

Abstract
Extensive DNA fragmentation that generates a multitude of DNA double-strand breaks (DSBs) is a hallmark of apoptosis. A widely used approach to identify apoptotic cells relies on labeling DSBs in situ with fluorochromes. Flow or image cytometry is then used to detect and quantify apoptotic cells labeled this way. We developed several variants of the methodology that is based on the use of exogenous terminal deoxynucleotidyl transferase (TdT) to label 3′-OH ends of the DSBs with fluorochromes, defined as the TUNEL assay. This chapter describes the variant based on DSBs labeling using 5-Bromo-2′-deoxyuridine-5′-triphosphate (BrdUTP) as a TdT substrate and the incorporated BrdU is subsequently detected immunocytochemically with anti-BrdU antibody. We also describe modifications of the protocol that allow using other than BrdUTP deoxyribonucleotides to label DSBs. Concurrent differential staining of cellular DNA and multiparameter analysis of cells by flow- or image cytometry enables one to correlate the induction of apoptosis with the cell cycle phase. Examples of the detection of apoptotic cells in cultures of human leukemic cell lines treated with TNF-α and DNA topoisomerase I inhibitor topote-can are presented. The protocol can be applied to the cells growing in vitro, treated ex vivo with cytotoxic drugs as well as to clinical samples.
Keywords: Apoptosis, DNA damage, Flow cytometry, Laser scanning cytometry, Cell cycle, Immuno-fluorescence, BrdU
1. Introduction
During apoptosis, DNA undergoes extensive fragmentation at the internucleosomal sections which generates a multitude of DNA double-strand breaks (DSBs) (1). Their presence is considered to be one of the most characteristic markers of apoptotic cells. A widely used approach to identify apoptotic cells, thus, relies on labeling DSBs in situ either with fluorochromes (2–4) or absorption dyes (5). We have developed several variants of the methodology that is based on the use of exogenous terminal deoxynucleotidyl transferase (TdT) to label 3′-OH termini of the DSBs either indirectly or directly with fluorochrome-tagged deoxyribonucleotides, commonly defined as the TUNEL assay (2–4, 6–8). In this chapter, we describe the variant based on DSBs labeling with 5-Bromo-2′-deoxyuridine-5′-triphosphate (BrdUTP) that subsequently is detected immunocytochemically with BrdU antibody (Ab). The BrdUTP labeling assay offers much greater sensitivity than other TUNEL variants (8). However, modifications of the protocol that allow one to use deoxyribonucleotides other than BrdUTP also are described. Concurrent staining of cellular DNA with propidium iodide (PI) or 4′,6-diamidino-2-phenylindole (DAPI) and multiparameter analysis of cells by flow or image cytometry enables one to correlate the induction of apoptosis with the cell cycle phase (9). The protocol can be applied to cells growing in vitro, treated ex vivo with cytotoxic drugs as well as to clinical samples (see Note 1).
The method presented in this chapter can be applied to cells measured by flow cytometry, and its modification, also included, to cells attached on microscope slides. The latter can be analyzed by image cytometry, e.g., using an instrument, such as the laser scanning cytometer (LSC). LSC is the microscope-based cyto-fluorometer that allows one to measure rapidly, with high sensitivity and accuracy, fluorescence of individual cells (10). Cells staining on slides eliminates their loss that otherwise occurs during repeated centrifugations in sample preparation for flow cytometry. Therefore, the procedure offers an advantage when applied to samples with paucity of cells, such as fine needle aspirate or spinal fluid tap (see Note 2). Another advantage of LSC is that it offers a possibility of electronic selection (gating) of cells of interest during the initial measurement for their subsequent analysis by imaging or staining with other fluorochromes. Imaging and visual examination are of particular importance because the characteristic changes in cell morphology are considered the gold standard for positive identification of apoptotic cells (3, 4). Furthermore, the cell attributes measured by LSC on live cells can be correlated with the attributes that to be measured require cell fixation (11). For example, the activation of caspases (11–13), DNA replication (14), translocation of Bax to mitochondria (15), or activation of NF-κB transcription factor (16), and the key events associated with apoptosis can be correlated, in the very same cells, with the presence of apoptosis-associated DSBs.
Fixation and permeabilization of the cells are the initial essential steps to successfully label DSBs. Cells are briefly fixed with the cross-linking fixative formaldehyde and then permeabilized by suspending in ethanol or using detergents in the subsequent rinses. By cross-linking small DNA fragments to other cell constituents, formaldehyde prevents their extraction, which otherwise occurs during repeated centrifugations and rinses (17). The 3′OH-termini of the DSBs serve as primers and become labeled in this procedure with BrdU when incubated with BrdUTP in a reaction catalyzed by exogenous TdT (2). The incorporated BrdU is immunocytochemically detected by BrdU Ab conjugated to fluorochromes of a desired emission wavelength (8). The BrdU Ab is a widely available reagent, also used in studies of cell proliferation to detect BrdU incorporated during DNA replication (18). The sensitivity of DSBs detection is higher and overall cost of reagents is significantly lower when BrdUTP is used, as compared to the alternative labeling with biotin- (or digoxigenin-) (2) or directly fluorochrome-tagged deoxyribonucleotides (7). The alternate procedures, utilizing digoxigenin, biotin, or directly labeled deoxynucleotides, however, are also described in the chapter.
2. Materials
2.1. Reagents and Glassware
Phosphate-buffered saline (PBS), pH 7.4.
1% Formaldehyde (methanol-free, “ultrapure”) (Polysciences, Warrington, PA), in PBS, pH 7.4.
70% Ethanol.
TdT (Roche Diagnostics, Indianapolis, IN). Supplied in storage buffer: (60 mM potassium phosphate at pH 7.2, 150 mM KCl, 1 mM 2-mercaptoethanol and 0.5% Triton X-100, 50% glycerol). The 5× TdT reaction buffer (Roche Diagnostics) contains: 1 M potassium (or sodium cacodylate), 125 mM HCl, pH 6.6, and 1.25 mg/mL bovine serum albumin (BSA).
BrdUTP stock solution (50 μL): 2 mM BrdUTP (Sigma) in 50 m mM Tris–HCl, pH 7.5.
10 mM CoCl2 (Sigma).
Rinsing buffer: 0.1% Triton X-100 (Sigma) and 5 mg/mL BSA dissolved in PBS.
Alexa Fluor 488-conjugated anti-BrdU monoclonal antibody (mAb): Dissolve 1.0 μg of Alexa Fluor 488-conjugated anti-BrdU Ab in 100 μL of PBS containing 0.3% Triton X-100 and 1% (w/v) BSA. Alternatively, use Fluorescein- (FITC)- or Alexa Fluor 647-conjugated anti-BrdU Ab. These Abs are available from Phoenix Flow System (San Diego, CA) or from Invitrogen/Molecular Probes (Eugene, OR).
PI staining buffer: 5 μg/mL PI (Invitrogen/Molecular Probes) and 10 μg/mL of RNase A (DNase-free) (Sigma) in PBS. Alternatively, use 1 μg/mL solution of DAPI (Invitrogen/Molecular Probes) in PBS.
Microscope slides or single- or multichambered Falcon CultureSlides (BD Biosciences) (to be used in conjunction with analysis by LSC/iCys® Research Imaging Cytometer).
Coplin jars (to be used in conjunction with analysis by LSC/iCys® Research Imaging Cytometer).
Parafilm “M” (American National Can, Greenwich, CT) (to be used in conjunction with LSC/iCys).
Glycerol (to be used in conjunction with analysis by LSC/iCys® Research Imaging Cytometer).
2.2. Commercial Kits
Several kits for labeling DSBs are commercially available. The APO-BRDU kit (Phoenix Flow Systems, San Diego, CA) uses a BrdUTP methodology similar to that described in this chapter. As mentioned, this methodology offers the most sensitive means of DNA strand break detection (8). The APO-DIRECT kit (also from Phoenix) offers a single-step labeling of DNA strand breaks with the fluorochrome-tagged deoxynucleotide. Its virtue is simplicity, but it is less sensitive than the APO-BRDU. Of importance, the positive and negative control cells are supplied with each of these Phoenix kits. It should be noted that the kits developed by Phoenix Flow Systems are provided also by other vendors. The kits utilizing biotin- or digoxigenin-tagged dUTP are also commercially available.
2.3. Instrumentation
Flow cytometers of different types, offered by several manufacturers, can be used to measure cell fluorescence following staining according to the procedures described below. The manufacturers of the most common flow cytometers are Coulter/Beckman Corporation (Miami, FL), BD Biosciences (formerly Becton Dickinson Immunocytometry Systems; San Jose, CA), iCyt (Urbana-Champain, IL) and PARTEC GmbH (Zurich, Switzerland). The multiparameter LSC (iCys® Research Imaging Cytometer) is available from CompuCyte, Inc., (Westwood, MA). Cytospin centrifuge, which is used in conjunction with LSC/iCys, is provided by Shandon (Pittsburgh, PA).
The software to deconvolute the DNA content frequency histograms, to analyze the cell cycle distributions, is available from Phoenix Flow Systems or Verity Software House (Topham, MA).
3. Methods
3.1. DNA Strand Break Labeling with BrdUTP for Analysis by Flow Cytometry
Suspend 1–2 × 106 cells in 0.5 mL PBS. With a Pasteur pipette transfer this suspension into a 5 mL polypropylene tube (see Note 2) containing 4.5 mL of ice-cold 1% formaldehyde (see Note 3). Keep the tube for 15 min on ice.
Centrifuge at 300 × g for 5 min and resuspend cell pellet in 5 mL of PBS. Centrifuge again and resuspend cell pellet in 0.5 mL of PBS. With a Pasteur pipette transfer the suspension to a tube containing 4.5 mL of ice-cold 70% ethanol. The cells can be stored in ethanol, at −20°C for several weeks.
Centrifuge at 200 × g for 3 min, remove ethanol, resuspend cells in 5 mL of PBS, and centrifuge at 300 × g for 5 min.
-
Resuspend the pellet in 50 μL of a solution containing:
10 μL TdT 5× reaction buffer.
2.0 μL of BrdUTP stock solution.
0.5 μL (12.5 units) TdT.
5 μL of 10 mM CoCl2 solution.
33.5 μL distilled H2O.
Incubate the cells in this solution for 40 min at 37 °C (see Notes 4 and 5).
Add 1.5 mL of the rinsing buffer, and centrifuge at 300 × g for 5 min.
Resuspend cell pellet in 100 μL of Alexa Fluor 488-conjugated anti-BrdU Ab solution. (Alternatively you may use the Ab conjugated either with fluorescein (FITC) or Alexa Fluor 647).
Incubate at room temperature for 1 h.
Add 1 mL of PI staining solution (alternatively you may add 1 mL of the DAPI staining solution).
Incubate for 30 min at room temperature, or 20 min at 37 °C, mL of the DAPI staining solution). in the dark.
-
Analyze cells by flow cytometry.
Illuminate with blue light (488 nm laser line or BG12 excitation filter).
Measure green fluorescence of FITC (or Alexa Fluor 488)-conjugated anti-BrdU Ab at 530 ± 20 nm.
Measure intensity of red fluorescence of PI at >600 nm. Alternatively, if DNA was stained with DAPI instead of PI use UV light as an excitation source and measure the intensity of blue fluorescence (480 ± 20 nm).
The bivariate (DSBs versus cellular DNA content) distributions (scatterplots) illustrating the cell populations containing a fraction of apoptotic cells labeled according to the method described in the protocol and analyzed by flow cytometry are shown in Fig. 1 and analyzed by LSC (iCys® Research Imaging Cytometer) are shown in Fig. 2. A correlation between the induction of apoptosis and cell position in the cell cycle is clearly evident: in the case of topotecan treated HL-60 cells, nearly all apoptotic cells are S-phase cells (Fig. 1) while the apoptotic U-932 cells treated with TNF-α are predominantly G1-cells.
Fig. 1.
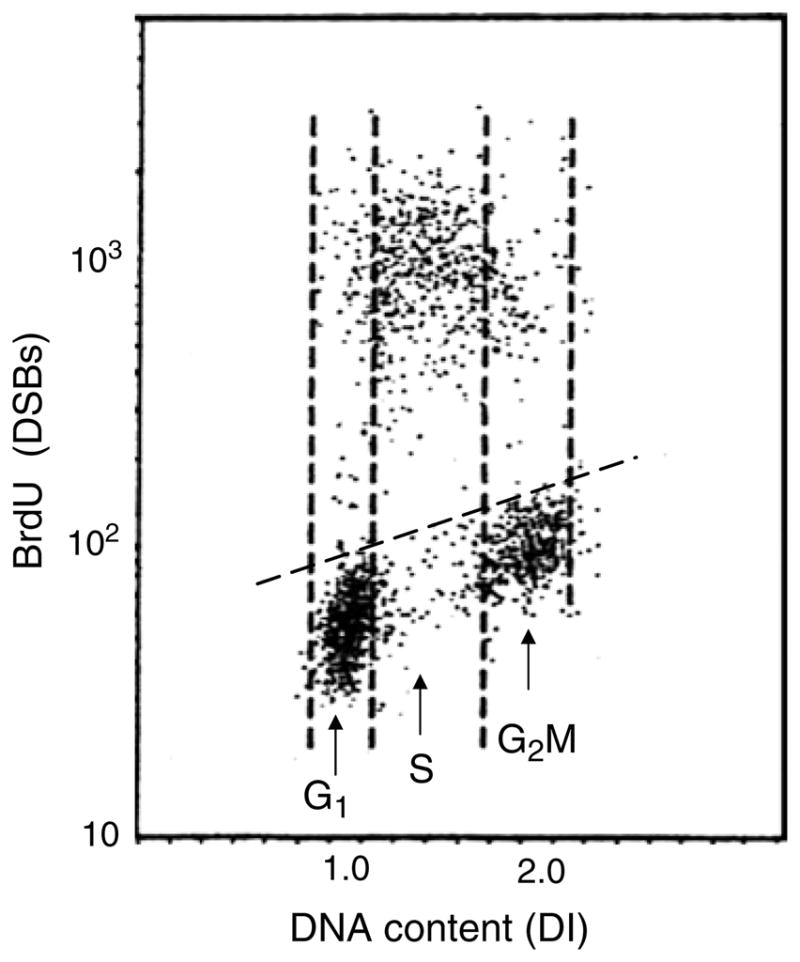
Detection of apoptotic cells after DSBs labeling with BrdUTP and fluorescence analysis by flow cytometry. To induce apoptosis leukemic HL-60 cells were treated in culture with DNA topoisomerase I inhibitor topotecan (0.15 μM) for 4 h. The cells were then subjected to DSBs labeling with BrdUTP as described in the protocol using fluorescein-tagged BrdU Ab and staining DNA with PI. Cellular fluorescence was measured by flow cytometry. The data are presented as the bivariate distributions (scatterplots) illustrating cellular DNA content (DNA index, DI) versus DSBs labeled with BrdU Ab. Note that essentially only S-phase cells underwent apoptosis as shown by high intensity of their BrdU-associated fluorescence, above the control level marked by the skewed dashed line. The leukemic cells treated with topoisomerase I inhibitors topotecan or camptothecin for 3–5 h present a convenient experimental model to assess whether the protocol of DSBs labeling is effective because in the same cell population there are DSBs positive (S-phase) and negative (G1 and G2M) cells.
Fig. 2.

Detection of apoptotic cells after DSBs labeling with BrdUTP and analysis by LSC. U-937 cells were untreated (a) or treated with tumor necrosis factor-α (TNF-α) in the presence of cycloheximide (b, (22, 23)). The cells were then subjected to DNA strand break labeling and DNA staining as described in the protocol using fluorescein-tagged BrdU Ab and staining DNA with PI. Cell fluorescence was measured by iCys® Research Imaging Cytometer. The bivariate distributions (scatterplots) allow one to identify apoptotic cells as the cells with labeled DSBs (strong green fluorescence intensity), and also reveal the cell cycle position of cells in either apoptotic or nonapoptotic population. Note pre-dominance of G1 cells among apoptotic cells. The cells with strong DSBs labeling were relocated, imaged, and their representative images are presented as shown. These cells show nuclear fragmentation and chromatin condensation, the typical features of apoptosis (3, 4).
3.2. DSBs Labeling with Other Markers for Analysis by Flow Cytometry
As mentioned in the Subheading 1 DNA strand breaks can be labeled with deoxynucleotides tagged with variety of other fluorochromes. For example, the Molecular Probes, Inc., catalog lists several types of dUTP conjugates, including BODIPY dyes (e.g., BODIPY-FL-X-dUTP), fluorescein, Cascade Blue and Texas Red. Several cyanine dyes conjugates (e.g., CY-3-dCTP) are available from Biological Detection Systems (Pittsburgh, PA). Indirect labeling, via biotinylated- or digoxigenin-conjugated deoxyribo-nucleotides offers a multiplicity of commercially available fluorochromes (fluorochrome-conjugated avidin or streptavidin, as well as digoxigenin antibodies) with different excitation and emission characteristics. DNA strand breaks, thus, can be labeled with a dye of any desired fluorescence color and excitation wavelength.
The procedure described in Subheading 3.1 can be adopted to utilize any of these fluorochromes. In the case of the direct labeling (7), the fluorochrome-conjugated deoxynucleotide is included in the reaction solution (0.25–0.5 nmol per 50 μL) instead of BrdUTP, as described in step 4 of Subheading 3.1. Following the incubation step (step 5), omit steps 6–8, and stain cells directly with PI (step 9). In the case of the indirect labeling, instead of BrdUTP, digoxygenin- or biotin-conjugated deoxy-nucleotides are included into the reaction buffer (0.25–0.5 nmol per 50 μL) at step 4. The cells are then incubated either with the fluorochrome-conjugated antidigoxigenin MAb (0.2–0.5 μg per 100 μL of PBS containing 0.1% Triton X-100 and 1% BSA), or with fluorochrome conjugated avidin or streptavidin (0.2–0.5 μg per 100 μL, as above) at step 7 and then processed through steps 8–10 as described in the protocol. Analysis by flow cytometry is carried out with excitation and emission wavelengths appropriate to the used fluorochrome.
3.3. DNA Strand Break Labeling for Analysis by LSC (iCys® Research Imaging Cytometer)
Transfer 300 μL of cell suspension (in tissue culture medium, with serum) containing approximately 20,000 cells into a cytospin chamber. Cytocentrifuge at 1,000 rpm (~150 × g) for 6 min to deposit the cells on a microscope slide. (Alternatively, to analyze cells that grow attached to surface maintain them in cultures in single- or multichambered Falcon CultureSlides (BD Biosciences). When harvested, remove the walls of the chambers, rinse cells with PBS and fix in formaldehyde as described in step 2 of Subheading 3.3.).
Without allowing the cytospin to completely dry, prefix the cells by transferring the slides for 15 min to a Coplin jar containing 1% formaldehyde in PBS, cooled to ice temperature.
Rinse the slides in PBS and transfer to 70% ethanol; fix in ethanol for at least 1 h; the cells can be stored in ethanol for weeks at −20°C.
Follow steps 4–8 of Subheading 3.1 as described for flow cytometry. Carefully layer small volumes (approximately 100 μL) of the respective buffers, rinses or staining solutions on the cytospin area (or over the sites of individual chambers if the cells were grown on Chamber Slides) of the horizontally placed slides. At appropriate times, remove these solutions with Pasteur pipette (or vacuum suction pipette). To prevent drying, place a 2 × 4 cm strip of Parafilm over the site where the cells are present atop of the solutions used for cell incubations (see Note 8).
Replace the PI staining solution with a drop of a mixture of glycerol and PI staining solution (9:1) and mount under the coverslip. To preserve the specimen for longer period of time or transport, seal the coverslip with nail polish or melted paraffin.
-
Measure cell fluorescence on LSC.
Excite fluorescence with 488 nm laser line.
Measure green fluorescence of Alexa Fluor 488 or fluorescein anti BrdU Ab at 530 ± 20 nm.
-
Measure red fluorescence of PI at >600 nm.
(Alternatively, if DSBs are labeled with Alexa Fluor 647 excite fluorescence with red diode laser and measure fluorescence intensity at far-red wavelength. If DAPI is used to stain DNA, excite DAPI fluorescence with UV laser and measure fluorescence intensity at 480 ± 20 wavelength).
The typical results are shown in Figs. 1 and 2 (see Notes 6 and 7).
3.4. Controls
The procedure of DNA strand break labeling is rather complex and involves many reagents. Negative results, therefore, may not necessarily mean the absence of DNA strand breaks (see Note 7) but may be due to methodological problems, such as the loss of TdT activity, degradation of BrdUTP, etc. It is necessary, therefore, to include the positive and negative control. An excellent control is to use HL-60 cells treated (during their exponential growth) for 3–4 h with 0.2 μM of the DNA topoisomerase I inhibitor camptothecin (CPT). Because CPT under these conditions induces apoptosis selectively during S phase, cells in G1 and G2/M may serve as negative control populations, while the S phase cells in the same sample, represent the positive control.
Another negative control consist cells processed identically as described in Subheading 3.1 except that TdT is excluded from step 4.
Acknowledgments
Supported by NCI grant RO1 28 704.
Footnotes
This method is also useful for clinical material, such as that obtained from in leukemias, lymphomas, and solid tumors (19, 20), and that can be combined with surface immunophenotyping. In such instance, the cells are first immunophenotyped, then fixed with 1% formaldehyde (which stabilizes the antibody bound on the cell surface) and subsequently subjected to the DSBs detection assay using other color fluorochrome (see Subheading 3.1) than the one used for immunophenotyping. The percent of apoptotic (DSBs-positive) cells is then estimated within the gated-immunophenotype cell population.
If the sample initially contains small number of cells, cell loss during repeated centrifugations is a problem. To minimize cell loss, polypropylene, or siliconized glass tubes are recommended. Since transferring cells from one tube to another one results in electrostatic attachment of a large fraction of cells to the surface of each new tube all steps of the procedure (including fixation) should be done in the same tube. Addition of 1% (w/v) BSA into rinsing solutions also decreases cell loss. When the sample contains very few cells, the carrier cells, which later can be recognized based on differences in DNA content (e.g., chick erythrocytes) may be included. Because there is no cell loss during processing for analysis by LSC the samples with paucity of cells can easily be measured.
Cell pre-fixation with a cross-linking agent, such as formaldehyde is required to prevent the extraction of the fragmented DNA from apoptotic cells (17). This ensures that despite the repeated cell washings during the procedure, the DNA content of apoptotic cells (and with it the number of DSBs) is not markedly diminished.
Alternatively, incubate at room temperature overnight.
Control cells may be incubated in the same solution, but without TdT.
It is generally easy to identify apoptotic cells, due to their intense labeling with Alexa Fluor 488, fluorescein, or Alexa Fluor 647-conjugated anti-BrdU Ab. The high fluorescence intensity often requires the use of the exponential scale (logarithmic amplifiers of the flow cytometer or LSC) for data acquisition and display (Figs. 1 and 2). As it is evident in these figures, because cellular DNA content of each, apoptotic and nonapoptotic cell population is measured, the cell cycle distribution and/or DNA ploidy of these both populations can be estimated.
While the presence of extensive DNA breakage, detected following strand break labeling, by the strong fluorescence, is a very characteristic feature of apoptosis, a weak fluorescence may not necessarily mean the lack of apoptosis. In some cell systems, DNA fragmentation stops at 300–50 kb size DNA fragments and does not progress into internucleosomal sections (21).
It is essential that the incubations are carried out in a humidified chamber. Even minor drying produces severe artifacts.
Contributor Information
Zbigniew Darzynkiewicz, Email: darzynk@nymc.edu, Brander Cancer Research Institute and Department of Pathology, New York Medical College, Valhalla, NY, USA.
Hong Zhao, Brander Cancer Research Institute and Department of Pathology, New York Medical College, Valhalla, NY, USA.
References
- 1.Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 2.Gorczyca W, Bruno S, Darzynkiewicz RJ, Gong J, Darzynkiewicz Z. DNA strand breaks occurring during apoptosis: Their early in situ detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int J Oncol. 1992;1:639–648. doi: 10.3892/ijo.1.6.639. [DOI] [PubMed] [Google Scholar]
- 3.Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- 4.Darzynkiewicz Z, Juan G, Li X, Gorczyca W, Murakami T, Traganos F. Cytometry in cell necrobiology: analysis of apoptosis and accidental cell death (necrosis) Cytometry. 1997;27:1–20. [PubMed] [Google Scholar]
- 5.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorczyca W, Tuziak T, Kram A, Melamed MR, Darzynkiewicz Z. Detection of apoptosis in fine-needle aspiration biopsies by in situ end-labeling of fragmented DNA. Cytometry. 1994;15:169–175. doi: 10.1002/cyto.990150211. [DOI] [PubMed] [Google Scholar]
- 7.Li X, Traganos F, Melamed MR, Darzynkiewicz Z. Single step procedure for DNA strand break labeling. Detection of apoptosis and DNA replication. Cytometry. 1995;20:172–180. doi: 10.1002/cyto.990200210. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Darzynkiewicz Z. Labelling DNA strand breaks with BrdUTP. Detection of apoptosis and cell proliferation. Cell Prolif. 1995;28:571–579. doi: 10.1111/j.1365-2184.1995.tb00045.x. [DOI] [PubMed] [Google Scholar]
- 9.Gorczyca W, Gong J, Ardelt B, Traganos F, Darzynkiewicz Z. The cell cycle related differences in susceptibility of HL-60 cells to apoptosis induced by various antitumor drugs. Cancer Res. 1993;53:3186–3192. [PubMed] [Google Scholar]
- 10.Pozarowski P, Holden E, Darzynkiewicz Z. Laser scanning cytometry: Principles and applications. Methods Mol Biol. 2006;319:165–192. doi: 10.1007/978-1-59259-993-6_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Darzynkiewicz Z. The Schrödinger’s cat quandary in cell biology: integration of live cell functional assays with measurements of fixed cells in analysis of apoptosis. Exp Cell Res. 1999;249:4–412. doi: 10.1006/excr.1999.4525. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Du L, Darzynkiewicz Z. Caspases are activated during apoptosis independently of dissipation of mitochondrial electrochemical potential. Exp Cell Res. 2000;257:290–297. doi: 10.1006/excr.2000.4901. [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Okafuji M, Traganos F, Luther E, Holden E, Darzynkiewicz Z. Assessment of histone H2AX phosphorylation induced by DNA topoisomerase I and II inhibitors topotecan and mitoxantrone and by DNA crosslinking agent cisplatin. Cytometry A. 2004;58A:99–110. doi: 10.1002/cyto.a.20018. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Melamed MR, Darzynkiewicz Z. Detection of apoptosis and DNA replication by differential labeling of DNA strand breaks with fluorochromes of different color. Exp Cell Res. 1996;222:28–37. doi: 10.1006/excr.1996.0004. [DOI] [PubMed] [Google Scholar]
- 15.Bedner E, Li X, Kunicki J, Darzynkiewicz Z. Translocation of Bax to mitochondria during apoptosis measured by laser scanning cytometry. Cytometry. 2000;41:83–88. [PubMed] [Google Scholar]
- 16.Deptala A, Bedner E, Gorczyca W, Darzynkiewicz Z. Simple assay of activation of nuclear factor kappa B (NF-κB) by laser scanning cytometry (LSC) Cytometry. 1998;33:376–382. doi: 10.1002/(sici)1097-0320(19981101)33:3<376::aid-cyto13>3.0.co;2-q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong J, Traganos F, Darzynkiewicz Z. A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal Biochem. 1994;218:314–319. doi: 10.1006/abio.1994.1184. [DOI] [PubMed] [Google Scholar]
- 18.Dolbeare F, Selden JR. Immunochemical quantitation of bromode-oxyuridine: Application to cell cycle kinetics. Methods Cell Biol. 1994;41:297–316. [PubMed] [Google Scholar]
- 19.Gorczyca W, Bigman K, Mittelman A, Ahmed T, Gong J, Melamed MR, Darzynkiewicz Z. Induction of DNA strand breaks associated with apoptosis during treatment of leukemias. Leukemia. 1993;7:659–670. [PubMed] [Google Scholar]
- 20.Li X, Gong J, Feldman E, Seiter K, Traganos F, Darzynkiewicz Z. Apoptotic cell death during treatment of leukemias. Leuk Lymph. 1994;13:65–72. doi: 10.3109/10428199409052678. [DOI] [PubMed] [Google Scholar]
- 21.Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Wakeling AE, Walker PR, Sikorska M. Apoptotic death in epithelial cells: Cleavage of DNA to 300 and 50 kb fragments prior to or in the absence of internucleosomal degradation of DNA. EMBO J. 1993;12:3679–3684. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Darzynkiewicz Z. Cleavage of poly(ADP-ribose) polymerase measured in situ in individual cells: relationship to DNA fragmentation and cell cycle position during apoptosis. Exp Cell Res. 2000;255:125–132. doi: 10.1006/excr.1999.4796. [DOI] [PubMed] [Google Scholar]
- 23.Bedner E, Smolewski P, Amstad P, Darzynkiewicz Z. Activation of caspases measured in situ by binding of fluorochrome-labeled inhibitors of caspases (FLICA): correlation with DNA fragmentation. Exp Cell Res. 2000;260:308–313. doi: 10.1006/excr.2000.4955. [DOI] [PubMed] [Google Scholar]