

Abstract
Per-Arnt-Sim (PAS) domain-containing protein kinase (PASK) is an evolutionary conserved protein kinase that coordinates cellular metabolism with metabolic demand in yeast and mammals. The molecular mechanisms underlying PASK regulation, however, remain unknown. Herein, we describe a crystal structure of the kinase domain of human PASK, which provides insights into the regulatory mechanisms governing catalysis. We show that the kinase domain adopts an active conformation and has catalytic activity in vivo and in vitro in the absence of activation loop phosphorylation. Using site-directed mutagenesis and structural comparison with active and inactive kinases, we identified several key structural features in PASK that enable activation loop phosphorylation-independent activity. Finally, we used combinatorial peptide library screening to determine that PASK prefers basic residues at the P-3 and P-5 positions in substrate peptides. Our results describe the key features of the PASK structure and how those features are important for PASK activity and substrate selection.
Keywords: Cell Metabolism, Crystal Structure, Protein Kinases, Protein Phosphorylation, Protein-serine/threonine Kinase, Activation Loop Phosphorylation, Kinase Structure, PAS Domain, PASK, Metabolism
Introduction
Nutrient sensing and signaling systems play an important role in coordinating growth, development, and physiology with nutrient availability (1, 2). These systems are required to maintain a dynamic balance between energy intake and expenditure. Failure to do so lies at the heart of metabolic disorders, including diabetes and obesity (3). The mammalian target of rapamycin and 5′-AMP-activated protein kinase, two of the best studied nutrient-responsive protein kinases; regulate cellular energy balance by coordinating energy expenditure with nutrient intake and availability (4–6). Although mammalian target of rapamycin is activated under conditions of nutrient abundance and promotes growth and anabolic processes (7), 5′-AMP-activated protein kinase is activated by increased levels of 5′-AMP, an indicator of low energy balance, and triggers growth cessation and catabolism (3). In addition to mammalian target of rapamycin and 5′-AMP-activated protein kinase, Per-Arnt-Sim (PAS)2 domain-containing kinase (PASK), an evolutionarily conserved protein-serine/threonine kinase (8), coordinates glucose utilization in response to cellular energy and metabolic demand (9, 10).
We previously demonstrated important roles for the Saccharomyces cerevisiae PASK orthologs, Psk1 and Psk2, in nutrient sensing and metabolic signaling (9–12). In response to cellular demand, Psk1 and Psk2 dictate the partitioning of glucose between storage as glycogen and utilization for cell growth and division (10–12). Mammalian PASK is also regulated by cellular nutrient status (13). Insight into the physiological role of PASK as a metabolic regulator came from in vivo studies of PASK-deficient mice. Mice lacking PASK were resistant to high fat diet-induced lipid accumulation and insulin resistance (14). Thus, both in yeast and mammals, PASK coordinates the utilization of nutrients and energy in response to metabolic state.
The major mode by which PASK signals to regulate energy utilization is almost certainly via phosphorylation of protein substrates catalyzed by a canonical C-terminal serine/threonine kinase domain. In contrast, the mechanism by which PASK senses cellular metabolic status is unknown, but likely relates to its N-terminal PAS domain. PAS domains serve as sensory modules for a variety of intracellular cues, including light, oxygen, redox state, and various metabolites (15). Coordinated with this sensing function, PAS domains trigger appropriate cellular responses via regulation of attached functional domains (15). Consistent with other PAS domains, NMR-based structural studies of the PAS domain of PASK showed that it can bind specific small molecules (16). The PAS domain of PASK also directly binds to and inhibits the catalytic activity of the kinase domain either in cis or in trans (16, 17). Together, these data and precedents set by the studies of other PAS domains provide support for the hypothesis that a cellular PAS domain ligand regulates PASK kinase activity through modulation of the PAS domain-kinase domain interaction.
Most protein kinases require phosphorylation on serine, threonine, or tyrosine residues within the activation loop (or P loop) of the kinase domain to achieve full activation. Here, we provide biochemical and crystallographic evidence that activation loop phosphorylation does not play a major role in PASK activation. We also describe structural adaptations that enable kinase activity in the absence of phosphorylation. Finally, we describe the peptide substrate preference of PASK and the structural features that dictate substrate selection.
EXPERIMENTAL PROCEDURES
Cloning and Protein Expression
Residues 977–1300 of human PASK (numbered relative to GenBank sequence AAK69752) were amplified by PCR and cloned into a custom TOPO-adapted baculovirus expression vector (Invitrogen) based on pFastBac. The NYSGXRC target and clone name are NYSGXRC-9501a and 9501a6KWg1h1, respectively, and the plasmid is available through the National Institutes of Health-funded PSI Material Repository. The expressed protein is fused to a noncleavable C-terminal His6 tag so the final protein is: MAL + PASK (977–1300, C1266F) + EGHHHHHH. (Several published PASK sequences have a cysteine at position 1266, however our clone and the GenBank Refseq have a phenylalanine at this position. Phe1266 is among the last residues visible in our x-ray crystal structure. cDNA from 9501a6KWg1h1 was isolated and sequence verified prior to expression.)
Recombinant baculovirus incorporating the kinase cDNA construct was generated by transposition with the Bac-to-Bac system (Invitrogen catalog no. 10359-016). PASK was expressed in Sf9 cells in a 48-h 12-liter fermentation at 29 °C. The cells were harvested by centrifugation and frozen at −80 °C.
Protein Purification and Characterization
Cell pellets produced from 3 liters of Sf9 insect cell culture expressing the C-terminal His-tagged human PAS kinase domain (residues 977–1300, C1266F) were suspended in 250 ml of lysis buffer (50 mm Tris-HCl, pH 7.7, 250 mm NaCl, and six tablets of Roche Complete, EDTA-free protease inhibitor mixture (catalog no. 1873580)). The suspension was stirred for 1 h, and insoluble debris was removed by centrifugation for 30 min (39,800 × g). The supernatant was collected and incubated with 10 ml of a 50% slurry of nickel-nitrilotriacetic acid-agarose (Qiagen catalog no. 30250), preequilibrated with the wash buffer (20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 10% glycerol, and 25 mm imidazole), for 30 min with gentle stirring. The sample was then poured into a drip column and washed with 50 ml of wash buffer to remove unbound proteins. PAS kinase was eluted using 25 ml of elution buffer (wash buffer with 500 mm imidazole). The eluted fraction was further purified by gel filtration chromatography on a GE Healthcare HiLoad 16/60 Superdex 200 preparation grade column (catalog no. 17-1069-01) preequilibrated with gel filtration buffer (10 mm HEPES, pH 7.5, 150 mm NaCl, 10% glycerol, and 5 mm DTT). Fractions containing PAS kinase were combined and concentrated to 10 mg/ml by centrifugation in an Amicon Ultra-15 10,000-Da MWCO centrifugal filter unit (catalog no. UFC901024). Electrospray mass spectrometry was used to obtain an accurate mass of the protein purified to confirm its identity and to define the phosphorylation status of the protein (∼50% was unphosphorylated, 40% singly phosphorylated, and 10% double phosphorylated).
Crystallization
The PASK protein (NYSGXRC clone 9501a6KWg1h1, PID 17274-3), concentrated to 9 mg/ml, was incubated with 2 mm ATP and 4 mm MgCl2 on ice for 15 min, centrifuged for 10 min at 13,000 rpm, then filtered through a 0.2-μm spin filter. Diffraction quality crystals were obtained via sitting drop vapor diffusion from 18% PEG 3350, 275 mm calcium acetate, and 4% butanediol (1 μl of protein plus 1 μl of crystallization solution). Crystals were frozen by direct immersion in liquid nitrogen following cryoprotection via addition of 15% 2-methyl-2,4-pentanediol.
Structure Determination
X-ray diffraction data to 2.3 Å resolution was obtained from frozen crystals at the Advanced Photon Source beamline LRL-CAT 31-ID. Data were processed using the CCP4 program package (20). The structure was determined via molecular replacement using Phaser (18) with a search model derived from the structure of human MAP microtubule affinity-regulating kinase MARK2 (Protein Data Bank (PDB) code 1ZMU). The atomic model was built using Xfit (19), and the structure was refined to convergence using Refmac (20). NCS restraints were used in the initial rounds of refinement and model building but were released in the final stages of refinement. The diffraction spots were streaky in one direction, resulting in a high Rmerge, and leading to somewhat higher refinement R-factors. Structure determination statistics are provided in Table 1. We also calculated root mean square deviation values among the six chains of PASK within the asymmetric unit. The six independent copies of the polypeptide adopt very similar tertiary structures, with the exception of the region surrounding the disordered loop near residues 1034–1040 in chain B–F. The root mean square deviation for 276 CA atoms varies from 0.23 to 0.50 Å between chain A and each of the other chains. All structural figures in the manuscripts are prepared using PyMOL three-dimensional visualization software from DeLano Scientific (The PyMOL Molecular Graphics System, Version 0.99rc6; Schrödinger, LLC).
TABLE 1.
Structure statistics for the catalytic domain of human PASK (PDB code 3DLS)
| Data collection and processing | |
| Beamline | LRL-CAT, Advanced Photon Source |
| Wavelength (Å) | 0.97929 |
| Space group | P1 |
| Cell dimensions (Å, °) | a = 85.8, b = 85.8, c = 94.2, α = 77.3, β = 77.5, γ = 60.1 |
| Molecules/asymmetric unit | 6 |
| Resolution (Å) | 31.2-2.3 (2.42-2.30) |
| Observed reflections | 368,548 |
| Unique reflections | 96,811 |
| Mean Rmerge (last shell) | 0.127 (0.528) |
| Mean redundancy | 3.8 |
| Completeness (%) (last shell) | 97.2 (96.4) |
| Mean I/σ(I) (last shell) | 7.1 (2.1) |
| Structure refinement | |
| Rwork | 0.241 |
| Rfree | 0.297 |
| Ramachandran plot (%) | |
| Most favored regions | 90.9 |
| Disallowed regions | 0.0 |
| Amino acid residues | 1693 |
| rmsda bond length (Å) | 0.009 |
| rmsd angle (°) | 1.256 |
| Mean B value protein/ADP per Mg per solvent (Å2) | 37.5/30.4/26.1/33.8 |
| Nucleotides | 6 |
| Magnesium ions | 19 |
a rmsd, root mean square deviation.
Constructs and Cell Lines
Cloning of human WT and K1028R PASK into a V5-tagged mammalian expression vector (pcDNA3.1-V5/His) was described previously (17). All mutations were performed using the QuikChange site-directed mutagenesis kit, and sequences were verified. HEK293T cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS and 1% penicillin/streptomycin. Transfections were done using FuGENE 6 (Roche) reagents according to the manufacturer's protocol.
Immunoprecipitation Kinase (IP Kinase) Assay
For IP kinase assays, HEK293T cells transfected with various PASK-V5 constructs were lysed in lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5% sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 μg/ml leupeptin). The lysates were centrifuged, and V5-tagged PASK was immunoprecipitated from the supernatant using anti-V5 antibody (Invitrogen) conjugated with protein G-Sepharose beads. The immune complexes were washed three times with lysis buffer, followed by twice with kinase assay buffer (18 mm HEPES, pH 7.5, 10 mm MgCl2, 50 μm ATP, 1 mm DTT). Kinase reactions were performed by adding 150 ng of bacterially purified Ugp1 and 1 μCi/reaction of [γ-32P]ATP (PerkinElmer Life Sciences). Reactions were terminated by adding SDS-buffer, and samples were separated by SDS-PAGE, transferred to nitrocellulose membrane followed by autoradiography.
In Vivo Metabolic 32P Labeling
HEK293T cells were transfected with various PASK plasmids. At 24 h after transfection, cells were washed twice with phosphate-free DMEM (Invitrogen) followed by incubation with 1.0 mCi of 32P. Cells were washed with phosphate-free DMEM to remove unincorporated 32P, lysed using lysis buffer, and immunoprecipitated as described above. Immunocomplexes were washed with buffer (20 mm Na2HPO4, 0.5% Triton X-100, 0.1% SDS, 0.02% NaN3) containing high salt (1 m NaCl and 0.1% BSA) followed by low salt (150 mm NaCl). Immunoprecipitated PASK was released by SDS-PAGE sample loading buffer, separated by SDS-PAGE, followed by transfer to nitrocellulose membrane, and imaged by autoradiography.
Determination of Substrate Specificity
PASK substrate specificity was determined by screening a combinatorial peptide library, essentially as described (33). The peptide library consisted of a set of 198 peptides having the general sequence YAXXXXXS/TXXXXAGKK (biotin), where X is an equimolar mixture of the 17 amino acids (excluding cysteine, serine, and threonine), and S/T is an even mixture of serine and threonine. In each peptide, one of the X positions was fixed as one of the 20 unmodified amino acids or phosphothreonine or phosphotyrosine. Peptides were arrayed in 384-well plates and incubated with PASK and [γ-32P]ATP for 2 h at 30 °C. Aliquots (2 μl) of the reactions were transferred to a streptavidin-coated membrane, which was washed as described, dried, and exposed to a phosphor storage screen.
RESULTS
PASK Activity Is Independent of Activation Loop Phosphorylation
To achieve full activity, most protein kinases require phosphorylation of one or more amino acids in the activation loop within the kinase domain (21). PASK possesses a threonine residue (Thr1161) in the site analogous to the canonical phosphorylation site in many other kinases. To determine the significance of activation loop phosphorylation for PASK activity in vivo, we mutated Thr1161 to alanine in V5-tagged human PASK and studied kinase activity in vitro and in vivo. The activity of the T1161A mutant was compared with wild-type (WT) PASK in an immunoprecipitation kinase assay using the in vitro PASK substrate, UDP-glucose pyrophosphorylase (Ugp1) (9). As shown in Fig. 1A, WT PASK exhibited strong Ugp1 phosphorylation and autophosphorylation activity. As expected, a K1028R mutant of PASK, which lacks a conserved lysine in the ATP binding pocket, showed no autophosphorylation or Ugp1 phosphorylation activity. Surprisingly, the T1161A mutation did not affect PASK activity as assessed by either autophosphorylation or Ugp1 phosphorylation. To determine whether the T1161A mutation affects PASK phosphorylation kinetics, we measured Ugp1 phosphorylation over a 30-min time course (Fig. 1B). Again, the catalytic activity of the T1161A PASK mutant was indistinguishable from WT PASK. We conclude that, unlike many protein kinases, in vitro activity of PASK does not require activation loop phosphorylation.
FIGURE 1.
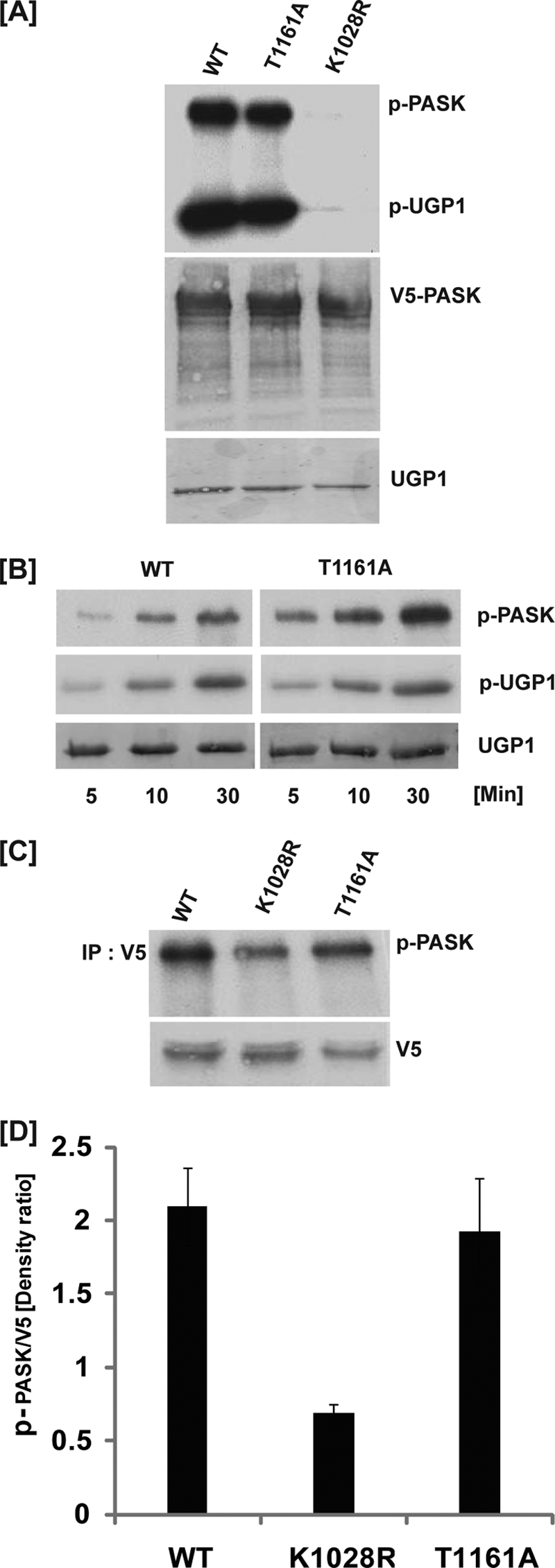
PASK activity is independent of activation loop phosphorylation. A, HEK293T cells were transfected with plasmids expressing the indicated variants of V5-tagged human PASK. At 24 h after transfection, cells were lysed, PASK was immunoprecipitated using anti-V5 antibody, and an in vitro kinase activity assay was performed using Ugp1 and [γ-32P]ATP as substrates. Samples were separated by SDS-PAGE and subjected to autoradiography (top panel), anti-V5 Western blotting (middle panel) and Ponceau S staining (bottom panel). B, HEK293T cells were transfected with plasmids expressing either WT or T1161A mutant V5-tagged human PASK as indicated. At 24 h after transfection, cells were lysed, PASK was immunoprecipitated using anti-V5 antibody, and an in vitro kinase activity assay was performed using Ugp1 and [γ-32P]ATP as substrates for the indicated time, followed by addition of SDS buffer to terminate the reaction. Samples were separated by SDS-PAGE and subjected to autoradiography (top and middle panels), and Ponceau S staining (bottom panel). C, HEK293T cells were transfected with plasmids expressing the indicated variants of V5-tagged human PASK. At 24 h after transfection, metabolic 32P labeling was performed for 4.0 h. Following cell lysis, PASK was immunoprecipitated using anti-V5 antibody, and samples were separated by SDS-PAGE and subjected to autoradiography (top panel) and anti-V5 Western blotting (bottom panel). D, three separate experiments were quantified as described in C using Scion Image software. Mean ± S.D. (error bars) are shown.
The in vivo targets of mammalian PASK remain unknown. PASK does phosphorylate itself in cells, however, and we have used autophosphorylation as a measure of in vivo PASK activity. As illustrated in Fig. 1, C and D, WT PASK is robustly phosphorylated in cultured HEK293T cells. Phosphorylation of the catalytically inactive K1028R mutant was decreased by ∼70% relative to WT. This reduction in phosphorylation is indicative of impaired autophosphorylation. In contrast, the T1161A mutation had no effect on PASK phosphorylation in vivo. These findings demonstrate that activation loop phosphorylation is not required for kinase activity of PASK in vitro or in vivo. In addition, mass spectrometry analysis of human PASK purified either from insect cells or human HEK293T cells did not show the presence of phosphothreonine 1161 within PASK (data not shown). These findings suggest that PASK has adapted structural features that enable kinase activity in the absence of activation loop phosphorylation.
Major Structural Features of the PASK Kinase Domain Crystal Structure
To understand the structural features that enable PASK activity in the absence of activation loop phosphorylation, we determined the x-ray crystal structure of a portion of human PASK encompassing the kinase domain (residues 977–1300) bound to ADP at 2.3 Å resolution (Table 1). As shown in Fig. 2, the kinase domain of human PASK adopts the classical two-lobe structure typical of eukaryotic protein kinases. The N-terminal lobe consists primarily of β-sheets, whereas the C-terminal lobe is predominantly α-helical. Within the PASK active site, two Mg2+ ions, and ADP, are clearly visible in the electron density map (supplemental Fig. 3A). ADP appears to be the product of hydrolysis of ATP, which was included in the crystallization solution.
FIGURE 2.

Crystal structure of the kinase domain of PASK. A, secondary structure elements of PASK kinase domain in the presence of ADP are shown. The α-helices are depicted in red, β-strands in yellow, loops in green, and ADP and Mg2+ ions in blue. The strands and helices are named according to the conventional nomenclature for PKA. The N and C termini of the kinase structure are indicated as N and C, respectively. B, PASK three-dimensional structure is rotated 180° along the vertical axis to show the positions of βH1 and βH2 helices (marked). See “Results” for details.
The N-terminal lobe of protein kinases is typically composed of five β-strands and the αC helix. In contrast, the PASK domain contains an additional two-stranded β-hairpin (termed βH1 and βH2) and a truncated αC helix (Fig. 2B). To understand the sequence-structure relationship leading to the unusual secondary structure of the PASK N-terminal lobe, we performed a structure-based sequence alignment of human protein kinases against PASK using DaliLite (22) (supplemental Fig. 2). Interestingly, the PIM1 proto-oncogene protein kinase showed the highest degree of similarity with PASK and, like PASK, also contains an additional β-hairpin, which is absent in PKA (supplemental Figs. 1 and 2) (23). Our structure-based alignment showed that PIM1 (and its close relatives PIM2 and PIM3) and PASK share a tryptophan residue at the beginning of the βH1 strand (Fig. 3A; PASK, Trp1040; PIM1, Trp168) that is absent in kinases lacking the β-hairpin structural motif (supplemental Fig. 2, shaded green). The tryptophan residue is well ordered in the crystal structure and is clearly visible in the electron density map (supplemental Fig. 3B). It is possible that the bulky aromatic side chain of tryptophan may destabilize the presumptive αC helix, thereby shortening the αC helix and stabilizing the β-hairpin structure. Although the side chains of Lys1032, Val1035, and Thr1050 participate, a phenylalanine (PASK, Phe1077; PIM1, Phe207) from the β5 strand contributes much of the hydrophobic environment surrounding the βH1 tryptophan (Fig. 3A). This phenylalanine is structurally conserved among PASK and all three PIM isoforms (supplemental Fig. 2, shaded blue). The importance of this unusual secondary structure in regulating kinase activity or substrate binding remains unclear (see “Discussion”). It does not, however, affect the highly conserved position, orientation or Lys1028 salt bridge of the catalytically important Glu1052 (Fig. 3B).
FIGURE 3.

Unusual N-terminal lobe structure in PASK is shared by PIM1. A, comparison of the βH1, βH2, and αC elements of the PASK and PIM1 N-terminal lobes is shown. The positions and orientations of the tryptophan (Trp1040 in PASK) and phenylalanine (Phe1077 in PASK) residues are shown in each structure. Side chains are colored by element. B, orientation and interactions of important and conserved ATP binding pocket residues are normal despite the insertion of the βH1/βH2 β-hairpin in PASK. Glu1052 from the αC helix forms an ionic interaction with Lys1028 and ADP (black) as typical of most active protein kinase structures.
Structural Features Stabilizing the Active Conformation of the Activation Loop in the Absence of Phosphorylation
Activation loop phosphorylation is a requirement for full activation of most eukaryotic protein kinases (21), but PASK activity was not affected by mutation of the activation loop threonine Thr1161 (Fig. 1). One important function of activation loop phosphorylation is to enable the formation of hydrogen bonds and salt bridges between the N-terminal and C-terminal lobes of the kinase domain (Fig. 4A, left) (21, 24, 25). This interlobe interaction is thought to be essential for stabilizing the active conformation and substrate-binding interface (24). In protein kinases that do not require activation loop phosphorylation, an alternative interlobe interaction must be enabled by sequence or structural changes. For example, Thr153 in the Mg2+-binding loop of the C-terminal lobe of CHK1 forms a hydrogen bond with Lys54 of the N-terminal lobe to stabilize the active conformation (Fig. 4A, right) (26). In our PASK structure, we found that Tyr1152 from the Mg2+-binding loop forms a hydrogen bond with Arg1058 from the αC-helix of the N-terminal lobe (Fig. 4B). To determine whether this interaction is required to stabilize the active conformation, we assayed PASK mutated at either Tyr1152 or Arg1058. The R1058A mutant, but not the R1058K mutant, showed a significant loss of PASK activity as measured by diminished Ugp1 phosphorylation and autophosphorylation (Fig. 4C). Similarly, the Y1152F mutant also exhibited significantly lower kinase activity. The residue corresponding to Y1152 is not found in most protein kinases, suggesting that the R1058-Y1152 interaction may represent a unique structural feature of PASK that stabilizes the active conformation in the absence of activation loop phosphorylation.
FIGURE 4.

Interaction of the PASK N-terminal lobe with the C-terminal lobe in the absence of Thr1161 phosphorylation. A, comparison of the interaction between the N- and C-terminal lobes in an activation loop phosphorylation-dependent RD kinase (PDK1, left panel, PDB code 1H1W) or activation loop phosphorylation-independent RD kinase (CHK1, right panel, PDB code 1IA8). B, stabilization of the interaction between the N- and C-terminal lobes of PASK in the absence of activation loop phosphorylation by an Arg1058-Tyr1152 hydrogen bond. Asp1043 from βH1 provides an additional anchor point for the Arg1058 side chain conformation. C, loss of the interaction between the PASK N- and C-terminal lobes resulting in decreased in vitro kinase activity. WT PASK and the indicated point mutants were analyzed as described in Fig. 1A.
In addition to promoting interlobe interaction, activation loop phosphorylation often plays a role in substrate binding and catalysis. Activation loop phosphoserine/threonine hydrogen bonds with side chains from catalytic residues, stabilizing a catalytically active conformation (27). Most eukaryotic protein kinases (referred to as RD kinases) contain a highly conserved HRD catalytic loop between the β6 and β7 strands, corresponding to His1126, Arg1127, and Asp1128 in PASK. Typically, the activation loop phospho-group interacts directly with the arginine of the HRD loop and stabilizes a conformation favorable for catalysis. In addition to the phosphorylated activation loop, the Mg2+-binding loop formed by Asp1146, Phe1147, and Gly1148 residues also interacts with the HRD catalytic loop. For many activation loop phosphorylation-dependent RD kinases, the position three residues C-terminal to the conserved DFG sequence (DFG+3) is often occupied by a lysine, which projects into the RD pocket (Fig. 5A, inverted triangle) (25). The negatively charged activation loop phosphate is thought to neutralize the positive charge repulsion of arginine from the RD pocket and the DFG+3 lysine, thereby stabilizing a catalytically active RD pocket conformation (25, 27). Interestingly, in the PASK and PIM1/2/3 sequences, this lysine is replaced by alanine (Ala1151 in PASK, marked by inverted triangle), possibly enabling the active conformation of the RD pocket arginine in the absence of activation loop phosphorylation. It is interesting to note that CHK1, another RD kinase that does not require activation loop phosphorylation, also has an uncharged residue (threonine) at this site. This difference is responsible for maintaining the active conformation of CHK1 in the absence of activation loop phosphorylation (26). To determine the importance of the corresponding alanine in PASK, we generated an A1151K mutant of PASK. As shown in Fig. 5B, the A1151K mutation significantly decreased Ugp1 phosphorylation and PASK autophosphorylation. Enzymatic impairment was almost completely reversed by combining it with a phosphomimetic substitution of the activation loop threonine (T1161E) (Fig. 5B). It is noteworthy that the T1161E single mutation did not affect PASK activity, consistent with our observation that Thr1161 phosphorylation is not a major regulator of PASK activity.
FIGURE 5.

Alanine 1151 enables PASK catalytic activity in the absence of activation loop phosphorylation. A, sequence alignment of PASK with several protein kinases is shown. The basic residue at the DFG+3 position is indicated by the inverted triangle. This basic residue is typically found in kinases regulated by activation loop phosphorylation (dependent), but not in activation loop phosphorylation-independent kinases (independent). B, A1151K substitution decreases PASK activity, which is partially rescued by a phosphomimetic T1161E substitution in the activation loop. WT PASK and the indicated point mutants were analyzed as described for Fig. 1A.
As seen in x-ray structures of other activated protein kinases (27), Asp1146 and the Gly1148 main chain of the PASK Mg2+-binding loop form a hydrogen bond, enabling Asp1146 to interact with Mg2+ and the nucleotide phosphates (Fig. 6A). In most kinases, arginine of the RD catalytic loop hydrogen bonds with the backbone of the DFG+1 residue, which is Ser1149 in PASK (Fig. 6A) (27). This interaction is not affected by activation loop phosphorylation status and the Arg1127-Ser1149 hydrogen bond is also seen in the PASK structure (Fig. 6A). In addition, we also observed that Arg1127 forms a hydrogen bond with the side chain of Tyr1179 located between the αEF and αF helices (Fig. 6B). In the structures of PIM1 and glycogen synthase kinase 3β, two examples of activation loop phosphorylation-independent kinases, the RD pocket arginine also forms a hydrogen bond with a tyrosine between αEF and αF as seen in the PASK structure (Fig. 6B). Although this interaction has been described previously (28), it is not clear whether this conserved tyrosine (supplemental Fig. 2, highlighted in orange) plays any role in enabling kinase activity. To determine whether the orientation and interaction of Tyr1179 with the RD pocket are specific for kinases in the active conformation, we examined structures of the active and inactive conformations of protein kinase B (PKB)/AKT (29, 30) and the extracellular signal-regulated kinase 1 (ERK1) (31, 32). As shown in Fig. 6C, this tyrosine residue hydrogen bonds with the RD pocket arginine only in the active conformations of AKT and ERK1. In the inactive forms of AKT and ERK1, this tyrosine is directed away from the RD pocket and interacts with adjacent residues (Fig. 6D). Activation loop phosphorylation has been shown to induce conformational changes in the αEF/αF region (25), possibly caused by interaction of this tyrosine with the activation loop phosphoserine/phosphothreonine (Fig. 6, C and D).
FIGURE 6.

Stabilization of catalytic core residues in PASK in the absence of activation loop phosphorylation. A, polar interactions of the catalytic loop and Mg2+-binding loop with ADP and αC helix residues. ADP and Mg2+ are indicated. C-α atoms of Gly1148 and Ser1149 that form hydrogen bonds with the Asp1146 and Arg1127 side chains, respectively, are shown. For simplicity, C-α of other amino acids not involved in hydrogen bond formation are not shown. B, structural comparison of the catalytic core of PASK, PIM1, and glycogen synthase kinase 3β (PDB code 1I09). Tyrosine analogous to Tyr1179 in all three kinases hydrogen bonds with the arginine from the catalytic loop, which in turn hydrogen bonds with the backbone of the serine in the Mg2+ binding loop. Tyr1179 of PASK and its structural homolog in PIM1 also forms a hydrogen bond with the main chain oxygen of the activation loop residue. See “Results” for details. C, active structures of AKT (PDB code 1O6K) and ERK1 (PDB code 2ERK) shown to illustrate the polar contacts involving the tyrosine, activation loop, and RD pocket. D, inactive structures of AKT (PDB code 1MRY) and ERK1 (PDB code 1ERK) showing a lack of hydrogen bonding between the tyrosine and RD pocket. In the AKT structure, the Mg2+-binding loop and activation loop are not ordered. Tyr327 of AKT interacts with Asn325 via a solvent molecule in the inactive conformation.
Analysis of PASK Substrate Specificity
Given the biological importance of PASK and the uniqueness of its structure, the mechanisms underlying substrate selectivity are of considerable interest. As the physiological protein substrates of human PASK remain unidentified, we screened an arrayed combinatorial peptide library to determine the consensus phosphorylation sequence for PASK (33). PASK displayed a strong preference for basic residues, primarily arginine, at the −3 position relative to the phosphorylation site, which is similar to other kinases in the CAMK group (Fig. 7A). PASK also showed a significant preference for basic residues at the −5 position. Although other protein kinases such as AKT/PKB, S6K, and the PIM kinases also phosphorylate sites having arginine at the −5 position (23, 33–35), PASK is unusual in that it is selective for both histidine and arginine. In addition, PASK demonstrated some preference for polar residues at the −2 position. Note that the higher level of phosphorylation observed for peptides with serine and threonine residues at fixed positions can be an artifact arising from the presence of multiple potential sites of phosphorylation in those peptides. The observed consensus sequence identified by peptide library screening conforms well, however, to the sequence of the best characterized PASK substrate, the S. cerevisiae Ugp1 protein (6-HTKTHS*-11), which is phosphorylated both in vivo and in vitro by yeast PASK (9–12) and in vitro by human PASK (Fig. 1). This sequence contains the −2 threonine, −3 lysine, and −5 histidine, which is consistent with the preference observed from the peptide library screen (Fig. 7A).
FIGURE 7.

Structural basis for the substrate specificity of the PASK kinase domain. A, full-length human PASK was used to phosphorylate a 198-member combinatorial peptide library. Peptides had the generic sequence YAXXXXXS/TXXXXAGKK (biotin), with the indicated residue (columns) fixed at the indicated position (row) relative to the central phosphoacceptor site. B, stereo view of the PASK kinase domain (blue) is superimposed on the PIM1 structure (red) bound to PIMTIDE substrate peptide (green; sequence KRRRHPS).
A comparison of the crystal structure of a PIM1-peptide substrate complex (23) with our PASK structure (Fig. 7B) suggests a similar mechanism of substrate recognition. Two PIM1 residues that form ionic interactions with the −3 arginine residue are conserved in PASK (Asp1089 and Glu1132) and presumably function in a similar manner. In addition, by analogy with PIM1, PASK is predicted to accommodate basic residues at the −5 position in a pocket composed of multiple acidic and polar residues (Asp1095, Asp1131, Thr1195, and Glu1200). Although three acidic residues and one threonine occupy analogous positions in PIM1, the precise arrangement of these residues differs from those in PASK (Fig. 7B). Such differences may enable accommodation of distinct basic residues at the −5 position by the two kinases.
DISCUSSION
We have demonstrated that PASK is an RD protein kinase that bypasses the requirement for activation loop phosphorylation to generate the active kinase conformation. PASK is unique among activation loop phosphorylation-independent RD kinases in that it has retained a phosphorylatable residue (threonine) in the activation loop. Other RD kinases that do not require activation loop phosphorylation typically have negatively charged residues like glutamate (PHK1) or aspartate (PIM1), or nonpolar residues (CHK1) in their activation loops. This distinction suggests that although phosphorylation may not be required for basal activity of the kinase, phosphorylation of the activation loop in PASK could modulate activity in specific contexts or regulate activity in a substrate-specific or otherwise noncanonical manner. It is interesting to note that although this activation loop threonine is conserved in most PASK orthologs, the two S. cerevisiae PASK orthologs, both of which are catalytically active (9, 10, 12), have a valine residue in this position. This difference provides additional evidence that phosphorylation at this site is not required for catalytic activity in this kinase family. It was observed previously that T1161A and T1165A mutants of PASK, which were expressed and purified from insect cells using recombinant baculovirus, had severely reduced catalytic activity (17). Based on in vivo and in vitro kinase assay data generated from PASK purified from mammalian cells (Fig. 1), we conclude that Thr1161 phosphorylation is not required for PASK catalytic activity. Further, we did not detect the presence of phosphothreonine 1161 or phosphothreonine 1165 in mass spectrometry experiments (17). It is possible that the previous results using baculovirus-expressed protein were the result of misfolding of the T1161A mutant in that heterologous expression system. Residues equivalent to Thr1165 in other kinases have been shown to play an important structural role in maintaining the orientation of the activation loop for substrate binding or catalysis. It is likely that this, rather than loss of phosphorylation, is the explanation for impaired activity of the T1165A mutant.
Our PASK structure shows several unusual features. The αC region of the N-terminal lobe is markedly different from the typical protein kinase fold due to a β-hairpin replacing part of the αC helix. Of the known human kinase domain structures, only the PIM1 protein kinase shows a structural arrangement similar to PASK (Fig. 3). The significance of this secondary structure distinction is not known for either of these kinases. One possible function is to anchor the αC helix in the absence of activation loop phosphorylation. In most kinases regulated by activation loop phosphorylation, the αC helix is stabilized in the active conformation by polar contacts with the phosphorylated activation loop residue. In the absence of phosphorylation and hence such an interaction, additional structural changes are likely required to orient and stabilize the αC helix. To this end, Asp1043 from the βH1 strand interacts with the αC helix residue Arg1058 in PASK (Fig. 4B). Arg1058 also hydrogen bonds with Tyr1152 of the C-terminal lobe, thereby maintaining the αC helix in an orientation amenable to kinase catalytic activity.
We identified several sequence-structure adaptations in PASK that allow the kinase to adopt the active conformation independent of activation loop phosphorylation. One important sequence difference is the presence of alanine at position 1151, which is occupied by lysine in many protein kinases that require activation loop phosphorylation (Fig. 5A). Mutation of this alanine to lysine lowered the activity of PASK (Fig. 5B), which was rescued by a phosphomimetic substitution in the activation loop (T1161E) to provide negative charge in the RD pocket. Some activation loop phosphorylation-independent kinases, such as glycogen synthase kinase 3β, have an arginine at this position (Fig. 5A, highlighted in yellow). In this case, the presence of a phosphorylated serine in the substrate peptide provides necessary negative charge to neutralize the RD pocket, allowing for glycogen synthase kinase 3β activation (supplemental Fig. 4) (39).
The PASK catalytic domain is also unusual from the standpoint of substrate specificity. The structurally related PIM1 kinase has similar peptide substrate specificity, in particular at the −3 and −5 positions, and it is likely that analogous binding pockets are used by these two kinases to accommodate basic residues in these substrate positions. Interestingly, AKT/PKB, despite having similar specificity, uses a distinct −5 interacting pocket that shares only a single residue with that of either PIM1 or PASK (30). Selectivity for histidine at the −5 position is unusual in serine/threonine kinases, having been observed previously only for human LATS (36) and its yeast homolog Cbk1 (37). However, PASK residues in the −5 interacting pocket are distinct from LATS and Cbk1, which are more closely related to AKT/PKB. PASK, therefore, appears to use an unprecedented structural solution for recognition of a −5 histidine in peptide substrates. Interestingly, the S. cerevisiae PASK ortholog Psk2 shows similar preference for −5 histidine in its peptide substrates (38). The phosphorylation site of the major bona fide Psk2 substrate Ugp1 (6-HTKTHS*-11) matches the consensus observed for human PASK, suggesting that PASK targeting to authentic phosphorylation sites in vivo uses similar mechanisms to that observed in the crystal structure and in in vitro experiments.
Supplementary Material
Acknowledgments
We thank Chris Hill for critical reading of the manuscript, Frank Whitby for help generating electron density maps for PASK, and Khadija Khan for technical assistance.
This work was funded by National Institutes of Health Grants U54 GM074945 (to New York SGX Research Center for Structural Genomics, principal investigator, S. K. Burley), DK071962 (to J. R.) and GM079498 (to B. E. T.). Use of the Advanced Photon Source was supported by the United States Dept. of Energy, Office of Basic Energy Sciences. Use of the LRL-CAT beam line facilities at Sector 31 of the APS (formerly SGX-CAT) was provided by Eli Lilly and Company, which operates the beamline (formerly SGX Pharmaceuticals).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4 and Methods.
- PAS
- Per-Arnt-Sim
- PASK
- PAS domain-containing kinase
- PDB
- Protein Data Bank
- Ugp1
- UDP-glucose pyrophosphorylase.
REFERENCES
- 1.Lindsley J. E., Rutter J. (2004) Comp Biochem. Physiol. B. Biochem. Mol. Biol. 139, 543–559 [DOI] [PubMed] [Google Scholar]
- 2.Marshall S. (2006) Sci. STKE 2006, re7. [DOI] [PubMed] [Google Scholar]
- 3.Zhang B. B., Zhou G., Li C. (2009) Cell Metab. 9, 407–416 [DOI] [PubMed] [Google Scholar]
- 4.Sarbassov D. D., Ali S. M., Sabatini D. M. (2005) Curr. Opin. Cell Biol. 17, 596–603 [DOI] [PubMed] [Google Scholar]
- 5.Martin D. E., Hall M. N. (2005) Curr. Opin. Cell Biol. 17, 158–166 [DOI] [PubMed] [Google Scholar]
- 6.Luo Z., Saha A. K., Xiang X., Ruderman N. B. (2005) Trends Pharmacol. Sci. 26, 69–76 [DOI] [PubMed] [Google Scholar]
- 7.Mamane Y., Petroulakis E., LeBacquer O., Sonenberg N. (2006) Oncogene 25, 6416–6422 [DOI] [PubMed] [Google Scholar]
- 8.Hao H. X., Rutter J. (2008) IUBMB Life 60, 204–209 [DOI] [PubMed] [Google Scholar]
- 9.Smith T. L., Rutter J. (2007) Mol. Cell 26, 491–499 [DOI] [PubMed] [Google Scholar]
- 10.Grose J. H., Smith T. L., Sabic H., Rutter J. (2007) EMBO J. 26, 4824–4830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grose J. H., Sundwall E., Rutter J. (2009) Cell Cycle 8, 1824–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutter J., Probst B. L., McKnight S. L. (2002) Cell 111, 17–28 [DOI] [PubMed] [Google Scholar]
- 13.da Silva Xavier G., Rutter J., Rutter G. A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8319–8324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao H. X., Cardon C. M., Swiatek W., Cooksey R. C., Smith T. L., Wilde J., Boudina S., Abel E. D., McClain D. A., Rutter J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 15466–15471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor B. L., Zhulin I. B. (1999) Microbiol. Mol. Biol. Rev. 63, 479–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amezcua C. A., Harper S. M., Rutter J., Gardner K. H. (2002) Structure 10, 1349–1361 [DOI] [PubMed] [Google Scholar]
- 17.Rutter J., Michnoff C. H., Harper S. M., Gardner K. H., McKnight S. L. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8991–8996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McRee D. E. (1999) J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 20.CCP4 (1994) Acta Cryst. D50, 760–763 [DOI] [PubMed] [Google Scholar]
- 21.Johnson L. N., Noble M. E., Owen D. J. (1996) Cell 85, 149–158 [DOI] [PubMed] [Google Scholar]
- 22.Holm L., Kääriäinen S., Rosenström P., Schenkel A. (2008) Bioinformatics 24, 2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bullock A. N., Debreczeni J., Amos A. L., Knapp S., Turk B. E. (2005) J. Biol. Chem. 280, 41675–41682 [DOI] [PubMed] [Google Scholar]
- 24.Komander D., Kular G., Deak M., Alessi D. R., van Aalten D. M. (2005) J. Biol. Chem. 280, 18797–18802 [DOI] [PubMed] [Google Scholar]
- 25.Nolen B., Taylor S., Ghosh G. (2004) Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 26.Chen P., Luo C., Deng Y., Ryan K., Register J., Margosiak S., Tempczyk-Russell A., Nguyen B., Myers P., Lundgren K., Kan C. C., O'Connor P. M. (2000) Cell 100, 681–692 [DOI] [PubMed] [Google Scholar]
- 27.Kornev A. P., Haste N. M., Taylor S. S., Eyck L. F. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 17783–17788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krupa A., Preethi G., Srinivasan N. (2004) J. Mol. Biol. 339, 1025–1039 [DOI] [PubMed] [Google Scholar]
- 29.Huang X., Begley M., Morgenstern K. A., Gu Y., Rose P., Zhao H., Zhu X. (2003) Structure 11, 21–30 [DOI] [PubMed] [Google Scholar]
- 30.Yang J., Cron P., Good V. M., Thompson V., Hemmings B. A., Barford D. (2002) Nat. Struct. Biol. 9, 940–944 [DOI] [PubMed] [Google Scholar]
- 31.Zhang F., Strand A., Robbins D., Cobb M. H., Goldsmith E. J. (1994) Nature 367, 704–711 [DOI] [PubMed] [Google Scholar]
- 32.Canagarajah B. J., Khokhlatchev A., Cobb M. H., Goldsmith E. J. (1997) Cell 90, 859–869 [DOI] [PubMed] [Google Scholar]
- 33.Hutti J. E., Jarrell E. T., Chang J. D., Abbott D. W., Storz P., Toker A., Cantley L. C., Turk B. E. (2004) Nat. Methods 1, 27–29 [DOI] [PubMed] [Google Scholar]
- 34.Alessi D. R., Caudwell F. B., Andjelkovic M., Hemmings B. A., Cohen P. (1996) FEBS Lett. 399, 333–338 [DOI] [PubMed] [Google Scholar]
- 35.Miller M. L., Jensen L. J., Diella F., Jørgensen C., Tinti M., Li L., Hsiung M., Parker S. A., Bordeaux J., Sicheritz-Ponten T., Olhovsky M., Pasculescu A., Alexander J., Knapp S., Blom N., Bork P., Li S., Cesareni G., Pawson T., Turk B. E., Yaffe M. B., Brunak S., Linding R. (2008) Sci. Signal. 1, ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao Y., Chun A., Cheung K., Rashidi B., Yang X. (2008) J. Biol. Chem. 283, 5496–5509 [DOI] [PubMed] [Google Scholar]
- 37.Mazanka E., Alexander J., Yeh B. J., Charoenpong P., Lowery D. M., Yaffe M., Weiss E. L. (2008) PLoS Biol. 6, e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mok J., Kim P. M., Lam H. Y., Piccirillo S., Zhou X., Jeschke G. R., Sheridan D. L., Parker S. A., Desai V., Jwa M., Cameroni E., Niu H., Good M., Remenyi A., Ma J. L., Sheu Y. J., Sassi H. E., Sopko R., Chan C. S., De Virgilio C., Hollingsworth N. M., Lim W. A., Stern D. F., Stillman B., Andrews B. J., Gerstein M. B., Snyder M., Turk B. E. (2010) Sci. Signal. 3, ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ter Haar E., Coll J. T., Austen D. A., Hsiao H. M., Swenson L., Jain J. (2001) Nat. Struct. Biol. 8, 593–596 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.