullivan W. P., Fadden P., Haystead T. A., Clark J., Mimnaugh E., Krutzsch H., Ochel H. J., Schulte T. W., Sausville E., Neckers L. M., Toft D. O. (1997) J. Biol. Chem. 272, 23843–23850 [DOI] [PubMed] [Google Scholar]
- 8.Hessling M., Richter K., Buchner J. (2009) Nat. Struct. Mol. Biol. 16, 287–293 [DOI] [PubMed] [Google Scholar]
- 9.Prodromou C., Panaretou B., Chohan S., Siligardi G., O'Brien R., Ladbury J. E., Roe S. M., Piper P. W., Pearl L. H. (2000) EMBO J. 19, 4383–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richter K., Muschler P., Hainzl O., Buchner J. (2001) J. Biol. Chem. 276, 33689–33696 [DOI] [PubMed] [Google Scholar]
- 11.Stebbins C. E., Russo A. A., Schneider C., Rosen N., Hartl F. U., Pavletich N. P. (1997) Cell 89, 239–250 [DOI] [PubMed] [Google Scholar]
- 12.Ali M. M., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., Pearl L. H. (2006) Nature 440, 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richter K., Moser S., Hagn F., Friedrich R., Hainzl O., Heller M., Schlee S., Kessler H., Reinstein J., Buchner J. (2006) J. Biol. Chem. 281, 11301–11311 [DOI] [PubMed] [Google Scholar]
- 14.Richter K., Soroka J., Skalniak L., Leskovar A., Hessling M., Reinstein J., Buchner J. (2008) J. Biol. Chem. 283, 17757–17765 [DOI] [PubMed] [Google Scholar]
- 15.Dutta R., Inouye M. (2000) Trends Biochem. Sci. 25, 24–28 [DOI] [PubMed] [Google Scholar]
- 16.Frey S., Leskovar A., Reinstein J., Buchner J. (2007) J. Biol. Chem. 282, 35612–35620 [DOI] [PubMed] [Google Scholar]
- 17.Leskovar A., Wegele H., Werbeck N. D., Buchner J., Reinstein J. (2008) J. Biol. Chem. 283, 11677–11688 [DOI] [PubMed] [Google Scholar]
- 18.Johnson J. L., Brown C. (2009) Cell Stress Chaperones 14, 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richter K., Walter S., Buchner J. (2004) J. Mol. Biol. 342, 1403–1413 [DOI] [PubMed] [Google Scholar]
- 20.Siligardi G., Hu B., Panaretou B., Piper P. W., Pearl L. H., Prodromou C. (2004) J. Biol. Chem. 279, 51989–51998 [DOI] [PubMed] [Google Scholar]
- 21.Prodromou C., Siligardi G., O'Brien R., Woolfson D. N., Regan L., Panaretou B., Ladbury J. E., Piper P. W., Pearl L. H. (1999) EMBO J. 18, 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richter K., Muschler P., Hainzl O., Reinstein J., Buchner J. (2003) J. Biol. Chem. 278, 10328–10333 [DOI] [PubMed] [Google Scholar]
- 23.Siligardi G., Panaretou B., Meyer P., Singh S., Woolfson D. N., Piper P. W., Pearl L. H., Prodromou C. (2002) J. Biol. Chem. 277, 20151–20159 [DOI] [PubMed] [Google Scholar]
- 24.Roe S. M., Ali M. M., Meyer P., Vaughan C. K., Panaretou B., Piper P. W., Prodromou C., Pearl L. H. (2004) Cell 116, 87–98 [DOI] [PubMed] [Google Scholar]
- 25.Zhang W., Hirshberg M., McLaughlin S. H., Lazar G. A., Grossmann J. G., Nielsen P. R., Sobott F., Robinson C. V., Jackson S. E., Laue E. D. (2004) J. Mol. Biol. 340, 891–907 [DOI] [PubMed] [Google Scholar]
- 26.Panaretou B., Siligardi G., Meyer P., Maloney A., Sullivan J. K., Singh S., Millson S. H., Clarke P. A., Naaby-Hansen S., Stein R., Cramer R., Mollapour M., Workman P., Piper P. W., Pearl L. H., Prodromou C. (2002) Mol. Cell 10, 1307–1318 [DOI] [PubMed] [Google Scholar]
- 27.Lotz G. P., Lin H., Harst A., Obermann W. M. (2003) J. Biol. Chem. 278, 17228–17235 [DOI] [PubMed] [Google Scholar]
- 28.Meyer P., Prodromou C., Liao C., Hu B., Roe S. M., Vaughan C. K., Vlasic I., Panaretou B., Piper P. W., Pearl L. H. (2004) EMBO J. 23, 1402–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaughlin S. H., Smith H. W., Jackson S. E. (2002) J. Mol. Biol. 315, 787–798 [DOI] [PubMed] [Google Scholar]
- 30.Young J. C., Hartl F. U. (2000) EMBO J. 19, 5930–5940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chadli A., Bouhouche I., Sullivan W., Stensgard B., McMahon N., Catelli M. G., Toft D. O. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 12524–12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H., Richter K., Buchner J. (2010) Mol. Cell 37, 344–354 [DOI] [PubMed] [Google Scholar]
- 33.Koulov A. V., Lapointe P., Lu B., Razvi A., Coppinger J., Dong M. Q., Matteson J., Laister R., Arrowsmith C., Yates J. R., 3rd, Balch W. E. (2010) Mol. Biol. Cell 21, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith D. F., Toft D. O. (1992) J. Steroid Biochem. Mol. Biol. 41, 201–207 [DOI] [PubMed] [Google Scholar]
- 35.Smith D. F., Stensgard B. A., Welch W. J., Toft D. O. (1992) J. Biol. Chem. 267, 1350–1356 [PubMed] [Google Scholar]
- 36.Smith D. F. (1993) Mol. Endocrinol. 7, 1418–1429 [DOI] [PubMed] [Google Scholar]
- 37.Brugge J. S. (1986) Curr. Top. Microbiol. Immunol. 123, 1–22 [DOI] [PubMed] [Google Scholar]
- 38.Hunter T., Poon R. Y. (1997) Trends Cell Biol. 7, 157–161 [DOI] [PubMed] [Google Scholar]
- 39.Stepanova L., Leng X., Parker S. B., Harper J. W. (1996) Genes Dev. 10, 1491–1502 [DOI] [PubMed] [Google Scholar]
- 40.Terasawa K., Yoshimatsu K., Iemura S., Natsume T., Tanaka K., Minami Y. (2006) Mol. Cell. Biol. 26, 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartson S. D., Matts R. L. (1994) Biochemistry 33, 8912–8920 [DOI] [PubMed] [Google Scholar]
- 42.Pashtan I., Tsutsumi S., Wang S., Xu W., Neckers L. (2008) Cell Cycle 7, 2936–2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sreeramulu S., Jonker H. R., Langer T., Richter C., Lancaster C. R., Schwalbe H. (2009) J. Biol. Chem. 284, 3885–3896 [DOI] [PubMed] [Google Scholar]
- 44.Zhang T., Li Y., Yu Y., Zou P., Jiang Y., Sun D. (2009) J. Biol. Chem. 284, 35381–35389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harst A., Lin H., Obermann W. M. (2005) Biochem. J. 387, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silverstein A. M., Grammatikakis N., Cochran B. H., Chinkers M., Pratt W. B. (1998) J. Biol. Chem. 273, 20090–20095 [DOI] [PubMed] [Google Scholar]
- 47.Pratt W. B., Silverstein A. M., Galigniana M. D. (1999) Cell. Signal. 11, 839–851 [DOI] [PubMed] [Google Scholar]
- 48.Vaughan C. K., Mollapour M., Smith J. R., Truman A., Hu B., Good V. M., Panaretou B., Neckers L., Clarke P. A., Workman P., Piper P. W., Prodromou C., Pearl L. H. (2008) Mol. Cell 31, 886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abbas-Terki T., Briand P. A., Donzé O., Picard D. (2002) Biol. Chem. 383, 1335–1342 [DOI] [PubMed] [Google Scholar]
- 50.Gaiser A. M., Brandt F., Richter K. (2009) J. Mol. Biol. 391, 621–634 [DOI] [PubMed] [Google Scholar]
- 51.Demeler B., Saber H., Hansen J. C. (1997) Biophys. J. 72, 397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacGregor I. K., Anderson A. L., Laue T. M. (2004) Biophys. Chem. 108, 165–185 [DOI] [PubMed] [Google Scholar]
- 53.Kroe R. R., Laue T. M. (2009) Anal. Biochem. 390, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stafford W. F., 3rd (1992) Anal. Biochem. 203, 295–301 [DOI] [PubMed] [Google Scholar]
- 55.Hayes D. B., Stafford W. F. (2010) Macromol. Biosci. 10, 731–735 [DOI] [PubMed] [Google Scholar]
- 56.Chen S., Sullivan W. P., Toft D. O., Smith D. F. (1998) Cell Stress Chaperones 3, 118–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H., Hartl F. U., Moarefi I. (2000) Cell 101, 199–210 [DOI] [PubMed] [Google Scholar]
- 58.Johnson J. L., Toft D. O. (1994) J. Biol. Chem. 269, 24989–24993 [PubMed] [Google Scholar]
- 59.Grenert J. P., Johnson B. D., Toft D. O. (1999) J. Biol. Chem. 274, 17525–17533 [DOI] [PubMed] [Google Scholar]
- 60.Weikl T., Abelmann K., Buchner J. (1999) J. Mol. Biol. 293, 685–691 [DOI] [PubMed] [Google Scholar]
- 61.Wandinger S. K., Suhre M. H., Wegele H., Buchner J. (2006) EMBO J. 25, 367–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith D. F., Sullivan W. P., Marion T. N., Zaitsu K., Madden B., McCormick D. J., Toft D. O. (1993) Mol. Cell. Biol. 13, 869–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cunningham C. N., Krukenberg K. A., Agard D. A. (2008) J. Biol. Chem. 283, 21170–21178 [DOI] [PMC free article] [PubMed] [Google Scholar]