

Summary
Novel classes of broad-spectrum antibiotics are needed to treat multidrug resistant pathogens. The arylomycin class of natural products inhibits a promising antimicrobial target, type I signal peptidase (SPase), but upon initial characterization appeared to lack whole cell activity against most pathogens. Here, we show that Staphylococcus epidermidis, which is sensitive to the arylomycins, evolves resistance via mutations in SPase and that analogous mutations are responsible for the natural resistance of Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa. We identify diverse bacteria lacking these mutations and demonstrate that most are sensitive to the arylomycins. The results illustrate that the arylomycins have a broad-spectrum of activity and are viable candidates for development into therapeutics. The results also raise the possibility that naturally occurring resistance may have masked other natural product scaffolds that might be developed into therapeutics.
INTRODUCTION
The clinical and agricultural use of antibiotics impose a relentless selection pressure on bacteria that has driven the evolution of multidrug resistance in many pathogens and novel classes of antibiotics are needed (Fischbach and Walsh, 2009; Hancock, 2007). While the potency and spectrum of known antibiotics have been improved through derivatization, modern efforts to discover novel classes of broad-spectrum antibiotics have largely failed, highlighting the unique challenges associated with antibiotic development (Payne et al., 2007). First, bacterial pathogens are extremely diverse, and only a handful of proteins and biochemical processes are sufficiently conserved across all bacteria to warrant consideration as antibiotic targets (Bumann, 2008; Silver, 2007). Second, all bacteria possess at least one lipid bilayer, a peptidoglycan-based cell wall, and efflux systems that reduce the accessibility of many targets (Poole, 2005). Given these challenges, it is perhaps not surprising that most antibiotics that have reached the clinic belong to one of a small number of natural product classes, whose activities have likely been optimized over eons of evolution (Newman and Cragg, 2007; von Nussbaum et al., 2006). Unfortunately, it is believed that most, if not all, of the broad-spectrum scaffolds produced by the known culturable strains of bacteria have already been identified (Baltz, 2006).
The arylomycins and related lipoglycopeptides (Fig. 1) constitute a recently discovered class of natural product antibiotics that inhibit the type I signal peptidases (SPases) of both Gram-positive and Gram-negative bacteria in vitro (Kulanthaivel et al., 2004; Paetzel et al., 2004; Schimana et al., 2002). However, the initially reported spectrum of the arylomycins appeared prohibitively narrow for drug development, with no activity against most bacteria tested, including the important human pathogens Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa (Kulanthaivel et al., 2004; Roberts et al., 2007; Schimana et al., 2002). Indeed, antibiotic activity of the arylomycins was originally only demonstrated against Streptococcus pneumoniae (Kulanthaivel et al., 2004) and the soil bacteria Rhodococcus opacus and Brevibacillus brevis (Schimana et al., 2002). However, we recently demonstrated that the potencies of a natural arylomycin, arylomycin A2, and of a synthetic derivative, arylomycin C16 (Fig. 1), against S. epidermidis are equal to or greater than available clinical antibiotics (Hellmark et al., 2009), with minimum inhibitory concentrations (MICs) of 1.0 and 0.25 μg/ml, respectively (Roberts et al., 2007).
Figure 1. Chemical composition of the arylomycin class of natural product antibiotics.

Arylomycin A2 has the substituent pattern (R1 = H, R2 = H, R3 = H, R4 = isoC11) and Arylomycin C16 has the substituent patter (R1 = H, R2 = H, R3 = H, R4 = nC15).
Despite their potent activity against S. epidermidis, the known spectrum of arylomycin activity remained surprisingly narrow. SPase, which proteolytically removes the N-terminal signal sequence of proteins translocated across the cytoplasmic membrane, is conserved in all Eubacteria (Paetzel et al., 2000) and has been demonstrated to be essential in several key pathogens, including E. coli and S. aureus (Cregg et al., 1996; Date, 1983; Zhang et al., 1997). Moreover, although SPase genes have diverged considerably at the sequence level, they all share a common fold and catalytic mechanism, and there is significant sequence conservation in functionally important regions (denoted as Boxes B – E) (Dalbey et al., 1997), which form the hydrophobic core, substrate binding cleft, and active site. Finally, the catalytic domain of SPase is located on the extracellular face of the cytoplasmic membrane; thus, membrane penetration cannot explain the resistance of Gram-positive bacteria such as S. aureus, although previous reports suggest that the outer-membrane of Gram-negative bacteria prevents the arylomycins and lipoglycopeptides from reaching SPase (Kulanthaivel et al., 2004; Roberts et al., 2007). Here, taking advantage of the sensitivity of S. epidermidis, we use a combination of genetic, biochemical, and phylogenetic methods to investigate the origins of the narrow spectrum of arylomycin activity. We determine that S. epidermidis evolves arylomycin resistance via specific SPase mutations and that analogous mutations are responsible for the natural resistance of many other bacteria. This, along with the elucidation of a much broader spectrum than originally believed, which includes Gram-negative bacteria, suggests that the arylomycins are promising candidates for development into broad spectrum antibiotics. Our results also suggest that naturally occurring resistance may have prevented the identification of other natural product scaffolds with the potential for broad-spectrum antibiotic activity.
RESULTS
Point mutations in SPase confer arylomycin resistance
S. epidermidis is atypically sensitive to the arylomycins (Roberts et al., 2007). To begin to investigate whether S. epidermidis lacks specific resistance mechanisms inherent to other bacteria, we performed selection experiments to isolate mutants that are able to grow in the presence of 2 μg/ml arylomycin C16 (8× MIC). Mutants were obtained at a frequency of 4 per 109 viable cells and fell into two phenotypic classes. The majority (~75%) have a 32-fold elevated MIC compared to the wild type strain, and the remainder have a greater than 256-fold elevated MIC. Consistent with the low frequency of resistant mutants, we found that arylomycin resistance is strongly correlated with either of two, single point mutations in SpsIB, one of the two SPases found in S. epidermidis. The 32-fold increase in resistance is associated with a Ser to Pro mutation at position 29 (10/11 clones sequenced), while the >256-fold increase in resistance is associated with a Ser to Pro mutation at position 31 (9/11 clones sequenced). Neither class of mutants exhibits growth defects under the standard laboratory conditions employed (Fig. S1). These data demonstrate that the whole cell antibiotic activity of the arylomycins results from their inhibition of SPase and also that mutations in SPase are the dominant mechanism whereby S. epidermis evolves resistance.
Next, to investigate whether naturally resistant bacteria harbor the same mutations that confer resistance in S. epidermidis, we examined the amino acid sequence of SPases in the closely related Gram-positive organism S. aureus, as well as in the more distantly related Gram-negative organisms E. coli and P. aeruginosa (Table 1). At the position corresponding to residue 31 in S. epidermidis, Pro is not found in any of these SPase sequences. Moreover, we were unable to construct an E. coli strain with Pro at this position, suggesting that it is not tolerated in some organisms. In contrast, at the position corresponding to residue 29 in S. epidermidis, Pro is found in the single SPase of S. aureus, the single SPase of E. coli, and one of the two SPases of P. aeruginosa (Pro29 in S. aureus, Pro84 in E. coli and P. aeruginosa).
Table 1.
MICs of arylomycin C16 for S. epidermidis, S. aureus, E. coli, and P. aeruginosa with wild type (WT) or mutant SPases. Mutated SPase residues are boxed
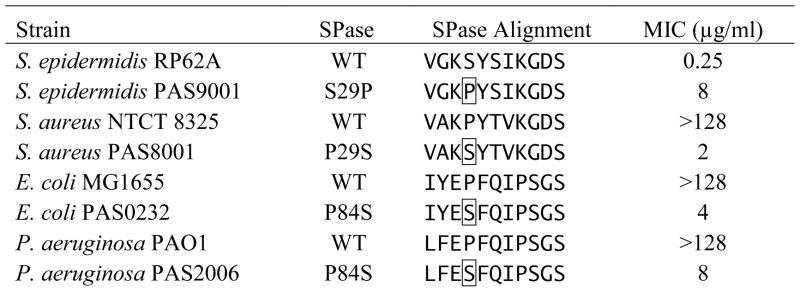 |
To determine whether the innate arylomycin resistance observed in E. coli, P. aeruginosa, and S. aureus results from the identified Pro residues, we constructed mutant strains of these bacteria in which Pro is replaced by Ser (the corresponding residue in wild type S. epidermidis SpsIB). In each organism, mutation of Pro to Ser is sufficient to confer a high degree of sensitivity to arylomycin C16 (Table 1). No growth defects are apparent in the mutant strains (Fig. 2 and S1), suggesting that the increased sensitivity does not result from decreased fitness under the growth conditions employed, although we cannot eliminate the possibility that the processing of some preprotein substrate(s) is affected. Importantly, the sensitivity of the E. coli and P. aeruginosa mutants suggests that the arylomycins penetrate the formidable outer-membrane of Gram-negative bacteria. Consistent with efficient outer-membrane penetration, we found that permeabilizing these bacteria with polymyxin B nonapeptide has only a negligible effect on the MICs (≤ 4-fold decrease).
Figure 2. Growth rates and arylomycin C16 sensitivities of E. coli strains harboring the indicated amino acid at SPase residue 84.
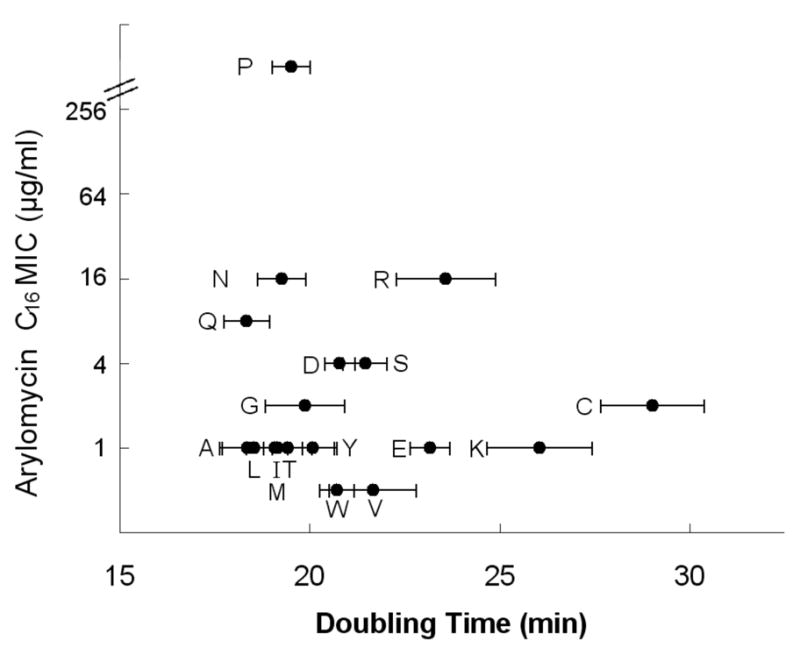
Horizontal bars indicate standard deviation of growth rates from three independent experiments. MICs varied less than 2-fold between experiments. His (MIC 4 μg/ml) and Phe (MIC 2 μg/ml) variants have a temperature sensitive phenotype and are therefore not shown. For Pro29, the MIC exceeds the detection limit of 256 μg/ml. See also Figure S1.
Next, to determine whether the identified Pro is unique in its ability to confer arylomycin resistance, we constructed mutant strains of E. coli in which each of the other 19 amino acids were introduced into SPase at the same position (residue 84). Based on the growth rates observed in arylomycin-free media, most amino acids at this position do not effect fitness under the conditions employed (Fig. 2), although a minor growth defect (Arg, Lys, Glu, and Cys) or a temperature sensitive phenotype (His and Phe) is observed in some strains. In contrast, the arylomycin C16 MICs are highly dependent on the identity of the amino acid at residue 84, and Pro is the only amino acid that imparts high-level arylomycin resistance (MIC > 256 μg/ml) (Fig. 2). All of the other amino acids confer sensitivity to arylomycin (MICs of ≤ 16 μg/ml), with hydrophobic amino acids conferring somewhat greater sensitivities.
Resistance-conferring mutations reduce the affinity of arylomycin for SPase
Based on the previously reported co-crystal structure (Paetzel et al., 2004), arylomycin A2 binds E. coli SPase in a manner that mimics that proposed for natural peptide substrates, and the resistance-conferring Pro residue (Pro84), is positioned within the substrate binding pocket, but distal to the catalytic residues (Fig. 3A). To test whether the resistance-conferring mutations directly interfere with arylomycin C16 binding in vitro, we determined equilibrium binding constants using recombinant SPase reconstituted in micelles that mimic a membrane environment. We first measured the affinity of arylomycin C16 for the wild type and P84S variants of a truncated E. coli SPase that lacks the N-terminal membrane helices, but still associates with micelles (Kuo et al., 1993) (Fig. S2). We found that arylomycin C16 binds the truncated wild type protein with a KD of 979 ± 69 nM, which is similar to the value reported for arylomycin A2 (Paetzel et al., 2004). In contrast, it binds the P84S variant with a KD of 39 ± 15 nM. Second, to control for artifacts associated with deletion of the N-terminal helices, which might interact with the lipid tail of the inhibitor or help to co-localize the protein and the inhibitor within the micelles, we determined the affinities of arylomycin C16 for the wild type and P84S variants of detergent-solubilized full-length E. coli SPase (Fig. 3B). While arylomycin C16 binds the full-length proteins with higher affinities than the corresponding soluble fragments, the affinity of arylomycin for the full-length Ser-variant (KD = 5.7 ± 1.0 nM) is again an order-of-magnitude higher than that for the corresponding Pro-variant (KD = 60 ± 16 nM). Lastly, to characterize a representative Gram-positive SPase, we measured the affinity of arylomycin C16 for the full-length wild type and P29S mutant of S. aureus SPase (Fig. 3C). As with the E. coli SPase, arylomycin C16 binds the Ser-variant of S. aureus SPase an order-of-magnitude more tightly than the Pro-variant, with KD values of 130 ± 53 and 1283 ± 278 nM, respectively. Thus, the Pro residues responsible for resistance in E. coli and S. aureus appear to act by interfering with arylomycin binding.
Figure 3. Physical and biochemical evidence for the proposed mechanism of arylomycin resistance.

(A) Crystal structure of E. coli SPase in complex with arylomycin A2 (PDB ID 1T7D) (Paetzel et al., 2004). Hydrogen-bonds observed in the crystal structure are shown in green, while the potential hydrogen bond prevented by Pro84 is shown in red. Equilibrium binding affinities of arylomycin for Pro- and Ser- variants of E. coli (B) and S. aureus (C) SPases. Data points and bars represent average values and standard deviations within a single experiment. KD values shown are the average of three independent experiments. See also Figure S2.
Distribution of resistance-conferring residues in nature
To better understand the distribution of this resistance determinant in nature, we determined the phylogenetic relationship of the fully sequenced bacteria from five phyla, as reflected by their 16S rRNA sequences. We then compared this phylogeny to the number of SPases in each organism and to the presence or absence of Pro at the position corresponding to residue 29 in S. epidermidis (unless otherwise specified, S. epidermidis numbering is used hereafter). In general, Gram-negative bacteria from the Chlamydiae/Verrucomicrobia, Proteobacteria, and Bacteroidetes phyla, have a single SPase, and in each phylum, Pro29 is present in a subset of organisms (Fig 4). Accordingly, almost all of the sequenced α-, β-, γ-Proteobacteria have SPases with Pro29 (115/123, 64/65, and 178/183 of the sequenced organisms, respectively), whereas most of the sequenced δ- and ε-Proteobacteria have SPases with Ala29 (32/35 and 27/29, respectively). Similarly, within the Bacteroidetes phylum, each of the sequenced Flavobacteria has one SPase and Pro is always present at position 29, whereas each of the Bacteroidia typically has an SPase with Asn29 and sometimes a second SPase with Ser29. Finally, among the few Chlamydiae/Verrucomicrobia that have been sequenced, each of the Chlamydia has one SPase with Leu29 (7/7), while each of the Verrucomicrobia has at least one SPase with Pro29 (8/8). The phylogeny of the SPase genes themselves largely mirrors that of the 16S rRNA sequences (Fig S3), indicating that relatively little horizontal transfer of SPase genes has occurred and confirming that Pro29 was installed independently into the SPases of these lineages. Interestingly, horizontal gene transfer is responsible for the scattered instances of Proteobacteria that do encode multiple SPases, including the second SPase of P. aeruginosa that has Leu29. These additional SPases are not closely related to any of the sequenced SPases examined in this analysis, and they may not be functionally equivalent to the other Proteobacterial SPases.
Figure 4. Distribution of SPase with Pro29 (or Pro31) residues based on bacterial 16S rRNA phylogenies.
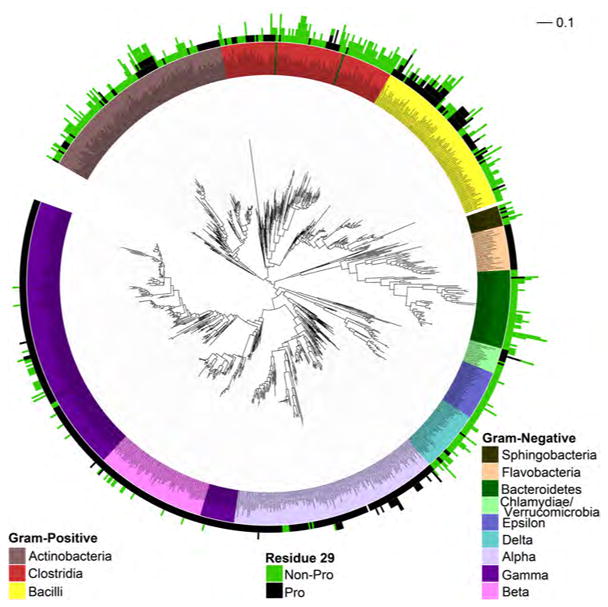
Phylogenies of Gram-positive Firmicutes and Actinobacteria and Gram-negative Proteobacteria, Bacteroidetes, and Verrucomicrobia/Chlamydiae based on 16S rRNA sequences. Species are color coded by phylogenetic class (inner color band). The number of SPases and the presence or absence Pro at the relevant positions is indicated by the length and color(s) of the outer color band. Scale bar corresponds to a 10% divergence in 16S rRNA sequence. See also Figure S3 and S4, and Table S1.
In contrast to the Gram-negative bacteria, the Gram-positive Firmicutes and Actinobacteria commonly encode multiple SPases, and comparison of the 16S rRNA and SPase phylogenies indicates that duplication of SPase genes has occurred multiple times in these lineages (Figs. 4 and S3). The distribution of Pro29 is also more irregular, which appears to have resulted from reduced conservation leading to the frequent introduction and removal of Pro at this position. Moreover, the region defined by residues 27–31 appears to be poorly conserved within the Gram-positive SPases relative to the same region in the Gram-negative proteins or to the regions that comprise the core and active site of the protein (Table S1). Nonetheless, Pro29 is particularly common among the SPases of the Bacillus, Listeria, and Staphylococcus genera of Firmicutes and the Streptomycetes genus of Actinobacteria. Interestingly, although SPases with Pro29 appear to have been present and maintained during speciation of the Bacilli, Streptomycetes, and Listeria, the common Staphylococcus ancestor appears to have had two SPases each with Ser29, as is still the case with S. epidermidis. S. aureus appears to have deleted one of these SPases and introduced Pro29 into the other (Fig S4).
The arylomycins have a broad spectrum of antibiotic activity
To further explore the spectrum of arylomycins and to test the contribution of the Pro29 to arylomycin resistance in a wider range of bacteria, we determined the arylomycin susceptibilities of representative organisms from the above phylogenetic analysis (Table 2). Bacteria from all five phyla were sampled, and when possible important human pathogens were included. Consistent with our expectations, arylomycin C16 is active against the ε-Proteobacteria H. pylori (whose SPase has Ala29) with an MIC of 4 μg/ml. Similarly, the intracellular Gram-negative pathogen C. trachomatis (Leu29) is eradicated from human HeLa 229 cells with an MIC of 6 μg/ml. Notably, no adverse effects on the human cells were observed up to 20 μg/ml of arylomycin, the highest concentrations examined. With Francisella tularensis (Asn29), a potential biological warfare agent and a member of the only genera of γ-Proteobacteria that does not have Pro29, we examined nineteen clinical isolates and found that 8 are inhibited with MICs of 4 to 16 μg/ml, one with an MIC of 32 μg/ml, and the remainder with MICs in excess of 64 μg/ml. Klebsiella pneumoniae encodes a single SPase that has Pro29 and is resistant to the arylomycins. Interestingly, although its single SPase has Pro29, Yersinia pestis, the causative agent of plague, is sensitive to arylomycin C16.
Table 2.
Associations between arylomycin C16 sensitivity and SPase genotype(s) among different wild type bacteria
| Species | Residue 29 | MIC (μg/ml) |
|---|---|---|
| Staphylococcus epidermidis | S,S | 0.25 |
| Staphylococcus haemolyticus | S,S | 2 |
| Rhodococcus opacus | V | 2 |
| Corynebacterium glutamicum | M | 2 |
| Helicobacter pylori | A | 4 |
| Yersinia pestis | P | 4 |
| Chlamydia trachomatis | L | 6 |
| Francisella tularensis | N | 4 –16, >64a |
| Streptococcus pneumoniae | N | 16 |
| Streptococcus pyogenes | A | 16 |
| Lactococcus lactis | L | 16, >128a |
| Rhodococcus equi | V,I | 16 |
| Corynebacterium efficiens | P | >64 |
| Staphylococcus aureus | P | 12–25, >128a |
| Brevibacillus brevis | P,P,P,P,V | >64 |
| Enterococcus faecalis | P,P,P,S | >64b |
| Bacillus subtilis | P,P,P,D | >128 |
| Streptococcus agalactiae | F,V | >128 |
| Escherichia coli | P | >128 |
| Pseudomonas aeruginosa | P,L | >128 |
| Klebsiella pneumoniae | P | >128 |
| Lactobacillus sakei | P,N | >128 |
| Lactobacillus gasseri | N,N | >128 |
| Lactobacillus acidophilus | N | >128 |
| Lactobacillus plantarum | M,M,V | >128 |
| Clostridium difficile | P,P,P | >16 |
| Clostridium bolteae | N,N,Q | >16 |
| Clostridium perfringens | K,K,K,I | >16 |
| Bacteroides fragilis | S,N | >16 |
| Prevotella copris | N | >16 |
Multiple values indicate heterogeneity within different strains of a species, as discussed in the text
Determined previously (Roberts et al., 2007)
The Gram-positive Firmicutes Streptococcus pneumoniae, Streptococcus pyogenes, and Staphylococcus haemolyticus, are all human pathogens that lack SPases with Pro29. As predicted, we find that each is sensitive to arylomycin C16. Also as predicted, we find that B. subtilis and E. faecalis, Firmicutes with multiple SPases with Pro29, are resistant. Interestingly we that find Brevibacillus brevis, which was previously reported to be moderately sensitive to the arylomycins (Schimana et al., 2002), grows in the presence of more than 64 μg/ml, as predicted from its four encoded SPases with Pro29. Within the Gram-positive Actinobacteria, Rhodococcus equi, like Rhodococcus opacus (Schimana et al., 2002) is sensitive to the arylomycins, and both lack Pro29. While the actinobacteria Corynebacterium glutamicum has a single SPase with Met29 and an arylomycin C16 MIC of 2 μg/ml, the related actinobacteria Corynebacterium efficiens has a single SPase with Pro29 and an MIC of greater than 64 μg/ml. However, we also found Gram-positive bacteria for which the model was not as predictive. For example, while Lactococcus lactis spp. cremonis has a single SPase with Leu29 and is sensitive to arylomycin C16, the highly related Lactococcus lactis spp. lactii also has a single SPase with Leu29 and is resistant. Additionally, we found that a variety of other Lactobacillales, and all investigated Clostridia and Bacteriodetes, are resistant to the arylomycins despite the fact that many lack SPases with Pro29 (MIC > 64 μg/ml for the Lactobacillales and > 16 μg/ml for the Clostridia and Bacteriodetes). Finally, a broader survey of S. aureus strains revealed two, COL, a tetracycline and penicillin resistant strain (MIC = 12 μg/ml), and Rosenbach 328, an MRSA strain (Pantosti and Venditti, 2009) (MIC = 25 μg/ml), are moderately sensitive despite having SPases with Pro29.
DISCUSSION
The golden age of antibiotic discovery, roughly covering the four decades following the discovery of penicillin, saw the development of almost all of the major classes of broad-spectrum antibiotics in use today. While these antibiotics revolutionized the treatment of infectious disease, the more recent decades have witnessed the systematic and relentless evolution of resistant bacteria, and discovery efforts have failed to produce even a single novel class for development. While many narrow-spectrum antibiotics have been identified, these compounds are typically assumed to inhibit targets that are poorly conserved, not universally essential, or not accessible, and therefore these compounds are not typically considered for commercial development (Lambert, 2002; Payne et al., 2007). One such class of novel natural products are the arylomycins, which despite inhibiting an essential target with a conserved active site, appeared to have a prohibitively narrow spectrum of whole cell activity. Although it was proposed that they do not penetrate the outer membrane of Gram-negative bacteria (Kulanthaivel et al., 2004; Roberts et al., 2007), their lack of activity against most Gram-positive bacteria has remained more enigmatic. We find that activity against the important human pathogens S. aureus, E. coli, and P. aeruginosa is not limited by target conservation, essentiality, or accessibility, but rather by the presence of a specific SPase residue, Pro29, which we initially identified by characterizing how the naturally sensitive organism S. epidermidis evolves resistance. For both the E. coli and S. aureus SPases, we also demonstrated that the presence of Pro at this position confers resistance by reducing the affinity with which SPase is bound by the arylomycins.
A physical basis for the observed resistance mechanism is suggested by the structure of arylomycin A2 bound to wild type E. coli SPase solved by Paetzel et al. (Paetzel et al., 2004) (Fig. 3A). The macrocyclic core packs into the deep SPase active-site cleft and forms critical contacts with the catalytic residues and with the residues that comprise the P3 substrate binding pocket. Moreover, each hydrogen-bond donor/acceptor of the peptide backbone portion of the macrocycle engages the protein. In contrast, the three residues N-terminal to the macrocycle interact only with a shallow groove and form only two hydrogen-bonds, each between backbone residues of the inhibitor and protein. Residue 84 (corresponding to residue 29 in S. epidermidis SpsIB) is located within this region, and reduced inhibitor-protein interactions are consistent with our finding that many amino acids at this position result in similar arylomycin sensitivities. However, the backbone amide of residue 84 appears to be positioned to form a stabilizing hydrogen-bond with the carbonyl oxygen of the arylomycin fatty acid tail, an interaction that is uniquely ablated by mutation to Pro, which lacks an amide proton. Similarly, the hydrogen-bond between Gln86 (corresponding to residue 31, the alternative resistance conferring residue in S. epidermidis) and arylomycin Gly4 would be ablated by mutation to Pro, likely explaining why it also confers resistance in S. epidermidis, while the proximity of this mutation to the catalytic residues may explain why it is not tolerated in some organisms. Moreover, hydrophobic amino acids at position 84 might promote desolvation of the backbone amide in the unbound state, which would favor arylomycin binding and account for the greater sensitivity conferred by these amino acids relative to their more polar counterparts.
The degree of conservation of the region surrounding residue 29 varies from low within Gram-positive bacteria, to relatively high within the Proteobacteria, and contrasts with the extreme conservation of the residues that more immediately surround the catalytic site and form the critical P1 and P3 substrate binding pockets. Interestingly, the sequence of the preprotein substrates cleaved by SPases are strongly conserved only at positions −1 and −3 relative to the cleavage site, where Ala residues are almost exclusively encoded (Choo and Ranganathan, 2008; Paetzel et al., 2002). Thus, as with the arylomycins, the natural substrates do not appear to make side chain-specific interactions with SPase in the region surrounding residue 29, which likely contributes to the observed sequence divergence in this region of enzyme. The presence of multiple paralogous SPases in many Gram-positive bacteria may further reduce the constraints on SPase divergence, and contribute to the sporadic distribution of Pro29 in the SPases of these bacteria. Interestingly, the relatively high sequence conservation within the region surrounding residue 29 of SPase from Gram-negative Proteobacteria suggests that this residue plays an important role in SPase function, despite the results of our model studies with E. coli where most residues are well tolerated at this position. Thus, whatever the role of Pro29, it is not manifest under laboratory conditions and must instead be unique to the demands of protein secretion in the organism’s natural environments (Papp et al., 2004).
Using Pro29 as a sequence signature of arylomycin resistance, we identified many diverse bacteria, including important human pathogens, that lack this residue and then confirmed that many of these bacteria have MICs less than or equal to 16 μg/ml (Table 2), potencies reasonable from the perspective of drug development. While the absence of Pro29 is often predictive of arylomycin sensitivity, the absolute sensitivity varies between different species. For example, while R. opacus and R. equi are closely related and lack SPases with Pro29, their sensitivities to arylomycin C16 differ by 8-fold. Similarly, wild type S. epidermidis, which its natural Ser29 SPase, is 8-fold more sensitive than the Ser29 mutant of S. aureus. Furthermore, some organisms are highly resistant to the arylomycins in the absence of SPases with Pro29, including many of the Lactobacillales, Clostridia and Bacteriodetes. Conversely, Y. pestis and several strains of S. aureus, including a clinical isolate of MRSA, which were predicted to be resistant, are in fact sensitive. Taken together, this data suggests that other factors also contribute to the absolute level of arylomycin sensitivity, possibly including other variations in SPase sequence, differences in SPase expression levels, differences in the levels of protein secretion required for growth, or the presence of yet to be identified modifying enzymes or efflux pumps. Alternatively, certain strains could be hyper-susceptible to the inhibition of secretion as a result of the accumulation of specific toxic proteins.
The arylomycins are active against a wide range of bacteria, including both Gram-negative and Gram-positive organisms, and are therefore not narrow-spectrum. The term “narrow-spectrum” is typically reserved for antibiotics that inhibit targets that are only accessible, essential, and sufficiently conserved within specific bacterial taxa. These limitations preclude the expansion of the scaffold’s spectrum to entire groups of other bacteria. For example, polymyxin B selectively destabilizes Gram-negative membranes (Evans et al., 1999), daptomycin selectively destabilizes Gram-positive membranes (Tally and DeBruin, 2000), and fosmidomycin inhibits a component of a metabolic pathway that is not present in many bacteria (Jomaa et al., 1999). Moreover, large or excessively hydrophobic antibiotics, such as vancomycin, are unable to penetrate the outer-membranes of Gram-negative bacteria (Nikaido, 2003). In contrast, the arylomycins penetrate permeability barriers including the lipid rich cell wall of Corynebacterium (Lambert, 2002) and the outer-membranes of Gram-negative bacteria (Nikaido, 2003), interact primarily with a highly conserved region of SPase, and consequently have activity against a remarkably diverse range of bacteria. This is reflected by the lack of correlation between arylomycin sensitivity and overall phylogenetic relatedness. The reduced spectrum of the arylomycins results instead from their interactions with a less conserved region of SPase where a specific resistance conferring mutation naturally occurs in some SPases. From an evolutionary perspective, this might suggest that the arylomycins were selected for activity against a specific competitor that lacked a resistance-conferring residue. Alternatively, arylomycin biosynthesis may have emerged early in bacterial evolution before this region of SPase diverged, and in this case, it is possible that selection by arylomycin contributed in some instances to the fixation of Pro29. Investigating the antiquity and prevalence of arylomycin biosynthesis should shed light on this fascinating possibility. From a medicinal chemistry perspective, the results suggest that an arylomycin derivative modified to bind with increased affinity to the conserved catalytic region of SPase, and to rely less on interactions with the more variable regions, would be active against most bacteria.
Importantly, our results also emphasize that specific resistance mechanisms in several bacteria commonly used to screen for novel antibiotics can prevent the identification of scaffolds with the potential for development into broad-spectrum therapeutics. The pharmaceutical industry discovered the arylomycin class of antibiotics only after screening a library of purified natural products for activity in an in vitro SPase assay, and they would not likely have been identified using a whole cell assay. Combined with the recently recognized high frequency of natural resistance to known antibiotics (D’Costa et al., 2007; D’Costa et al., 2006; Riesenfeld et al., 2004; Waters and Davies, 1997), our results raise the possibility that the broad-spectrum potential of other hits identified from in vitro assay may have been obfuscated and that novel classes of antibiotics may have been missed entirely in whole cell screens. Thus, additional “latent antibiotics” might be identified by more careful examination of known apparently narrow-spectrum antibiotics, especially where resistance patterns are uneven across phylogenetically related species, or by expanding the set of bacteria tested during drug discovery efforts to include a broader range of phylogenetically diverse bacteria. Given the well established precedent for optimizing suitable antibiotic scaffolds to overcome specific mechanisms of resistance, as evidenced by the successful development of many “next generation” variants of antibiotics that possess broader spectra of activity (Essack, 2001; Gootz, 1990; Hammerschlag and Sharma, 2008; Schneider et al., 2003), the discovery of additional latent antibiotics could provide invaluable scaffolds for optimization into broad-spectrum antibiotics.
EXPERIMENTAL PROCEDURES
Strains and Culture Conditions
Standard methods were used to culture bacteria for all experiments and to construct mutant strains; details and a description of the strains used in this study are provided in SI Methods.
Selection of Arylomycin Resistant S. epidermidis and Sequencing of Signal Peptidase Genes
S. epidermidis (~1 × 109 cfu) were plated on tryptic soy agar (TSA) containing 2 μg/ml arylomycin C16. Resistant colonies visible at 24 hours were re-streaked onto TSA containing 2 μg/ml arylomycin C16 to confirm the resistant phenotype. Isolation of genomic DNA and sequencing of SPases genes is described in SI Methods.
Minimum Inhibitory Concentration Experiments
With the exception of C. trachomatis and H. pylori, minimum inhibitory concentrations of arylomycin C16 were determined by a modified CLSI micro-broth dilution method in 100 μL of media containing 2-fold dilutions of arylomycin C16. Media and growth conditions used for MIC experiments, as well as a detailed description of the determination of the arylomycin MICs for C. trachomatis and H. pylori are described in SI Methods.
In Vitro KD Measurements
Construction of expression vectors and the subsequent production of the various SPase variants used in this study are described in SI Methods. Steady state binding of arylomycin C16 was determined by measuring the previously described increase in arylomycin fluorescence (λex = 320 nm, λem = 410 nm) upon binding E. coli Δ2-75 SPase (Paetzel et al., 2004). The binding buffer for full length and truncated E. coli proteins was as follows: 100 mM NaCl, 20 mM Tris-HCl pH 7.4, 1 mM EDTA, 1% n-octyl-β-glucopyranoside (Anatrace). This buffer was supplemented with 10% glycerol for experiments with S. aureus SPase protein.
SPase Sequence Analysis
The amino acid sequences of the SPases from E. coli, S. aureus, B. fragilis and C. efficiens were concatenated and used as the query sequence in a BLAST against all of the fully sequenced genomes of Bacteriodetes, Actinobacteria, Firmicutes, Proteobacteria, and Chlamydiae/Verrucomicrobia available in the NCBI Microbial Genome Database. The amino acid sequence of BLAST hits with an E-value less than 0.1 were aligned using MUSCLE (Edgar, 2004), and all sequences lacking the catalytic Ser or Lys residues were removed. Poorly aligned regions were removed using the “Block Mapping and Gathering using Entropy” program found at http://mobyle.pasteur.fr/cgi-bin/portal.py, with Gap Rate Cutoff 0.3 and the Entropy Cutoff of 0.7. Phylogenetic analysis was conducted using PhyML 3.0 with 20 rate categories and SPR branch improvement (Guindon and Gascuel, 2003). SPases from Gram-positive and Gram-negative organisms were kept separate during alignment and phylogenetic analysis to improve the quality of these analyses. Also several SPases from the Gram-negative Proteobacteria were removed prior to analysis of SPase phylogeny, since they did not show an obvious relation to any of the other Gram-negative or Gram-positive SPases examined. Phylogenetic trees were displayed using the Interactive Tree of Life (Letunic and Bork, 2007).
16S rRNA Sequence Analysis
Aligned 16S rRNA sequences were analyzed were obtained from the Ribosomal Database Project (Cole et al., 2009). The “Block Mapping and Gathering using Entropy” program was used with a Gap Rate Cutoff of 0.7 and an Entropy Cutoff of 0.7 to remove poorly aligned regions. Phylogenetic analysis was performed using PhyML 3.0, with the HKY85 substitution model with 20 rate categories and SPR branch improvement, and the resulting tree was displayed using the Interactive Tree of Life (Letunic and Bork, 2007).
Highlights.
The arylomycins have broad-spectrum antibiotic activity
Resistance in key pathogens is mediated by naturally occurring target mutations
The arylomycin scaffold is promising for development as a broad-spectrum antibiotic
Natural resistance may prevent the identification of novel classes of antibiotics
SIGNIFICANCE
Natural products have proven to be the most fruitful source of antibiotics, but discovering novel natural products with broad-spectrum activity has proven difficult, and it has been almost fifty years since a novel class of broad-spectrum antibiotics was introduced into the clinic. The arylomycin class of natural products originally elicited significant interest due to their in vitroinhibition of type I signal peptidase (SPase), a promising antibiotic target. However, the arylomycins were soon found to be inactive against many important pathogens, and like many antibiotics with a narrow-spectrum of activity, they were then dismissed as potential therapeutics. Herein, we show that when challenged with arylomycin, the naturally sensitive organism Staphylococcus epidermidis evolves resistance via mutations in SPase. Unexpectedly, we find that the analogous mutations are naturally present in SPases of many bacteria, including those of the important human pathogens Staphylococcus aureus, Escherichia coli, and Pseudomonas aeruginosa, and we confirm that they are responsible for the natural resistance of these pathogens. We identify diverse bacteria lacking SPases with these mutations and demonstrate that most are sensitive to the arylomycins, and in doing so we greatly expand the known activity spectrum of the arylomycins to include organisms from four distinct phyla. Thus, the arylomycins are bona fide broad-spectrum antibiotics. Although the presence of resistance conferring mutation in key pathogens currently limits the therapeutic utility of these molecules, historical precedent suggests that natural product scaffolds can be modified to overcome specific resistance mechanisms. Thus, the arylomycins are valuable leads for antibiotic development. Importantly, our results also demonstrate that resistance mechanisms in the bacteria commonly used to screen for novel antibiotics can prevent the identification of promising scaffolds and consequently that valuable leads for antibiotic development may still lie undetected among available natural products.
Supplementary Material
Acknowledgments
We thank Prof. Erguang Li (Nanjing University School of Medicine) for help in determining the MICs of C. trachomatis, Michael Powers and Prof. Marygorret Obonyo (UCSD) for help in determining the MICs of H. pylori, Dr. Hank Heine (USAMRIID) for help in determining the MICs F. tularensis, and Prof. Sydney Finegold (West L.A. VA Medical Center) for help in determining the MICs of the Clostridia and Bacteroidetes. This work was supported by the Office of Naval Research (Awards N000140310126 and N000140810478) and the National Institutes of Health (AI081126).
Footnotes
Supplemental information includes 4 figures, 1 table, and supplemental experimental procedures and can be found with this article online at doi:10.1016/j.chembiol.####.##.###.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baltz RH. Marcel Faber Roundtable: is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol. 2006;33:507–513. doi: 10.1007/s10295-005-0077-9. [DOI] [PubMed] [Google Scholar]
- Bumann D. Has nature already identified all useful antibacterial targets? Curr Opin Microbiol. 2008;11:387–392. doi: 10.1016/j.mib.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Choo KH, Ranganathan S. Flanking signal and mature peptide residues influence signal peptide cleavage. BMC Bioinformatics. 2008;9(Suppl 12):S15. doi: 10.1186/1471-2105-9-S12-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37:D141–145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg KM, Wilding I, Black MT. Molecular cloning and expression of the spsB gene encoding an essential type I signal peptidase from Staphylococcus aureus. J Bacteriol. 1996;178:5712–5718. doi: 10.1128/jb.178.19.5712-5718.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Costa VM, Griffiths E, Wright GD. Expanding the soil antibiotic resistome: exploring environmental diversity. Curr Opin Microbiol. 2007;10:481–489. doi: 10.1016/j.mib.2007.08.009. [DOI] [PubMed] [Google Scholar]
- D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- Dalbey RE, Lively MO, Bron S, van Dijl JM. The chemistry and enzymology of the type I signal peptidases. Protein Sci. 1997;6:1129–1138. doi: 10.1002/pro.5560060601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date T. Demonstration by a novel genetic technique that leader peptidase is an essential enzyme of Escherichia coli. J Bacteriol. 1983;154:76–83. doi: 10.1128/jb.154.1.76-83.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essack SY. The development of beta-lactam antibiotics in response to the evolution of beta-lactamases. Pharm Res. 2001;18:1391–1399. doi: 10.1023/a:1012272403776. [DOI] [PubMed] [Google Scholar]
- Evans ME, Feola DJ, Rapp RP. Polymyxin B sulfate and colistin: old antibiotics for emerging multiresistant gram-negative bacteria. Ann Pharmacother. 1999;33:960–967. doi: 10.1345/aph.18426. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Antibiotics for emerging pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootz TD. Discovery and development of new antimicrobial agents. Clin Microbiol Rev. 1990;3:13–31. doi: 10.1128/cmr.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hammerschlag MR, Sharma R. Use of cethromycin, a new ketolide, for treatment of community-acquired respiratory infections. Expert Opin Investig Drugs. 2008;17:387–400. doi: 10.1517/13543784.17.3.387. [DOI] [PubMed] [Google Scholar]
- Hancock REW. The end of an era? Nat Rev Drug Discov. 2007;6:28–28. [Google Scholar]
- Hellmark B, Unemo M, Nilsdotter-Augustinsson A, Soderquist B. Antibiotic susceptibility among Staphylococcus epidermidis isolated from prosthetic joint infections with special focus on rifampicin and variability of the rpoB gene. Clin Microbiol Infect. 2009;15:238–244. doi: 10.1111/j.1469-0691.2008.02663.x. [DOI] [PubMed] [Google Scholar]
- Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- Kulanthaivel P, Kreuzman AJ, Strege MA, Belvo MD, Smitka TA, Clemens M, Swartling JR, Minton KL, Zheng F, Angleton EL, et al. Novel lipoglycopeptides as inhibitors of bacterial signal peptidase I. J Biol Chem. 2004;279:36250–36258. doi: 10.1074/jbc.M405884200. [DOI] [PubMed] [Google Scholar]
- Kuo DW, Chan HK, Wilson CJ, Griffin PR, Williams H, Knight WB. Escherichia coli leader peptidase: production of an active form lacking a requirement for detergent and development of peptide substrates. Arch Biochem Biophys. 1993;303:274–280. doi: 10.1006/abbi.1993.1283. [DOI] [PubMed] [Google Scholar]
- Lambert PA. Cellular impermeability and uptake of biocides and antibiotics in Gram-positive bacteria and mycobacteria. J App Microbiol. 2002;92:46s–54s. [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2007;23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzel M, Dalbey RE, Strynadka NC. The structure and mechanism of bacterial type I signal peptidases. A novel antibiotic target. Pharmacol Ther. 2000;87:27–49. doi: 10.1016/s0163-7258(00)00064-4. [DOI] [PubMed] [Google Scholar]
- Paetzel M, Goodall JJ, Kania M, Dalbey RE, Page MG. Crystallographic and biophysical analysis of a bacterial signal peptidase in complex with a lipopeptide-based inhibitor. J Biol Chem. 2004;279:30781–30790. doi: 10.1074/jbc.M401686200. [DOI] [PubMed] [Google Scholar]
- Paetzel M, Karla A, Strynadka NC, Dalbey RE. Signal peptidases. Chem Rev. 2002;102:4549–4580. doi: 10.1021/cr010166y. [DOI] [PubMed] [Google Scholar]
- Pantosti A, Venditti M. What is MRSA? Eur Respir J. 2009;34:1190–1196. doi: 10.1183/09031936.00007709. [DOI] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD. Metabolic network analysis of the causes and evolution of enzyme dispensability in yeast. Nature. 2004;429:661–664. doi: 10.1038/nature02636. [DOI] [PubMed] [Google Scholar]
- Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Poole K. Efflux-mediated antimicrobial resistance. J Antimicrob Chemother. 2005;56:20–51. doi: 10.1093/jac/dki171. [DOI] [PubMed] [Google Scholar]
- Riesenfeld CS, Goodman RM, Handelsman J. Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environ Microbiol. 2004;6:981–989. doi: 10.1111/j.1462-2920.2004.00664.x. [DOI] [PubMed] [Google Scholar]
- Roberts TC, Smith PA, Cirz RT, Romesberg FE. Structural and initial biological analysis of synthetic arylomycin A2. J Am Chem Soc. 2007;129:15830–15838. doi: 10.1021/ja073340u. [DOI] [PubMed] [Google Scholar]
- Schimana J, Gebhardt K, Holtzel A, Schmid DG, Sussmuth R, Muller J, Pukall R, Fiedler HP. Arylomycins A and B, new biaryl-bridged lipopeptide antibiotics produced by Streptomyces sp Tü 6075 I Taxonomy, fermentation, isolation and biological activities. J Antibiot (Tokyo) 2002;55:565–570. doi: 10.7164/antibiotics.55.565. [DOI] [PubMed] [Google Scholar]
- Schneider P, Hawser S, Islam K. Iclaprim, a novel diaminopyrimidine with potent activity on trimethoprim sensitive and resistant bacteria. Bioorg Med Chem Lett. 2003;13:4217–4221. doi: 10.1016/j.bmcl.2003.07.023. [DOI] [PubMed] [Google Scholar]
- Silver LL. Multi-targeting by monotherapeutic antibacterials. Nat Rev Drug Discov. 2007;6:41–55. doi: 10.1038/nrd2202. [DOI] [PubMed] [Google Scholar]
- Tally FP, DeBruin MF. Development of daptomycin for gram-positive infections. J Antimicrob Chemother. 2000;46:523–526. doi: 10.1093/jac/46.4.523. [DOI] [PubMed] [Google Scholar]
- von Nussbaum F, Brands M, Hinzen B, Weigand S, Habich D. Antibacterial natural products in medicinal chemistry--exodus or revival? Angew Chem Int Ed Engl. 2006;45:5072–5129. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]
- Waters B, Davies J. Amino acid variation in the GyrA subunit of bacteria potentially associated with natural resistance to fluoroquinolone antibiotics. Antimicrob Agents Chemother. 1997;41:2766–2769. doi: 10.1128/aac.41.12.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YB, Greenberg B, Lacks SA. Analysis of a Streptococcus pneumoniae gene encoding signal peptidase I and overproduction of the enzyme. Gene. 1997;194:249–255. doi: 10.1016/s0378-1119(97)00198-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.