

Abstract
Junctional adhesion molecule-C (JAM-C) is an adhesion molecule expressed by endothelial cells that plays a role in tight junction formation, leukocyte adhesion, and trans-endothelial migration. In the present study, we investigated whether JAM-C is found in soluble form and if soluble JAM-C (sJAM-C) mediates angiogenesis. We found that JAM-C is present in soluble form in normal serum and elevated in rheumatoid arthritis (RA) serum. The concentration of sJAM-C is also elevated locally in RA synovial fluid compared to RA serum or osteoarthritis synovial fluid. sJAM-C was also present in the culture supernatant of human microvascular endothelial cells (HMVECs) and immortalized human dermal microvascular endothelial cells (HMEC-1s), and its concentration was increased following cytokine stimulation. In addition, sJAM-C cleavage from the cell surface was mediated in part by a disintegrin and metalloproteinase 10 (ADAM10) and ADAM17. In functional assays, sJAM-C was both chemotatic and chemokinetic for HMVECs, and induced HMVEC tube formation on Matrigel in vitro. Neutralizing anti-JAM-C antibodies inhibited RA synovial fluid induced HMVEC chemotaxis and sJAM-C induced HMVEC tube formation on Matrigel. sJAM-C also induced angiogenesis in vivo in the Matrigel plug and sponge granuloma models. Moreover, sJAM-C mediated HMVEC chemotaxis was dependent on Src, p38, and PI3K. Our results show that JAM-C exists in soluble form, and suggest that modulation of sJAM-C may provide a novel route for controling pathological angiogenesis.
Introduction
Angiogenesis is a highly regulated process of new blood vessel formation from pre-existing vessels. It is important in a number of physiological processes including reproduction, development, and wound healing; and is dysregulated in disease states such as cardiovascular disease, rheumatoid arthritis (RA), and tumor growth (1). The initiation of angiogenesis depends upon the release of proangiogenic mediators which activate endothelial cells (ECs) and initiate their proliferation and migration (2). Several types of proangiogenic mediators have been identified including growth factors, cytokines, chemokines, and cellular adhesion molecules (1).
Adhesion molecules play a central role in angiogenesis. ECs utilize adhesion molecules for homophilic and heterophilic adhesion, and adhesion to and migration through the extracellular matrix, a key step in the progression of angiogenesis (3). In addition, stimulated increase of adhesion molecule expression results in their shedding or release from ECs (4). Several EC adhesion molecules have been found in soluble form including ICAM-1, VCAM-1, and E-selectin (5). Previously our laboratory has shown that the soluble forms of E-selectin and VCAM-1 are angiogenic (6). Both adhesion molecules induce EC chemotaxis, as well as angiogenic responses in vivo (6).
Junctional adhesion molecules (JAMs) are a recently described subfamily of the immunoglobulin supergene family that localize to tight junctions between epithelial cells and between ECs (7). To date five members of the JAM family have been identified; JAM-A (8), JAM-B (9, 10), JAM-C (11, 12), JAM4 (13), and JAML (14). On the surface of ECs, JAMs control tight junction maintenance by engaging in homophilic and heterophilic interactions with neighboring JAM molecules (11, 15, 16). In addition to binding interactions between family members, JAMs can be redistributed to the apical surface of ECs and bind specific leukocyte integrins (17-20). By undergoing an upregulation and redistribution to the cell surface from the junctional interface, JAMs mediate the influx of leukocytes during inflammation and injury. We have previously shown that JAM-C is overexpressed on RA synovial fibroblasts and mediates myeloid cell adhesion and retention in the RA synovium (21).
Recent studies have begun to demonstrate the role that JAMs play in angiogenesis. JAM-A has been shown to interact with integrin αvβ3 to mediate basic fibroblast growth factor (bFGF) induced angiogenesis (22-24). In addition, recent work has suggested an indirect role for JAM-C in angiogenesis (25). In this study, a neutralizing anti-JAM-C antibody abolished angiogenesis ex vivo and in vivo, and reduced tumor growth and vascularization (25). Currently, we hypothesized that JAM-C may be a soluble mediator of angiogenesis. We report that JAM-C is present in soluble form and elevated in RA serum and synovial fluid. Soluble JAM-C (sJAM-C) was also found to be present in the culture supernatant of cytokine stimulated ECs. Moreover, we demonstrate that sJAM-C stimulates human microvascular endothelial cell (HMVEC) migration in vitro, in a process that requires Src, p38, and PI3K. In addition, using the Matrigel plug and sponge granuloma models, we found that sJAM-C stimulated angiogenesis in vivo. Our results show that JAM-C exists in soluble form and that sJAM-C is a potent proangiogenic mediator.
Methods
Patients
Synovial fluids were isolated from patients meeting the ACR criteria for psoriatic arthritis (PsA), RA, and OA by arthrocentesis. RA and normal peripheral blood was obtained by venipuncture. All specimens were obtained following approval from the University of Michigan Institutional Review Board.
Animals
All experiments performed with animals were done with approval from the University of Michigan Committee on Use and Care of Animals. C57BL/6 mice (National Caner Institute, Bethesda, MD) were used for both the Matrigel plug and sponge granuloma experiments.
Cell culture
HMVECs (2 × 106/well) were grown in complete EBM-2 media with EGM-2 SingleQuots (Lonza, Switzerland). Twenty four hours prior to the experiment, the media was changed to serum free EBM-2 with or without TNF-α (25 ng/ml, R&D Systems, Minneapolis, MN). Cell culture supernatants were collected 48 hours after the stimulation, cells and cellular debris were removed by centrifugation, and the supernatants were concentrated using Amicon Ultra centrifugal filters (3,000 molecular weight cut off, Millipore, Billerica, MA) following the manufacturer’s instructions.
In addition, SV-40 immortalized human dermal microvascular endothelial cells (HMEC-1s, 2 × 106/well) were grown in complete EBM-2 media with EGM-2 SingleQuots (Lonza, Switzerland). In one series of experiments, the media was changed to serum free EBM-2 24 hours prior to the experiment and then the HMEC-1s were untreated or stimulated with the following mediators (all from R&D Systems) at concentrations previously established (26, 27): TNF-α (25 ng/ml), IL-1β (25 ng/ml), IFN-γ (25 ng/ml), IL-18 (25 ng/ml), IL-17 (25 ng/ml), LPS (50 μg/ml), macrophage inhibitory factor (MIF, 300 ng/ml), basic fibroblast growth factor (bFGF, 50 ng/ml), acidic FGF (aFGF, 50 ng/ml), or PMA (50 ng/ml). Cell culture supernatants were collected at 48 hours and treated as above.
In a second series of experiments, HMEC-1s were cultured as above and stimulated with TNF-α (25 ng/ml) in the presence of DMSO (solvent control), TAPI-2 (a broad spectrum inhibitor of matrix metalloproteinases (MMPs), TNF-α converting enzyme (TACE), and a disintegrin and metalloproteinases (ADAMs), 20 μM, Calbiochem), GM 1489 (an inhibitor of MMPs-1, -2, -3, -8, and -9, 20 μM, Calbiochem), aprotinin (a serine protease inhibitor, 300 nM, Sigma Aldrich), leupeptin (an inhibitor of serine and cysteine proteases, 5 mM, Sigma Aldrich), pepstatin A (an aspartic protease inhibitor, 3 μM, Sigma Aldrich), and phenylmethylsulphonyl fluoride (PMSF, an inhibitor of serine proteases, 575 μM, Sigma Aldrich). Cell culture supernatants were collected at 48 hours and treated as above.
To confirm the above results, HMVECs were cultured as described above. After reaching approximately 70% confluence, the cells were transfected with siRNAs using TranisIT-TKO (Mirus, Madison, WI) following the manufacturer’s instructions. Control, ADAM10, and ADAM17 siRNAs were purchased from Invitrogen (Carlsbad, CA). The transfection was allowed to proceed for 72 hours, at which point cell culture supernatants were collected and treated as described above. Transfection efficiency was monitored with the use of fluorescence-labeled control siRNA and was found to be greater than 90%. Specific knockdown of ADAM10 and ADAM17 was confirmed by Western blotting using antibodies specific for either ADAM10 or ADAM17 (both from Abcam, Cambridge, MA).
ELISA
An ELISA was designed to determine the concentration of sJAM-C in biological fluids and cell culture supernatants. 96-well plates (Nalge Nunc Inc., Rochester, NY) were coated overnight at 4°C with goat anti-human JAM-C antibody (R&D Systems). The plates were then washed and blocked with Starting Block blocking buffer (Thermo Scientific, Waltham, MA), and incubated overnight with serum, synovial fluids or supernatants at 4°C. Prior to use, serum and synovial fluids were immunodepleted of rheumatoid factor to avoid interference with the assay (28). The plates were then washed and biotinylated goat anti-human JAM-C antibody (R&D Systems) was added, followed by Streptavidin-HRP (BD Bioscience, San Jose, CA). The plates were developed using tetramethylbenzidine (TMB) and were read on a microplate reader. A standard curve was prepared using sJAM-C/Fc (R&D Systems). PBS served as the negative control.
Expression and purification of sJAM-C
sJAM-C was produced as previously described (12). Briefly, a pcDNA vector containing murine JAM-C in frame with a Flag tag sequence was expanded in chemically competent DH5α cells (Invitrogen) following the manufacturer’s protocol. BOSC23 cells were then transfected with the vector using a calcium-phosphate technique (12). Flag tagged sJAM-C was purified using an anti-Flag M2 affinity gel column (Sigma-Aldrich). The purity and specificity of sJAM-C was confirmed by silver staining, Western blot and ELISA. The amount of endotoxin in the purified sJAM-C was determined using the QCL-1000 Endpoint Chromogenic LAL assay following the manufacturer’s instructions (Lonza).
Western blot
Western blotting was performed as described previously (21). To determine the presence of sJAM-C in HMVEC supernatants, a mouse monoclonal anti-JAM-C antibody was used (R&D Systems). For cell signaling experiments, antibodies against phosphorylated and total Src family kinases, p38, and PI3K (Cell Signaling Technology, Danvers, MA) were used. As a loading control, the blots were stripped and probed with an anti-actin antibody (Sigma Aldrich).
In vitro HMVEC chemotaxis assays
HMVEC chemotaxis assays were preformed as previously described (26). sJAM-C was diluted in PBS and used as a test substance at concentrations ranging from 1 μM to 10 pM. bFGF (60 nM) was used as a positive control and PBS was the negative control.
To determine if the sJAM-C present in RA synovial fluid contributes to RA synovial fluid mediated HMVEC chemotaxis, we neutralized sJAM-C and performed HMVEC chemotaxis. RA synovial fluids were first depleted of rheumatoid factor and then incubated with neutralizing anti-JAM-C antibodies F26 and H33 (each at 25 μg/ml) or rat IgG (50 μg/ml, negative control) for 15 minutes prior to the assay. The depleted RA synovial fluids were then used as test substances in the assay.
Checkerboard analysis was performed to determine if sJAM-C was chemotatic and/or chemokinetic for HMVECs. HMVEC chemotaxis was performed with concentrations of sJAM-C in the upper chamber ranging from 0 - 100 nM and concentrations of sJAM-C in the lower chamber ranging from 0 - 100 nM. PBS was used as a negative control and bFGF (60 nM) was used as a positive control.
To determine which kinases were required for sJAM-C mediated HMVEC chemotaxis, cells were incubated with chemical signaling inhibitors. HMVECs were preincubated with chemical signaling inhibitors for 2 hours prior to the assay, and the inhibitors were present in the lower chamber with the HMVECs during the assay. The following inhibitors were purchased from and used at concentrations recommended by Calbiochem (La Jolla, CA): PD98059 (Erk1/2 inhibitor, 10 μM), LY294002 (PI3K inhibitor, 10 μM), PP2 (Src inhibitor, 1 μM), and SB203580 (p38 MAPK inhibitor, 10 μM), and suramin (G protein inhibitor, 40 μM).
In vitro Matrigel tube formation assays
Matrigel tube formation assays using growth factor reduced Matrigel (BD Bioscience) were performed (26). Test substances used were sJAM-C (10 nM), bFGF (60 nM, R&D Systems, positive control), and PBS (negative control). After an overnight incubation at 37°C, the cells were fixed and counterstained. Photographs (100x) were taken and tubes were counted by a blinded observer. Tubes were defined as elongated connecting branches between two identifiable HMVECs.
A second series of experiments was performed to determine if depletion of sJAM-C from the sJAM-C protein preparation inhibited the ability of sJAM-C to induce HMVEC tube formation on Matrigel. Matrigel tube formation assays were performed with sJAM-C (50 nM) incubated with either rat IgG (50 nM, isotype control) or anti-JAM-C antibodies (F26 and H33, 25 μg/ml each). After an overnight incubation at 37°C, the cells were fixed and counterstained. Photographs (100x) were taken and tubes were counted by a blinded observer.
In vivo Matrigel plug angiogenesis assays
Matrigel plug assays were performed as previously described (26). C57BL/6 mice were anesthetized and injected subcutaneouslywith 500 μL of growth factor reduced Matrigel containing either sJAM-C (100 nM), aFGF (R&D Systems, 62.5 pM, positive control), or PBS (negative control). Mice were euthanized afterday 7, plugs dissected, and angiogenesis was analyzed by hemoglobin measurement using the TMB method (26). Hemoglobin measurements were normalized to plug weight. Alternatively, some of the plugs removed were frozen in optimal cutting temperature (OCT) media for immunohistological examination. Sections were cut and immunohistochemistry was performed as previously described (21). Polyclonal rabbit anti-von Willebrand factor (vWF, Dako, Denmark) was used as a primary antibody, with rabbit IgG serving as a negative control. Fluorescein isothiocyanate (FITC) labeled anti-rabbit IgG (Invitrogen) was used as a secondary antibody, and 4’,6-diamidino-2-phenylindole (DAPI, Invitrogen) was used to observe nuclear staining. Photographs were taken with a fluorescence microscope (Olympus).
In vivo sponge granuloma angiogenesis assays
The in vivo sponge granuloma assay is a model of inflammatory angiogenesis, and was performed as previously described (29). Briefly, 1 cm2 sponge discs were cut from 2-mm-thick polyvinyl alcohol foam sponges (M-pact, Eudora, KS). A 2-mm pellet was cut into the disc center to serve as a depot for sJAM-C (250 nM), aFGF (62.5 pM, positive control), or PBS (negative control). After adding the test substances to the center pellet, the sponges were sealed with Millipore filters (0.45 μm) using Millipore glue number 1 (Millipore, Bedford, MA). C57BL/6 mice were then anesthetized and the sponges were inserted subcutaneously into the back of each mouse. After 7 days, the animals were sacrificed, and the sponge discs were harvested. The sponges were then cut, minced, homogenized, and analyzed for the amount of hemoglobin per sponge using the TMB method. Hemoglobin values were normalized to sponge weight.
Statistical Analysis
Data were analyzed using Student’s t-test assuming equal variances. P values < 0.05 were considered statistically significant. Data are represented as the mean ± standard error of the mean (SEM).
Results
JAM-C is present in soluble form
To determine if JAM-C was present in soluble form we designed an ELISA. We found that JAM-C is soluble and detectable in normal serum (mean 0.7 ng/ml ± 0.1, figure 1), and that it is significantly elevated in RA serum (1.2 ng/ml ± 0.1, p<0.05). Moreover, we found that sJAM-C is upregulated locally in RA synovial fluid (2.4 ng/ml ± 0.3, p<0.05) and PsA synovial fluid (2.3 ng/ml ± 0.5, p<0.05), and that both are significantly elevated compared to noninflammatory osteoarthritis (OA) synovial fluid (1.3 ng/ml ± 0.1, p<0.05). This is the demonstration of JAM-C in soluble form in biological fluids.
Figure 1. JAM-C is present in soluble form and elevated in RA synovial fluid.
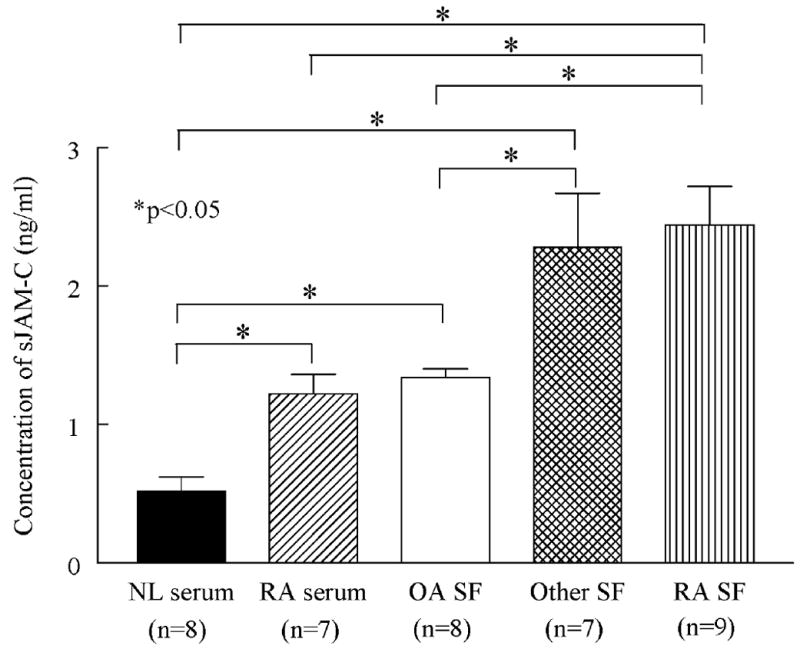
The amount of sJAM-C was determined in normal serum, RA serum, OA synovial fluid, PsA synovial fluid, and RA synovial fluid by ELISA. Means are given with SEM. Differences were determined using the student’s t test and p values less than 0.05 were significant. n=number of patients.
sJAM-C is present in EC supernatants
To determine the source and molecular weight of sJAM-C, HMVECs and HMEC-1s were cultured, the cell culture supernatants were collected and concentrated, and Western blotting and ELISAs were performed. Our results indicate that sJAM-C is present in the culture supernatant of HMVECs (mean 15.5 pg/ml ± 8.4) and HMEC-1s (80.0 pg/ml ± 50.0) (figure 2). In addition, sJAM-C was elevated in the culture supernatant of TNF-α stimulated HMVECs (43.4 pg/ml ± 13.9) compared to nonstimulated (figure 2a). The sJAM-C in HMVEC supernatants had an approximate molecular weight of 40 kDa, whereas the anti-JAM-C antibody identified two proteins of 40 kDa and 50 kDa, suggesting the presence of both soluble and full length JAM-C in HMVEC protein lysates (figure 2b).
Figure 2. sJAM-C is released from the surface of ECs following stimulation with inflammatory mediators.
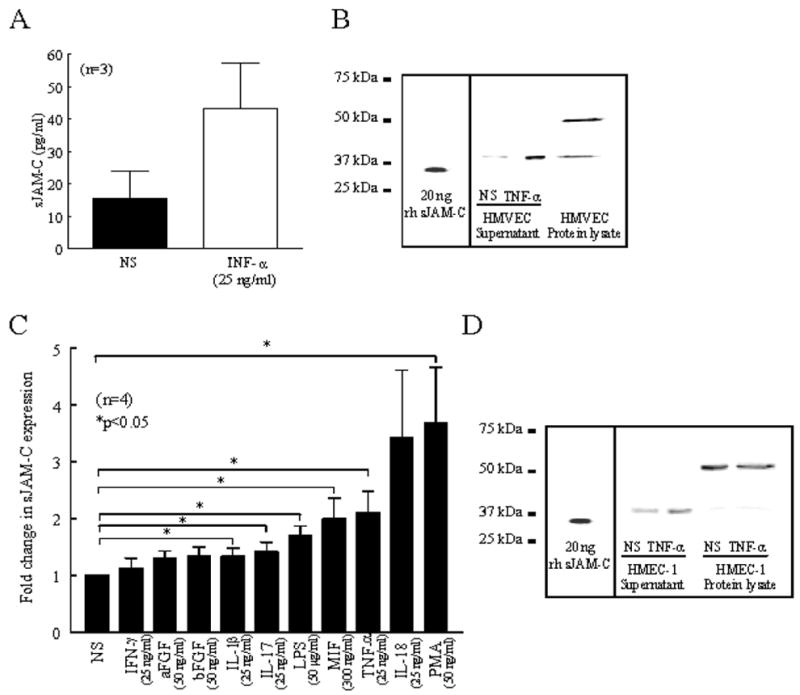
ELISAs and Western blotting were used to determine the concentration and molecular weight of sJAM-C in HMVEC and HMEC-1 cell culture supernatants. A) HMVECs were stimulated with or without TNF-α for 48 hours and cell culture supernatants were collected, concentrated, and assayed by ELISA. B) Western blotting was used to determine the presence and molecular weight of sJAM-C in nonstimulated (NS) or TNF-α stimulated HMVEC supernatants and protein lysate compared to recombinant human sJAM-C. C) HMEC-1s were stimulated with or without IFN-γ, aFGF, bFGF, IL-1β, IL-17, LPS, MIF, TNF-α, IL-18, or PMA for 48 hours and cell culture supernatants were collected, concentrated, and subjected to ELISA. D) Western blotting was used to determine the presence and molecular weight of sJAM-C in nonstimulated (NS) or TNF-α stimulated HMEC-1 supernatants and protein lysates compared to recombinant human sJAM-C. For the ELISA experiments, each sample was tested in duplicate and n=number of individual experiments. Means are given with SEM. Differences were determined using the student’s t test and p values less than 0.05 were significant.
We then determined if other proinflammatory or proangiogenic mediators were able to increase EC expression of sJAM-C. We found that sJAM-C was significantly elevated in the culture supernatant of IL-1β, IL-17, LPS, MIF, TNF-α, or PMA stimulated HMEC-1s compared to nonstimulated cells (all p<0.05) (figure 2c). As with the HMVEC supernatants, we again observed that the form of JAM-C present in HMEC-1 supernatants had an apparent molecular weight of 40 kDa, with no presence of the full length molecular weight protein (figure 2d). These results suggest that ECs release sJAM-C following stimulation with proinflammatory mediators.
ADAM10 and ADAM17 mediate release of sJAM-C
After finding that sJAM-C was present in the cell culture supernatant of HMEC-1s and upregulated in the presence of proinflammatory mediators, we sought to determine the mechanism for its release. First we performed quantitative PCR to determine if the increase of sJAM-C following TNF-α stimulation was the result of increased JAM-C production. However, we found that TNF-α stimulation had no effect on JAM-C mRNA expression in HMEC-1s (data not shown). Therefore, we determined if the increase in sJAM-C in TNF-α stimulated EC supernatants was a result of membrane bound JAM-C being shed from the cell surface. HMEC-1s were stimulated with TNF-α in the presence of various sheddase inhibitors (figure 3a). We found that the concentration of sJAM-C was significantly reduced in the cell culture supernatants of HMEC-1s stimulated with TNF-α in the presence of TAPI-2 (p<0.05), an inhibitor of MMPs, TACE, and ADAMs. Specific inhibitors of MMPs, serine, cysteine, or aspartic proteases had no effect on the release of JAM-C from the surface of these cells (figure 3a).
Figure 3. JAM-C is cleaved into soluble form by ADAM10 and ADAM17.

ELISAs were used to determine the concentration of sJAM-C in HMEC-1 or HMVEC cell culture supernatants. A) HMEC-1s were stimulated with TNF-α for 48 hours in the presence of DMSO, TAPI-2, Aprotinin, GM 1489, Leupeptin, Pepstatin A, or PMSF and cell culture supernatants were collected, concentrated, and subjected to ELISA. B) HMVECs were transfected with control, ADAM10, or ADAM17 siRNA for 72 hours and cell culture supernatants were collected, concentrated, and assayed by ELISA. Means are given with SEM. Differences were determined using the student’s t test and p values less than 0.05 were significant. Each sample was tested in duplicate. n=number of individual experiments.
We then performed siRNA experiments to further elucidate which type of protease inhibited by TAPI-2 is responsible for the release of JAM-C from the cell surface. We found that the amount of sJAM-C in the culture supernatants of HMVECs transfected with specific siRNA against either ADAM10 or ADAM17 was decreased compared to those transfected with control siRNA (figure 3B). Collectively, these results suggest that JAM-C is cleaved from the surface of ECs by ADAM10 and ADAM17.
Characteristics of purified sJAM-C
To determine the role of sJAM-C in angiogenesis, we purified sJAM-C from the culture supernatant of BOSC23 cells transfected with a vector containing the extracellular domain of JAM-C as previously described (12). We found that our purified sJAM-C was greater than 95% pure by Western blot and silver staining with a molecular weight of approximately 35 kDa (figure 4a). In addition, we found that 50 nM of sJAM-C contains 0.1 EU/ml of endotoxin, which equates to 10 pg/ml or less than 0.01 ng of endotoxin/1 μg of total protein (data not shown). Collectively, this data indicates that our purified sJAM-C is free of contaminating proteins and contains only trace amounts of endotoxin.
Figure 4. sJAM-C induces HMVEC chemotaxis and contributes to a significant portion of RA synovial fluid induced HMVEC chemotaxis.
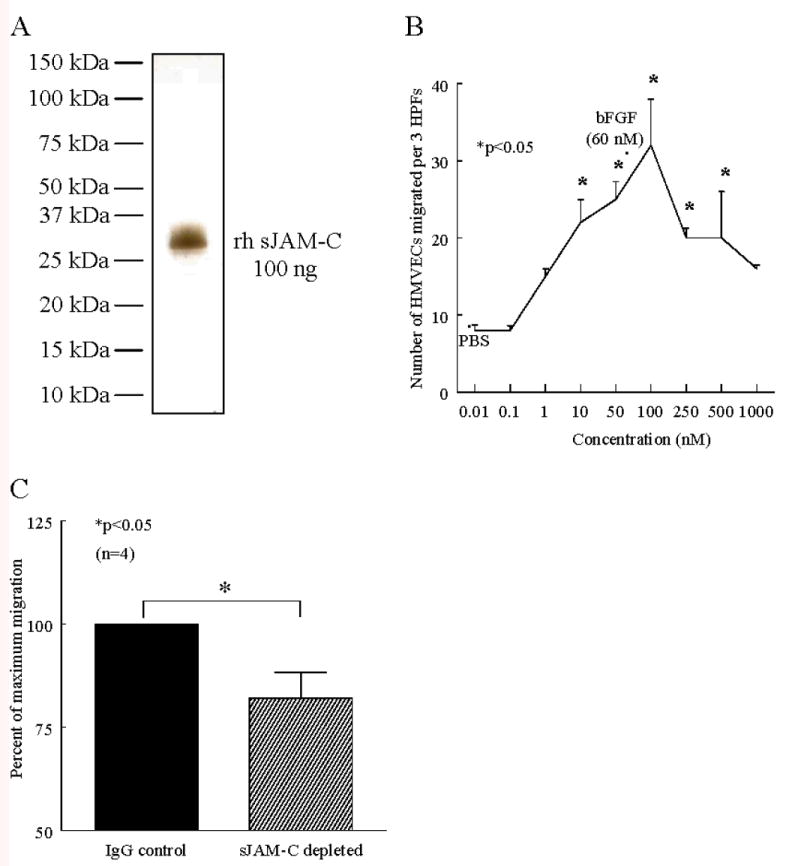
A) Our sJAM-C protein preparation (100 ng) was ran on an SDS-PAGE gel and silver stained. B-C) HMVEC chemotaxis assays were performed using a modified Boyden chamber. Three high power fields (HPF) were counted per well and the assay was performed in quadruplicate. B) sJAM-C was used as a stimulus, along with PBS as a negative control, and bFGF (60 nM) as a positive control. The assay was repeated on 3 separate occasions with similar results. C) RA synovial fluid was depleted of rheumatoid factor and either sJAM-C using anti-JAM-C antibodies F26 and H33, or IgG control antibody (all 25 μg/ml), and used as a stimulus for HMVEC chemotaxis. The percent of maximal migration was calculated by dividing the number of cells migrating in the IgG control group by the number migrating in the sJAM-C depleted group. n=the number of different RA synovial fluid samples used. For both chemotaxis assays, means are presented with SEM, and differences were determined using the student’s t test where p values less than 0.05 were significant.
sJAM-C mediates facets of angiogenesis in vitro and contributes the angiogenic potential of RA synovial fluid
We wondered weather sJAM-C may stimulate angiogenesis. Therefore, we performed in vitro HMVEC chemotaxis assays in the presence of sJAM-C, as EC chemotaxis is an initial step in the angiogenic process. Our results indicate that sJAM-C stimulates HMVEC chemotaxis in a dose dependent manner, with migration significantly greater than phosphate buffered saline (PBS), occurring from 10 nM to 500 nM (p<0.05) (figure 4a). In addition, a checkerboard analysis was performed to determine whether sJAM-C was chemotatic and/or chemokinetic for HMVECs. Our findings suggest that sJAM-C is both chemotatic and chemokinetic for HMVECs (table I).
Table I.
Checkerboard analysis of sJAM-C mediated HMVEC migration
| Lower chamber sJAM-C | Upper chamber sJAM-C1 |
||
|---|---|---|---|
| 0 nM | 10 nM | 100 nM | |
| 0 nM | 8.0 ± 0.6 | 11.0 ± 1.5* | 14.1 ± 1.2* |
| 10 nM | 8.4 ± 1.0 | 9.1 ± 0.7 | 8.0 ± 0.8 |
| 100 nM | 11.1 ± 0.9* | 10.4 ± 1.0* | 10.2 ± 1.1 |
sJAM-C was assayed for HMVEC migration using a modified Boyden chamber. Three HPFs were counted per well and the assay was performed in quadruplicate. The results represent 4 independent experiments. Results are given as means ± SEM. Positive control migration in response to bFGF (60 nM) was 13.8 ± 2.2.
p<0.05 vs PBS.
RA synovial fluid is rich in angiogenic mediators. Therefore we sought to determine if sJAM-C is a significant contributor to the chemotatic potential of RA synovial fluid for ECs in vitro. We found that depleting JAM-C from RA synovial fluids resulted in a significant decrease in the chemotatic potential of RA synovial fluid for HMVECs compared to RA synovial fluid sham depleted with an isotype matched IgG control antibody (18% decrease, p<0.05) (figure 4b). This result further demonstrates that sJAM-C is an angiogenic mediator, and that sJAM-C is a significant angiogenic component in RA synovial fluid.
As HMVEC chemotaxis is only one facet of angiogenesis, we determined whether sJAM-C can induce other aspects of new blood vessel development, and thus performed in vitro HMVEC tube formation assays. We found that sJAM-C induced a significantly greater number of HMVEC tubes on Matrigel compared to PBS (p<0.05) (figure 5a-d). Collectively, these results indicate that sJAM-C mediates angiogenesis in vitro.
Figure 5. sJAM-C induces HMVEC tube formation.
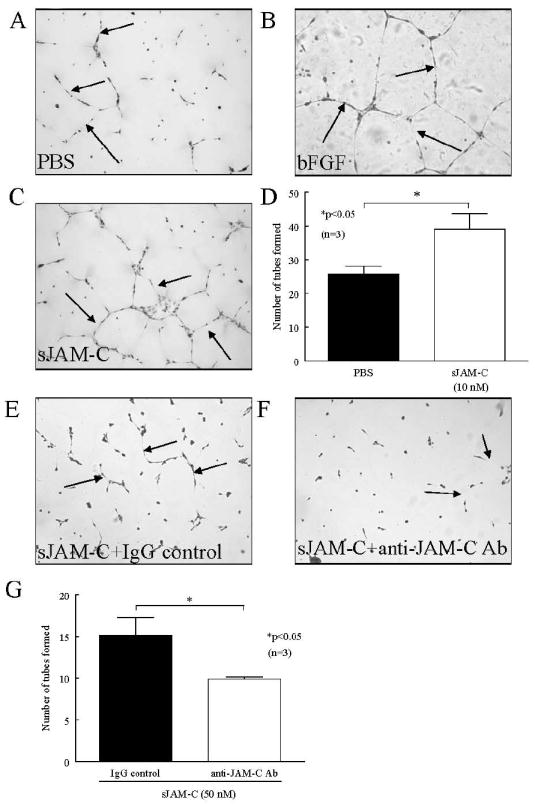
A-D) HMVEC tube formation on Matrigel in the presence of sJAM-C (10 nM) was assessed. Photographs were taken at 100x and tube formation was determined in a blinded fashion. A representative photograph of PBS (A), bFGF (B), and sJAM-C (C) treated HMVECs are shown and arrows indicate tube formation. D) HMVECs formed significantly greater number of tubes on Matrigel in response to sJAM-C compared to PBS. E-G) sJAM-C induced HMVEC tube formation on Matrigel in the presence of neutralizing anti-JAM-C antibody or an isotype control antibody was assessed. Photographs were taken at 100x and tube formation was determined in a blinded fashion. A representative photograph of sJAM-C (50 nM) with an IgG control antibody (E) and sJAM-C (50 nM) with anti-JAM-C antibodies F26 and H33 (F) treated HMVECs are shown and arrows indicate tube formation. G) The addition of neutralizing anti-JAM-C antibody significantly inhibited sJAM-C induced HMVEC tube formation on Matrigel. For both sets of experiments, means of the number of tubes per well are given with SEM. Differences were determined using the student’s t test and p values less than 0.05 were significant. Each assay was performed in triplicate. n=number of individual experiments.
To confirm that the observed tube formation on Matrigel was directly attributable to sJAM-C, and not other contaminating proteins or endotoxin, we performed Matrigel in vitro assays with neutralizing anti-JAM-C antibodies. HMVEC tube formation on Matrigel was significantly inhibited in chambers treated with sJAM-C and anti-JAM-C antibodies (F26 and H33) compared to those treated with sJAM-C and an isotype control antibody (figure 5e-g). These results suggest that the angiogenic potential of our sJAM-C preparation is specifically due to sJAM-C.
sJAM-C induces angiogenesis in vivo
To determine if sJAM-C has angiogenic properties both in vitro and in vivo we initially performed in vivo Matrigel plug angiogenesis assays. Matrigel was mixed with PBS, acidic fibroblast growth factor (aFGF), or sJAM-C and injected subcutaneously into C57BL/6 mice. Blood vessel formation was assessed by immunohistology and hemoglobin content, a measure of plug vascularity. We found that mixing Matrigel with either angiogenic aFGF or sJAM-C resulted in a greater number of blood vessels than PBS alone (figure 6a-c). Moreover, when plugs were removed and the amount of hemoglobin was determined, we observed that Matrigel plugs containing sJAM-C had a significantly greater amount of hemoglobin compared to plugs containing PBS (p<0.05) (figure 6d).
Figure 6. sJAM-C stimulates angiogenesis in vivo.
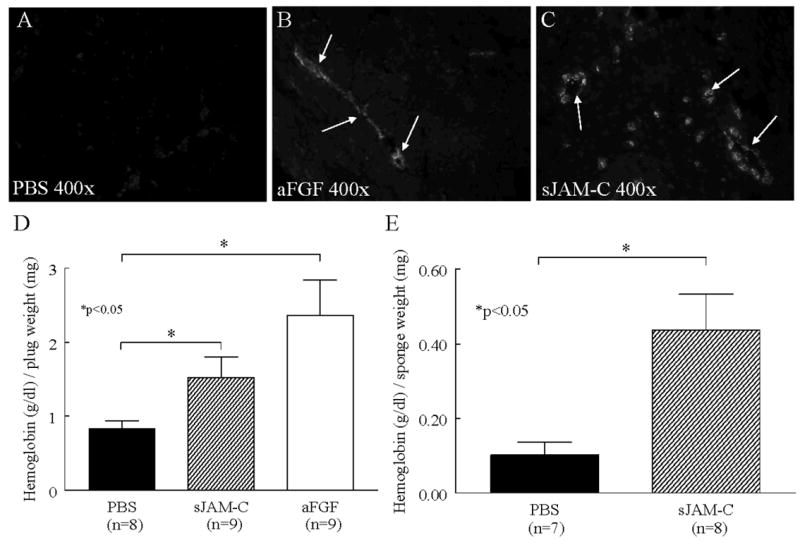
The ability of sJAM-C to mount an in vivo angiogenic response was assessed using Matrigel and sponge granuloma assays. A-D) Matrigel was mixed with either PBS, aFGF (63 pM), or sJAM-C (100 nM) and injected into C57BL/6 mice s.c. After 7 days the plugs were removed. Representative photographs of vWF staining in Matrigel plugs treated with PBS (A), aFGF (B), and sJAM-C (C) are shown at 400x. Arrows indicate positive vWF staining. D) In a separate experiment, the amount of hemoglobin per plug was determined. E) PBS or sJAM-C (250 nM) treated sponges were implanted in C57BL/6 mice. After 7 days, the sponges were removed, homogenized, and the amount of hemoglobin per sponge was determined. Means are given with SEM. Differences were determined using the student’s t test and p values less than 0.05 were significant. Each sample was tested in duplicate. n=number of mice.
In addition to the in vivo Matrigel plug angiogenesis assay, we performed an in vivo sponge granuloma angiogenesis assay, which is a model of inflammatory angiogenesis. Here we found that sponges containing sJAM-C induced a significantly greater angiogenic response compared to those with PBS (p<0.05) (figure 6e). Collectively, these findings suggest that sJAM-C is angiogenic in vivo.
Src, p38, and PI3K are required for sJAM-C mediated angiogenesis
To determine the EC signaling mechanism involved in sJAM-C mediated angiogenesis, we performed HMVEC chemotaxis assays in the presence of inhibitors to known signaling intermediates previously shown to be important in angiogenesis. Inhibitors targeting Src, p38, or PI3K kinases significantly reduced the ability of sJAM-C to induce HMVEC chemotaxis compared to sJAM-C with DMSO treated cells (all p<0.05) (figure 7a). In contrast, Erk 1/2 and G protein inhibitors had no effect on sJAM-C mediated HMVEC chemotaxis.
Figure 7. Src, p38, and PI3K are required for sJAM-C mediated HMVEC chemotaxis and are activated by sJAM-C in HMVECs.
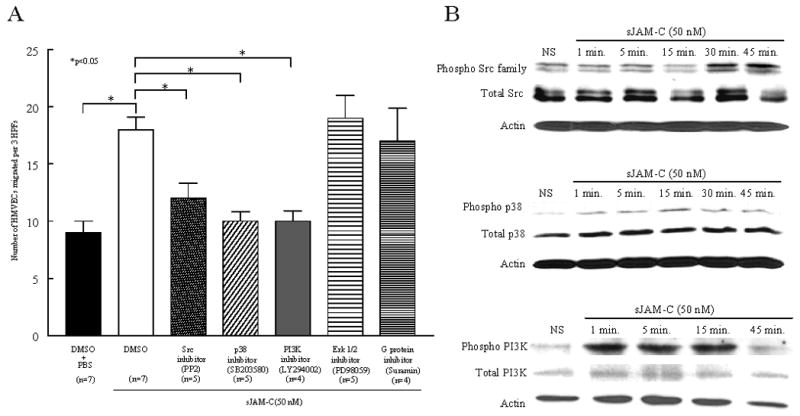
A) HMVEC chemotaxis assays were performed using a modified Boyden chamber. sJAM-C (50 nM) was used as a stimulus, with PBS serving as a negative control. HMVECs were pretreated with chemical inhibitors of Src (PP2), p38 (SB203580), PI3K (LY294002), Erk 1/2 (PD98059), G proteins (Suramin), or DMSO (vehicle control). The inhibitors were also present with the HMVECs during the assay. Three HPFs were counted per well and the assay was performed in quadruplicate. The 3 HPFs of each well were summed, the mean of the quadruplicates were calculated, and means are presented with SEM. Differences were determined using the student’s t test and p values less than 0.05 were significant. n=the number of individual experiments. B) Western blotting was performed to determine if sJAM-C stimulates the phosphorylation of Src family members, p38, or PI3K in a time dependent manner in HMVECs. Each experiment was performed on 3 separate occasions with similar results.
After observing that Src, p38, and PI3K are required for sJAM-C induced HMVEC chemotaxis, Western blots were performed to determine if stimulation with sJAM-C results in the phosphorylation of these mediators. We found that sJAM-C stimulated the phosphorylation of each of these mediators, with PI3K phosphorylation occurring first, followed by the phosphorylation of p38, and finally Src family kinases (figure 7b). Together, these results suggest that sJAM-C stimulates the phosphorylation of Src, p38, and PI3K, and that these pathways are required for sJAM-C mediated angiogenesis.
Discussion
Angiogenesis is a critical process in both physiological and pathological conditions, of which adhesion molecules are known to play key roles. Adhesion molecules regulate angiogenesis both indirectly and directly. They regulate angiogenesis by promoting the influx and retention of leukocytes capable of secreting proangiogenic factors, or by being cleaved or alternatively spliced into soluble form and directly stimulating ECs.
We hypothesized that JAM-C is present in soluble form and mediates angiogenesis. We found that JAM-C is present in soluble form and is detectable in normal serum. To date, the only other JAM family member to be found in soluble form is JAM-A, which is elevated in the blood of patients with cardiovascular diseases (30, 31). In addition, we found that sJAM-C is more highly expressed in RA serum and synovial fluid compared to normal serum and OA synovial fluid, respectively. sJAM-C was also elevated in the synovial fluid of patients with other inflammatory conditions, including those with psoriatic arthritis. OA synovial fluid was used as a noninflammatory synovial fluid control, as normal synovial fluid is present in small amounts and not readily obtainable. Previous studies have shown that the RA synovial fluid is rich in both proinflammatory and proangiogenic mediators (4). Moreover, soluble adhesion molecules such as E-selectin, P-selectin, VCAM-1, ICAM-1, and ICAM-3 have been shown to be upregulated in RA synovial fluid (4). However of these soluble adhesion molecules, only soluble E-selectin, ICAM-1, and VCAM-1 have been shown to be directly angiogenic (6, 32).
We and others have previously shown JAM-C to be present on the cell surface of several cell types, including fibroblasts, epithelial cells, and ECs (11, 12, 21). To determine the cellular source of sJAM-C, we cultured ECs and found sJAM-C in their unstimulated cell culture supernatants. Moreover, we found that the concentration of sJAM-C was increased in EC supernatants following stimulation with the proinflammatory mediators IL-1β, IL-17, LPS, MIF, TNF-α, IL-18, or PMA. Koenen et al. found similar results for JAM-A, as stimulation of human umbilical vascular endothelial cell (HUVECs) with PMA, a combination of IFN-γ and TNF-α, or platelet-activating factor resulted in the release of soluble JAM-A (sJAM-A) (33). In addition, previous studies have shown similar findings with members of the selectin and immunoglobulin adhesion molecule families (4). Pigott et al. found that cytokine stimulated ECs released soluble forms of E-selectin, ICAM-1, and VCAM-1 (5). Importantly these adhesion molecules, aside from E-selectin, are also secreted or released from a variety of other cell types in response to various conditions. Therefore, while we have established that ECs are a source of sJAM-C, they are likely not the only source, and further studies will be needed to determine which other cell types secrete or release sJAM-C.
After finding that ECs are a source of sJAM-C, we sought to determine how sJAM-C is produced by these cells. Adhesion molecules that are found in soluble form have previously been shown to be the result of cleavage from the cell surface or from alternative splicing events (4). Membrane bound E-selectin, ICAM-1, and VCAM-1 are proteolytically cleaved from the surface of ECs (5, 34). In addition however, splice variants of ICAM-1 and VCAM-1 without their transmembrane domains have also been observed (35-37). Moreover, Koenen et al. demonstrated that JAM-A is cleaved from the surface of ECs by ADAM17 and, to a lesser extent, ADAM10 (33). We found that JAM-C is proteolytically cleaved from the cell surface of ECs, as treatment with the inhibitor TAPI-2 decreased the amount of sJAM-C in the cell culture supernatant. In contrast, inhibitors of MMPs, serine, cysteine, or aspartic proteases had no effect on the release of JAM-C from the surface of these cells. As TAPI-2 is a broad spectrum inhibitor of MMPs, TACE, and ADAMs, these results suggested the release of JAM-C from the surface of ECs is dependent on TACE and ADAMS. To confirm these findings, we performed siRNA experiments and found that ADAM10 and ADAM17 (TACE) are able to cleave JAM-C from the cell surface. However, as we also found lesser amounts of sJAM-C in HMVEC and HMEC-1 protein lysates, it is likely that JAM-C is also alternatively spliced into soluble form.
Previous studies have shown that adhesion molecules, both membrane bound and soluble, play key roles in mediating angiogenesis. We have shown that that the soluble forms of E-selectin and VCAM-1 mediate angiogenesis directly (6). An indirect role for JAM-C in angiogenesis was established by Lamagna et al. (25). They found that an anti-JAM-C antibody abolished neovascularization of aortic rings in vitro, and reduced tumor volume and vascularization in vivo (25). Based on this study and our finding that JAM-C is present in soluble form following cleavage from the surface of ECs, we postulated that sJAM-C is a proangiogenic factor. Our results show that sJAM-C is angiogenic in vitro, and able to promote two facets of angiogenesis. We found that sJAM-C stimulates HMVEC chemotaxis, an initial event in angiogenesis, in the nanomolar range. Our previous studies have shown that soluble E-selectin and soluble VCAM-1 are chemotatic for HMVECs at a similar concentration range (6). Using a checkerboard analysis of sJAM-C induced HMVEC migration we found that sJAM-C is both chemotatic and chemokinetic for HMVECs. Moreover, we found that sJAM-C is a significant contributor to the chemotatic potential of RA synovial fluid for ECs. This result demonstrated that sJAM-C is a significant angiogenic factor present in RA synovial fluid.
In addition, we found that sJAM-C induces HMVEC tube formation on growth-factor reduced Matrigel. When ECs are grown on Matrigel in the presence of an angiogenic substance, robust EC tube formation occurs (38). This assay is a reliable in vitro model of angiogenesis, as we and others have previously shown a strong correlation between angiogenic factors that stimulate EC tube formation on Matrigel and those with the ability to promote in vivo angiogenesis (6, 39, 40). Additionally, we utilized this model to demonstrate that neutralizing anti-JAM-C antibodies block the angiogenic effect of our sJAM-C protein preparation. Collectively, these studies indicate that sJAM-C stimulates two different facets of angiogenesis in vitro.
After finding that sJAM-C induces HMVEC chemotaxis and tube formation in vitro, we assessed the effect of sJAM-C on angiogenesis in vivo using two different models. We first used an in vivo Matrigel angiogenesis assay in which liquid Matrigel is mixed with a potential angiogenic substance and injected s.c. into mice. The resulting solid plug, when supplemented with angiogenic factors, supports an intense vascular response (41). We found that sJAM-C induces the growth of blood vessels into the Matrigel plug and increases the amount of hemoglobin per plug. Previously, we have shown that the adhesion molecule soluble E-selectin elicits a robust angiogenic response in vivo using this method (39). In addition, we found that sJAM-C induced in vivo angiogenesis in a sponge granuloma assay. This model represents inflammatory angiogenesis, as fibroblasts, lymphocytes, mast cells, and macrophages have been observed to be present at sites of neovascularization (42). Thus it is possible that sJAM-C also mediates inflammatory cell migration, an avenue that warrants further investigation. In all, these results suggest that sJAM-C is a proangiogenic mediator in vivo.
To date little is known regarding signaling cascades initiated by membrane bound JAM-C, and this is the first study to show the presence of sJAM-C and to begin to examine the signaling pathways initiated by sJAM-C. Previous reports have demonstrated that JAMs can engage in homophilic and heterophilic interactions with neighboring JAM molecules (11, 15, 16). Therefore, it is possible that sJAM-C may bind membrane bound JAM-B or JAM-C on the surface of ECs to initiate angiogenesis. It is also possible that as yet unidentified additional receptors for sJAM-C are present on ECs. We observed that sJAM-C initiates the phosphorylation of Src, p38, and PI3K in HMVECs. In addition, we found that these kinases are required for sJAM-C mediated HMVEC chemotaxis. Other studies have shown that VEGF also uses Src and PI3K pathways to mediate angiogenesis (43, 44). In addition, we have previously shown that Src and Erk 1/2 are required for soluble E-selectin mediated angiogenesis (39). Our results further demonstrate that Src and PI3K play key roles in mediating angiogenesis through a variety of proangiogenic mediators.
We have previously shown that JAM-C is overexpressed on ECs in the RA synovium (21). In this study we report the novel finding of sJAM-C, and demonstrate that that sJAM-C is elevated in the serum and synovial fluid of patients with RA. In addition, we have shown that ECs are a source of sJAM-C, and that proinflammatory cytokines, which are elevated in RA synovial fluid, upregulate the expression of sJAM-C. Moreover, it is now evident that sJAM-C mediates facets of angiogenesis in vitro, and in vivo angiogenesis. Thus, in the case of RA, it is likely that the local overexpression of JAM-C combined with the presence of inflammatory cytokines results in the production of sJAM-C, which acts to promote angiogenesis in the synovium. Collectively, these findings suggest that therapies aimed at modulating the release or function of sJAM-C may be beneficial to combating angiogenic diseases such as RA, cardiovascular disease, and tumor growth.
Acknowledgments
The authors thank Drs. Edwin Ades of the Centers for Disease Control and Thomas Lawley of Emory University for providing the HMEC-1 cells.
Grant support: This work was supported by the National Institute of Health (grants HL094017 to B.J.R., AR052482 to M.A.A., and AR48267 to A.E.K.), the Office of Research and Development, Medical Research Service, Department of Veterans Affairs, and the Frederick G. L. Huetwell and William D. Robinson, MD Professorship in Rheumatology.
Abbreviations used in this paper
- RA
Rheumatoid arthritis
- ECs
endothelial cells
- bFGF
basic fibroblast growth factor
- JAM
junctional adhesion molecule
- HMVECs
human dermal microvascular endothelial cells
- OA
osteoarthritis
- PsA
psoriatic arthritis
- HMEC-1s
immortalized human microvascular endothelial cells
- MIF
macrophage migration inhibitory factor
- aFGF
acidic fibroblast growth factor
- MMPs
matrix metalloproteinases
- TACE
TNF-α converting enzyme
- ADAMs
a disintegrin and metalloproteinases
- TMB
tetramethybenzidine
- EBM
EC basal media
- OCT
optimal cutting temperature media
- vWF
von Willebrand factor
- HUVECs
human umbilical vascular endothelial cells
References
- 1.Szekanecz Z, Koch AE. Mechanisms of Disease: angiogenesis in inflammatory diseases. Nat Clin Pract Rheumatol. 2007;3:635–643. doi: 10.1038/ncprheum0647. [DOI] [PubMed] [Google Scholar]
- 2.Szekanecz Z, Gaspar L, Koch AE. Angiogenesis in rheumatoid arthritis. Front Biosci. 2005;10:1739–1753. doi: 10.2741/1657. [DOI] [PubMed] [Google Scholar]
- 3.Szekanecz Z, Koch AE. Adhesion molecules: Potent inducers of endothelial cell chemotaxis. In: Zilla PP, Greisler H, editors. Tissue engineering of vascular grafts. R.G. Landes; Austin: 1999. pp. 271–277. [Google Scholar]
- 4.Volin MV. Soluble adhesion molecules in the pathogenesis of rheumatoid arthritis. Curr Pharm Des. 2005;11:633–653. doi: 10.2174/1381612053381972. [DOI] [PubMed] [Google Scholar]
- 5.Pigott R, Dillon LP, Hemingway IH, Gearing AJ. Soluble forms of E-selectin, ICAM-1 and VCAM-1 are present in the supernatants of cytokine activated cultured endothelial cells. Biochem Biophys Res Commun. 1992;187:584–589. doi: 10.1016/0006-291x(92)91234-h. [DOI] [PubMed] [Google Scholar]
- 6.Koch AE, Halloran MM, Haskell CJ, Shah MR, Polverini PJ. Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature. 1995;376:517–519. doi: 10.1038/376517a0. [DOI] [PubMed] [Google Scholar]
- 7.Weber C, Fraemohs L, Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467–477. doi: 10.1038/nri2096. [DOI] [PubMed] [Google Scholar]
- 8.Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham SA, Arrate MP, Rodriguez JM, Bjercke RJ, Vanderslice P, Morris AP, Brock TA. A novel protein with homology to the junctional adhesion molecule. Characterization of leukocyte interactions. J Biol Chem. 2000;275:34750–34756. doi: 10.1074/jbc.M002718200. [DOI] [PubMed] [Google Scholar]
- 10.Palmeri D, van Zante A, Huang CC, Hemmerich S, Rosen SD. Vascular endothelial junction-associated molecule, a novel member of the immunoglobulin superfamily, is localized to intercellular boundaries of endothelial cells. J Biol Chem. 2000;275:19139–19145. doi: 10.1074/jbc.M003189200. [DOI] [PubMed] [Google Scholar]
- 11.Arrate MP, Rodriguez JM, Tran TM, Brock TA, Cunningham SA. Cloning of human junctional adhesion molecule 3 (JAM3) and its identification as the JAM2 counter-receptor. J Biol Chem. 2001;276:45826–45832. doi: 10.1074/jbc.M105972200. [DOI] [PubMed] [Google Scholar]
- 12.Aurrand-Lions M, Duncan L, Ballestrem C, Imhof BA. JAM-2, a novel immunoglobulin superfamily molecule, expressed by endothelial and lymphatic cells. J Biol Chem. 2001;276:2733–2741. doi: 10.1074/jbc.M005458200. [DOI] [PubMed] [Google Scholar]
- 13.Hirabayashi S, Tajima M, Yao I, Nishimura W, Mori H, Hata Y. JAM4, a junctional cell adhesion molecule interacting with a tight junction protein, MAGI-1. Mol Cell Biochem. 2003;23:4267–4282. doi: 10.1128/MCB.23.12.4267-4282.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moog-Lutz C, Cave-Riant F, Guibal FC, Breau MA, Di Gioia Y, Couraud PO, Cayre YE, Bourdoulous S, Lutz PG. JAML, a novel protein with characteristics of a junctional adhesion molecule, is induced during differentiation of myeloid leukemia cells. Blood. 2003;102:3371–3378. doi: 10.1182/blood-2002-11-3462. [DOI] [PubMed] [Google Scholar]
- 15.Kostrewa D, Brockhaus M, D’Arcy A, Dale GE, Nelboeck P, Schmid G, Mueller F, Bazzoni G, Dejana E, Bartfai T, Winkler FK, Hennig M. X-ray structure of junctional adhesion molecule: structural basis for homophilic adhesion via a novel dimerization motif. EMBO J. 2001;20:4391–4398. doi: 10.1093/emboj/20.16.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamagna C, Meda P, Mandicourt G, Brown J, Gilbert RJ, Jones EY, Kiefer F, Ruga P, Imhof BA, Aurrand-Lions M. Dual interaction of JAM-C with JAM-B and alpha(M)beta2 integrin: function in junctional complexes and leukocyte adhesion. Mol Biol Cell. 2005;16:4992–5003. doi: 10.1091/mbc.E05-04-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cunningham SA, Rodriguez JM, Arrate MP, Tran TM, Brock TA. JAM2 interacts with alpha4beta1. Facilitation by JAM3. J Biol Chem. 2002;277:27589–27592. doi: 10.1074/jbc.C200331200. [DOI] [PubMed] [Google Scholar]
- 18.Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- 19.Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, Chavakis T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. 2002;196:679–691. doi: 10.1084/jem.20020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zen K, Babbin BA, Liu Y, Whelan JB, Nusrat A, Parkos CA. JAM-C is a component of desmosomes and a ligand for CD11b/CD18-mediated neutrophil transepithelial migration. Mol Biol Cell. 2004;15:3926–3937. doi: 10.1091/mbc.E04-04-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rabquer BJ, Pakozdi A, Michel JE, Gujar BS, Haines GK, 3rd, Imhof BA, Koch AE. Junctional adhesion molecule C mediates leukocyte adhesion to rheumatoid arthritis synovium. Arthritis Rheum. 2008;58:3020–3029. doi: 10.1002/art.23867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooke VG, Naik MU, Naik UP. Fibroblast growth factor-2 failed to induce angiogenesis in junctional adhesion molecule-A-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2005–2011. doi: 10.1161/01.ATV.0000234923.79173.99. [DOI] [PubMed] [Google Scholar]
- 23.Naik MU, Mousa SA, Parkos CA, Naik UP. Signaling through JAM-1 and alphavbeta3 is required for the angiogenic action of bFGF: dissociation of the JAM-1 and alphavbeta3 complex. Blood. 2003;102:2108–2114. doi: 10.1182/blood-2003-04-1114. [DOI] [PubMed] [Google Scholar]
- 24.Naik MU, Vuppalanchi D, Naik UP. Essential role of junctional adhesion molecule-1 in basic fibroblast growth factor-induced endothelial cell migration. Arterioscler Thromb Vasc Biol. 2003;23:2165–2171. doi: 10.1161/01.ATV.0000093982.84451.87. [DOI] [PubMed] [Google Scholar]
- 25.Lamagna C, Hodivala-Dilke KM, Imhof BA, Aurrand-Lions M. Antibody against junctional adhesion molecule-C inhibits angiogenesis and tumor growth. Cancer Res. 2005;65:5703–5710. doi: 10.1158/0008-5472.CAN-04-4012. [DOI] [PubMed] [Google Scholar]
- 26.Amin MA, Volpert OV, Woods JM, Kumar P, Harlow LA, Koch AE. Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ Res. 2003;93:321–329. doi: 10.1161/01.RES.0000087641.56024.DA. [DOI] [PubMed] [Google Scholar]
- 27.Rabquer BJ, Hou Y, Del Galdo F, Kenneth Haines G, 3rd, Gerber ML, Jimenez SA, Seibold JR, Koch AE. The proadhesive phenotype of systemic sclerosis skin promotes myeloid cell adhesion via ICAM-1 and VCAM-1. Rheumatology (Oxford) 2009;48:734–740. doi: 10.1093/rheumatology/kep091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruth JH, Shahrara S, Park CC, Morel JC, Kumar P, Qin S, Koch AE. Role of macrophage inflammatory protein-3alpha and its ligand CCR6 in rheumatoid arthritis. Lab Invest. 2003;83:579–588. doi: 10.1097/01.lab.0000062854.30195.52. [DOI] [PubMed] [Google Scholar]
- 29.Park CC, Morel JC, Amin MA, Connors MA, Harlow LA, Koch AE. Evidence of IL-18 as a novel angiogenic mediator. J Immunol. 2001;167:1644–1653. doi: 10.4049/jimmunol.167.3.1644. [DOI] [PubMed] [Google Scholar]
- 30.Cavusoglu E, Kornecki E, Sobocka MB, Babinska A, Ehrlich YH, Chopra V, Yanamadala S, Ruwende C, Salifu MO, Clark LT, Eng C, Pinsky DJ, Marmur JD. Association of plasma levels of F11 receptor/junctional adhesion molecule-A (F11R/JAM-A) with human atherosclerosis. J Am Coll Cardiol. 2007;50:1768–1776. doi: 10.1016/j.jacc.2007.05.051. [DOI] [PubMed] [Google Scholar]
- 31.Salifu MO, Kolff Q, Murty P, Haria DM, Zimpa M, Shakeel M, Lee H, Kornecki E, Babinska A. Relationship between the soluble F11 receptor and markers of inflammation in hemodialysis patients. J Investig Med. 2007;55:115–119. doi: 10.2310/6650.2007.06041. [DOI] [PubMed] [Google Scholar]
- 32.Gho YS, Kleinman HK, Sosne G. Angiogenic activity of human soluble intercellular adhesion molecule-1. Cancer Res. 1999;59:5128–5132. [PubMed] [Google Scholar]
- 33.Koenen RR, Pruessmeyer J, Soehnlein O, Fraemohs L, Zernecke A, Schwarz N, Reiss K, Sarabi A, Lindbom L, Hackeng TM, Weber C, Ludwig A. Regulated release and functional modulation of junctional adhesion molecule A by disintegrin metalloproteinases. Blood. 2009;113:4799–4809. doi: 10.1182/blood-2008-04-152330. [DOI] [PubMed] [Google Scholar]
- 34.Wellicome SM, Kapahi P, Mason JC, Lebranchu Y, Yarwood H, Haskard DO. Detection of a circulating form of vascular cell adhesion molecule-1: raised levels in rheumatoid arthritis and systemic lupus erythematosus. Clin Exp Immunol. 1993;92:412–418. doi: 10.1111/j.1365-2249.1993.tb03413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robledo O, Papaioannou A, Ochietti B, Beauchemin C, Legault D, Cantin A, King PD, Daniel C, Alakhov VY, Potworowski EF, St-Pierre Y. ICAM-1 isoforms: specific activity and sensitivity to cleavage by leukocyte elastase and cathepsin G. Eur J Immunol. 2003;33:1351–1360. doi: 10.1002/eji.200323195. [DOI] [PubMed] [Google Scholar]
- 36.Terry RW, Kwee L, Levine JF, Labow MA. Cytokine induction of an alternatively spliced murine vascular cell adhesion molecule (VCAM) mRNA encoding a glycosylphosphatidylinositol-anchored VCAM protein. Proc Natl Acad Sci U S A. 1993;90:5919–5923. doi: 10.1073/pnas.90.13.5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wakatsuki T, Kimura K, Kimura F, Shinomiya N, Ohtsubo M, Ishizawa M, Yamamoto M. A distinct mRNA encoding a soluble form of ICAM-1 molecule expressed in human tissues. Cell Adhes Commun. 1995;3:283–292. doi: 10.3109/15419069509081014. [DOI] [PubMed] [Google Scholar]
- 38.Staton CA, Stribbling SM, Tazzyman S, Hughes R, Brown NJ, Lewis CE. Current methods for assaying angiogenesis in vitro and in vivo. Int J Exp Pathol. 2004;85:233–248. doi: 10.1111/j.0959-9673.2004.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar P, Amin MA, Harlow LA, Polverini PJ, Koch AE. Src and phosphatidylinositol 3-kinase mediate soluble E-selectin-induced angiogenesis. Blood. 2003;101:3960–3968. doi: 10.1182/blood-2002-04-1237. [DOI] [PubMed] [Google Scholar]
- 40.Puxeddu I, Berkman N, Nissim Ben Efraim AH, Davies DE, Ribatti D, Gleich GJ, Levi-Schaffer F. The role of eosinophil major basic protein in angiogenesis. Allergy. 2009;64:368–374. doi: 10.1111/j.1398-9995.2008.01822.x. [DOI] [PubMed] [Google Scholar]
- 41.Auerbach R, Lewis R, Shinners B, Kubai L, Akhtar N. Angiogenesis assays: a critical overview. Clin Chem. 2003;49:32–40. doi: 10.1373/49.1.32. [DOI] [PubMed] [Google Scholar]
- 42.Norrby K. In vivo models of angiogenesis. J Cell Mol Med. 2006;10:588–612. doi: 10.1111/j.1582-4934.2006.tb00423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cells. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 44.Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A. 2000;97:1749–1753. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]