

Abstract
Methylmercury (MeHg) exposure from occupational, environmental, and food sources is a significant threat to public health. MeHg poisonings in adults may result in severe psychological and neurological deficits, and in utero exposures can confer embryonic defects and developmental delays. Recent epidemiological and vertebrate studies suggest that MeHg exposure may also contribute to dopamine (DA) neuron vulnerability and the propensity to develop Parkinson's disease (PD). In this study, we describe a Caenorhabditis elegans model of MeHg toxicity that shows that low, chronic exposure confers embryonic defects, developmental delays, decreases in brood size and animal viability, and DA neuron degeneration. Toxicant exposure results in the robust induction of the glutathione-S-transferases (GSTs) gst-4 and gst-38 that are largely dependent on the PD-associated phase II antioxidant transcription factor SKN-1/Nrf2. We also demonstrate that the expression of SKN-1, a protein previously localized to a small subset of chemosensory neurons and intestinal cells in the nematode, is also expressed in the DA neurons, and a reduction in SKN-1 gene expression increases MeHg-induced animal vulnerability and DA neuron degeneration. These studies recapitulate fundamental hallmarks of MeHg-induced mammalian toxicity, identify a key molecular regulator of toxicant-associated whole-animal and DA neuron vulnerability, and suggest that the nematode will be a useful in vivo tool to identify and characterize mediators of MeHg-induced developmental and DA neuron pathologies.
Keywords: methylmercury, Parkinson's disease, oxidative stress, C. elegans, nematode, genetics, Nrf2, SKN-1, dopamine neurodegeneration
Methylmercury (MeHg) is a pervasive environmental toxicant that primarily targets the central nervous system (Aschner and Aschner, 1990; Friberg and Mottet, 1989). Adults exposed to toxic levels of MeHg can display a range of symptoms that includes a loss of physical coordination, abnormal speech, and death. The developing fetus is particularly vulnerable to MeHg because of the toxicant's ease in crossing the placenta and several fold greater toxicant accumulation relative to the adult (Bakir et al., 1973; Clarkson and Magos, 2006; Myers and Davidson, 1998; Takeuchi, 1982). Children exposed to toxic levels either pre- or postnatally display developmental defects that are associated with motor and sensory deficits and mental retardation (Clarkson and Magos, 2006).
Although MeHg poisonings have been studied for decades, the molecular bases for the toxicities are largely ill defined. MeHg diffuses across most cellular membranes and intracellular compartments and disrupts a wide array of cellular functions including cell migration, neuronal axon guidance, microtubule integrity, and cell signaling (Bridges and Zalups, 2005; Kunimoto and Suzuki, 1997; Limke et al., 2004). Dopamine (DA) neurons are particularly vulnerable to the toxicant as exposure to MeHg increases DA release and decreases DA reuptake and catabolism (Dreiem et al., 2009; Newland et al., 2008). Intriguingly, anecdotal evidence suggests that MeHg may be an environmental toxicant that contributes to the development of Parkinson's disease (PD), a progressive neurodegenerative disorder that results in motor deficits, mitochondria dysfunction, and loss of DA neurons in the substantia nigra (Nass and Przedborski, 2008; Petersen et al., 2008). The mitochondria are also likely an intracellular target of MeHg as the toxicant has been shown to disrupt respiration, oxidative phosphorylation, and calcium regulation, as well as causing an increase in reactive oxygen species (ROS) and oxidative stress (Limke et al., 2004; Yu et al., 2010).
Vertebrate studies indicate that MeHg induces the phase II class of detoxification enzymes glutathione-S-transferases (GSTs) (Di Simplicio et al., 1993; Goering et al., 2000; Samali and Orrenius, 1998; Yu et al., 2010). GSTs catalyze the conjugation of a broad range of electrophilic substrates to the thiol group of reduced glutathione molecules which typically inactivates the electrophile and prepares it for excretion (Wilce and Parker, 1994). Nrf2, a leucine zipper class transcription factor, regulates a number of detoxifying enzymes including PD-associated proteins and GSTs (Kensler et al., 2007; Taylor et al., 2008; von Otter et al., 2010). MeHg also induces Nrf2 expression, overexpression of the transcription factor inhibits MeHg-induced cell death, and Nrf2-deficient hepatocytes and primary cortical neurons are vulnerable to MeHg and PD-associated toxicants (Lee et al., 2003; Toyama et al., 2007).
The nematode Caenorhabditis elegans is a powerful model system to explore the molecular basis of neurotoxicity and human disease (Nass and Hamza, 2007; Nass et al., 2008; Settivari et al., 2009). The molecular pathways and mechanisms underlying basic biological processes such as cell development and migration are highly conserved between the nematode and humans. Both organisms have virtually an identical number of genes, and almost all the known signaling and neurotransmitter systems, including the dopaminergic (DAergic) system, identified in humans have counterparts in the worm (Nass et al., 2008). The similarities in the DAergic system between the worm and humans has allowed for the successful recapitulation of both the familial PD-associated genetic as well as toxicant-induced DA neuron pathology found in vertebrate models of PD (Nass and Przedborski, 2008; Nass et al., 2008). Molecular pathways and components involved in cellular detoxification and cellular stress in eukaryotes are also conserved in C. elegans including GSTs, Heat Shock Proteins, cytochrome P450s, and the Nrf2 transcription factor orthologue SKN-1 (Oliveira et al., 2009; Park et al., 2009).
In this study, we describe a novel C. elegans model for MeHg toxicity and DA neuron degeneration. We show that that chronic low exposure to MeHg confers developmental defects, decrease in brood size, concentration-dependent animal death, and DA neurodegeneration. Exposure to the toxicant increases cellular ROS levels, induces the expression of specific GSTs, and whole-animal vulnerability to the toxicant is mitigated by the expression of the phase II transcription factor SKN-1. Furthermore, we show for the first time that SKN-1 is expressed in C. elegans DA neurons, and genetic knockdown renders the neurons vulnerable to MeHg-induced cell death. These studies indicate that the cellular and molecular response to MeHg-induced toxicity is likely conserved between the nematode and vertebrates and suggests that C. elegans will be a powerful in vivo model to explore the molecular basis of MeHg-induced developmental and DA neuron pathologies.
MATERIAL AND METHODS
Caenorhabditis elegans strains and maintenance.
The following strains were obtained from the C. elegans Genetics Center: wild-type (WT) Bristol N2; NL2099 rrf-3(pk1426); OD70 unc-119(ed3) III; ItIs44 [pie-1p-mCherry::PH(PLC1delta) + unc119 (+)]; RJ928, a genetic cross between BY250 (Pdat-1::GFP) and NL2099 (Settivari et al., 2009). The C. elegans strains were cultured on bacterial lawns containing either OP50 or NA22 bacteria on nematode growth medium (NGM) or 8P plates, respectively, at 20°C according to standard methods (Brenner, 1974; Hope, 1999).
Whole-animal vulnerability assay.
Synchronized L1 stage worms were obtained by hypochlorite treatment of gravid adults followed by incubation of the embryos in M9 buffer for 18 h and washed at least 3× in dH2O as described previously (Nass et al., 2002; Nass and Hamza, 2007). L1 staged worms were incubated on 8P/NA22 plates at 20°C for 48 h until they reached L4 stage. They were then transferred to 8P/NA22 plates containing various concentrations of MeHg (0–125μM) (CH3HgCl, 442534-5G-A; Sigma-Aldrich, St Louis, MO) and grown for 48 h at 20°C. Worms were assayed for viability by touching the nose of the animal with a metal pick. Worms that moved were considered alive. Each of the experiments was performed at least in triplicate, and the results were reported as mean ± SE.
Brood-size assay.
Single synchronized L4 stage N2 worms were each placed onto an 8P/NA22 plate containing various concentrations of MeHg (0–15μM). Each worm was transferred to a fresh plate after 24 h and then twice daily for 5 days. The total number of live worms on each plate was counted following a 48- to 72-h incubation.
Larvae development rate assay.
L1 synchronized N2 nematodes were incubated on 8P/NA22 plates containing 0 or 10μM MeHg at 20°C. The plates were examined every 12 h, and the time was recorded when the worms reached the L4 and adult stages. Each of the experiments was repeated at least four times.
Embryonic development assay.
Synchronized L1 stage OD70 worms were incubated on 8P/NA22 growth plates containing ± 10μM MeHg for 72 h. OD70 worms contain a fusion of the pleckstrin homology domain derived from mammalian PLC1δ1 to the red fluorescent protein mCherry, which binds to a phosphoinositide lipid on the plasma membrane. Use of the pie-1 promoter drives expression specifically in germ cells, allowing them to be visualized under an inverted microscope (Zeiss Axioplan 2, Jena, Germany) (Axiovision Release 4.5, Jena, Germany) (Audhya et al., 2005). Following the incubation period, at least 50 worms were immobilized on 2% agarose pads with 2% sodium azide, and the image of the embryos was captured within the young adult using an inverted fluorescent microscope. Each of the experiments was performed at least in triplicate.
RNA extraction and complementary DNA synthesis.
Synchronized L4 stage worms were incubated on 8P/NA22 media plates containing 25μM MeHg and collected by washing with water, Trizol added to pellet, and frozen at −80°C until RNA purification. Total RNA was isolated from a synchronized C. elegans population using Trizol reagent as described previously with minor modifications (Novillo et al., 2005). Briefly, the worm pellets were resuspended in Trizol (1 ml/100 μl compact worm pellet), the protein and other impurities were separated from nucleic acids using chloroform, and the RNA was precipitated with isopropyl alcohol. The RNA pellet was washed with 75% ethanol, air-dried and dissolved in RNase-free water, and stored at −80°C until used for further analysis. The RNA concentrations were measured using an ND-1000 spectrophotometer (Nanodrop Technology, Wilmington, DE). One microgram of total RNA was reverse transcribed to complementary DNA (cDNA) using the iScript cDNA Synthesis Kit and following the manufacturer's instructions (Bio-Rad, Hercules, CA). The cDNA was further purified using Microcon YM30 filters (Millipore Corp., Bedford, MA) and measured using an ND-1000 spectrophotometer.
qPCR measurements.
Gene-specific primers were designed using Primer3 software, and primers were designed to be exon spanning to avoid amplification of contaminating genomic DNA. Glyceraldehyde-3-dehydrogenase (GAPDH) was selected as the housekeeping gene as its expression does not change as a result of MeHg treatment. Primers used to determine changes in gene expression are found in Supplementary figure 1. Real-time PCR was performed using 2x SYBR Green PCR master mix and the ABI Prism 7500 sequence detection system (Applied Biosystems, Warrington, UK). Gene expression studies were performed in triplicate and the formation of a single PCR product was confirmed using dissociation curves. Negative controls with the primers consisted of all the components of PCR mix except cDNA. Relative fold change in gene expression for each gene was calculated using normalized CT values (the cycle number at which the fluorescence passes the threshold).
ROS analysis.
Total ROS formation was evaluated in the whole worm using 2,7-dichlorodihydro-fluorescein-diacetate (H2-DCF-DA). ROS oxidize the dye to the fluorescent 2,7-dichlorofluorescein, and the level of fluorescence corresponds to cellular ROS levels. ROS levels were determined following previously established protocols with slight modifications (Kampkotter et al., 2007; Schulz et al., 2007). Briefly, synchronized L4 worms were exposed to 25μM MeHg as described above for 8 h and washed 3× with M9 buffer. The animals were suspended in M9, and 50 μl of the suspension was incubated in 50 μl freshly prepared 100μM H2-DCF-DA (for a final concentration of 50μM) in four replicates in a 96-well plate. Control wells included worms from each treatment without H2-DCF-DA or wells containing H2-DCF-DA without worms. The basal fluorescent signal from each well was measured at excitation and emission wavelengths of 485 and 520 nm, respectively, using a Tecan spectrophotometer (Tecan Spectrafluor Plus, Durham, NC). Following the initial reading, plates were incubated on a shaker at room temperature for an hour and were then measured following earlier conditions. Fluorescence values from the control wells were subtracted from the corresponding signal value of each well after the final measurement and the change in fluorescence was determined by subtracting the initial value from the final value for each well. All assays were performed in triplicate.
Antibodies and Western analysis.
Antibodies to amino acids 6–92 from the putative C. elegans GST-38 protein (WP:CE15958) were generated using Genomic Antibody Technology at Strategic Diagnostics, Inc. (SDI) (Newark, DE). Rabbit polyclonal antibodies were further purified at SDI. GAPDH (ab36840 Abcam, Cambridge, MA) was used as a loading control for Western analysis. To prepare protein for Western blot analysis, synchronized L4 stage worms exposed to MeHg were pelleted from media plates as described above: 150 μl of mito buffer (20mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.5, 250mM sucrose, 1mM ethylene diamine tetraacetic acid, 1mM ethylene glycol tetraacetic acid, 10mM KCl, 1.5mM MgCl2, 1mM dithiothreitol, 0.1mM PMSF, 2 μg/ml leupeptin, 2 μg/ml pepstatin, 2 μg/ml aprotinin) was added to 300–400 μl of pelleted worms and the tubes frozen at −20°C until protein purification. Worm samples stored at −20°C were thawed and homogenized on ice with 50–60 strokes with a 2-ml glass homogenizer. The lysate was spun at 400 g at 4°C for 4 min, the supernatant collected in a sterile tube, and protein concentration determined using the Bradford assay with bovine gamma globulin as the standard. The samples were diluted in NuPAGE LDS buffer (Invitrogen, Carlsbad, CA), heated at 95°C for 20 min, and total cell lysates (50 μg protein) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to PVDF membranes (Bio-Rad). Membranes were blocked with 5% nonfat dry milk dissolved in TBST (Tris-buffered saline, 0.1% Tween-20) for 2 h at RT, followed by incubation with the appropriate primary antibody dilution (anti-GST-38 at 1:20,000; anti-GAPDH at 1:5,000) at 4°C overnight. The membranes were washed three times at RT for 15 min and incubated with horseradish peroxidase-conjugated secondary anti-rabbit IgG (611-1302; Rockland, Gilbertsville, PA). The membrane was developed using enhanced chemiluminescence (ECL) (Amersham Biosciences, Pittsburgh, PA), image captured using Bio-Rad ChemiDoc XRS, and total-protein intensities were measured using QuantityOne software (Bio-Rad).
RNA interference.
RNA-mediated interference (RNAi) using the sensitive strain NL2099 rrf-3(pk1426) were carried out on NGM plates containing 1mM isopropyl β-D-thiogalactoside and 100 μg/ml ampicillin and seeded with HT115 (DE3), an RNase III-deficient Escherichia coli strain carrying L4440 vector with the gene fragment (skn-1) (GeneService, Source BioScience, PLC, Nottingham, UK) or empty vector (Addgene, Cambridge, MA) (Timmons and Fire, 1998). Synchronized L1 stage NL2099 worms were transferred onto RNAi plates and the feeding protocol was followed with slight modifications (Kamath and Ahringer, 2003). As knockdown of skn-1 is embryonic lethal, assays were performed with first-generation synchronized L1 larvae incubated on RNAi plates at 20°C for 48 h. The L4 staged worms were then transferred onto RNAi plates containing various [MeHg]’s (1–25μM), and the number of live worms were counted 24 h later as described above. For DA neuron degeneration assays, animals were incubated on plates containing MeHg (0, 0.5, 1.0, and 2.0μM) at 20°C for 96 h, 50–60 animals at each concentration were immobilized on 2% agarose pads with 2% sodium azide, and were scored for DA neurodegeneration under fluorescent microscope (Leica MZ 16FA, Switzerland). Worms were scored positive for DA neuron degeneration when GFP in any part of the four cephalic dendrites (CEP; which run from nerve ring to tip of the nose) was absent (Nass et al., 2002). For mRNA and protein analysis, worms that had been growing on RNAi bacteria for 48 h were transferred to RNAi plates containing 25μM MeHg for 4 h. Worms were collected, and mRNA or protein was isolated for use in quantitative Polymerase Chain Reaction (qPCR) or Western blotting as described above.
Immunohistochemistry.
Primary C. elegans cultures were prepared as previously described with slight modifications (Bianchi and Driscoll, 2006; Settivari et al., 2009). Briefly, RNAi-sensitive (RJ928) L1 stage worms expressing GFP in the DA neurons were grown on RNAi plates containing control (HT115) or skn-1 (RNAi) bacteria until the gravid adult stage. The gravid adults were then lysed with the synchronization solution, and the egg pellet was washed using egg buffer (118mM NaCl, 48mM KCl, 2mM CaCl2, 2mM MgCl2, 25mM HEPES) (Settivari et al., 2009). The eggs were then separated from the debris using a 60% sucrose solution, digested using 4 mg/ml chitinase (Sigma), and the embryonic cells were dissociated using a syringe. The embryonic cells were then resuspended in L-15 medium (containing 10% fetal bovine serum and 1% pen/strep) and grown on polylysine-coated cover slips at 20°C. Following growth for 72 h, cells were fixed in 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, and 1% normal donkey serum was used as blocking buffer. The cells were then incubated with either SKN-1 primary antibodies (1:1000) (sc-9244; Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight (14 h), followed by incubation with a Cy5-conjugated donkey anti-goat secondary antibodies (Millipore) (1:1000) at room temperature for 1 h. Images were captured using confocal microscopy (Zeiss LSM 510 microscope). To further confirm the specificity of SKN-1 immunoreactivity, the antibody and its complementary antigenic peptide (sc-9244p; Santa Cruz Biotechnology) were mixed in a ratio of 1:5 and incubated at 4°C for 14 h. Following incubation, primary cultures were stained with the SKN-1 antibody-complementary peptide mixture for 4°C for 14 h and analyzed as described above.
RESULTS
MeHg Confers Concentration-Dependent Animal Death
Vertebrate studies indicate that chronic exposure to MeHg confers whole-animal and cellular death. In order to determine whether chronic exposure to low concentrations of MeHg affects C. elegans viability, we added synchronized WT N2 L4 larvae to agar media plates containing between 0 and 125μM MeHg and determined the number of live animals 48 h later. We find that MeHg causes a concentration-dependent loss of viability, with an LC50 of approximately 95μM (Fig. 1). MeHg exposure also appears to significantly reduce the number of viable eggs on the plate once the animals reach adulthood (data not shown). These results indicate that C. elegans are sensitive to low concentrations of MeHg in a concentration-dependent manner and suggest that the toxicant may affect embryo viability and development.
FIG. 1.
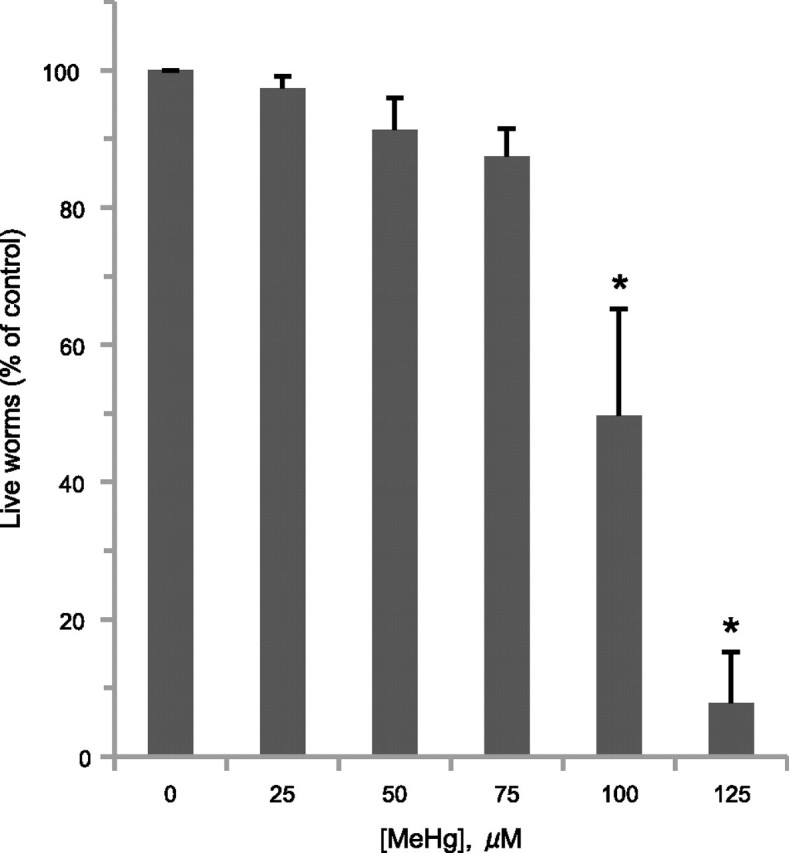
MeHg exposure confers concentration-dependent animal death. WT (N2) L4 nematodes (≥ 30 worms/condition, six replicates) were grown on 8P plates with various concentrations of MeHg, and the number of live animals was determined 48 h later. Data were analyzed using one-way ANOVA followed by Dunnett's multiple comparison test. The number of live animals was significantly different from control (0μM MeHg) at 100 and 125μM (*p < 0.01).
MeHg Exposure Decreases Brood Size
Mammalian studies indicate that MeHg exposures can cause a reduction in the number of offspring. Studies with rodents have shown a decrease in progeny relatively to nonexposed animals (Beyrouty et al., 2006; Hughes and Annau, 1976). To determine whether exposure to MeHg can cause a decrease in the number of progeny, we incubated L4 animals, the developmental stage immediately prior to self-fertilization, on media plates containing low concentrations of MeHg. Every 24 h, the adult animals were moved to a new plate until the adults ceased producing embryos, and the total number of progeny on each plate was counted. As can be seen in Figure 2A, exposure to low concentrations of MeHg dramatically reduced the brood size. Growth on media plates containing 2.5μM MeHg reduces the number of progeny by almost 20%, whereas growth on agar containing 10μM reduces the number of progeny by over 90%. These results indicate that exposure to MeHg decreases the number of viable progeny and, as in vertebrate studies, may affect maturation and cell division.
FIG. 2.

Chronic exposure to MeHg confers reproductive and developmental defects in Caenorhabditis elegans. MeHg exposure results in decreases in C. elegans brood size (A). L4 animals (≥ 15 worms/condition) were placed on 8P/NA22 plates and allowed to lay eggs, and every 24 h, the parent worm was transferred to a new plate until the animal terminated laying eggs (approximately 5 days). The total number of progeny from each plate was counted for each animal and combined to determine the animal's brood size. MeHg delays animal development (B). L1 nematodes (≥ 100 worms/condition) were placed on 8P/NA22 plates containing 10μM MeHg, and the time to reach adulthood was determined. Exposure to MeHg causes embryonic defects (D–F). L1 nematodes expressing the mCherry fluorophore behind the pie-1 promoter were grown to adulthood in the presence of water (C) or 10μM MeHg (D–F), and fluorescence (C, D, and F) or differential interference contrast (E) image was evaluated. Relative to control (C), MeHg causes abnormal cell division (D), and some cells contain several nuclei (E and F). Scale bar = 50 μm. Data were analyzed using one-way ANOVA followed by Dunnett's multiple comparison test. All individual treatments were statistically significant (*p < 0.01) compared with control group (0μM MeHg).
MeHg Confers Developmental Delays and Embryonic Defects
Mammalian and invertebrate studies indicate that exposure to MeHg can cause delays in animal development and confer embryonic defects (Carvalho et al., 2008; Curle et al., 1987; Weis, 2009). In order to determine if MeHg can affect normal development, we examined the rate of development of animals grown on a low concentration of MeHg from L1 to the L4 stage. L1 animals exposed to 10μM MeHg took approximately 30% longer to reach adulthood at 20°C relative to non-MeHg–exposed animals (56 vs. 78 h) (Fig. 2B). These results indicate that MeHg confer delays in morphogenesis and gonadogenesis and suggest that the toxicant may have a deleterious effect on embryogenesis and cellular division. To determine whether chronic MeHg exposure may affect early embryonic development, we examined embryos during the early stages of cell division in the adult hermaphrodite in vivo. Exposure of L1 animals to 10μM of MeHg until they reached adulthood results in the generation of embryos with significant developmental defects relative to control (Figs. 2C–F). Embryonic defects were seen in nearly all the animals evaluated; some embryos contain three nuclei within a single cell, suggesting defects in mitosis (Figs. 2D–F). Taken together, these results indicate that exposure of C. elegans to MeHg has deleterious effects on animal growth and development.
MeHg Exposure Increases ROS Levels
Although the origin of the cellular stress induced by MeHg is not well defined, the toxicant is believed to generate an increase in cellular ROS (InSug et al., 1997; Limke et al., 2004). To determine if MeHg may also increase ROS levels in C. elegans, we compared total ROS levels in control and MeHg-exposed animals. L4 animals were incubated on plates containing 25μM MeHg for 8 h, and ROS levels were determined using the ROS-dependent dye DCF as previously described (Settivari et al., 2009). We find that a brief exposure to MeHg confers over a twofold increase in cellular ROS relative to non–MeHg-exposed animals (Fig. 3A). These results suggest that oxidative stress may contribute to C. elegans cellular sensitivity to MeHg.
FIG. 3.

MeHg exposure increases ROS levels and GST mRNA expression. MeHg exposure increases the levels of ROS (A). Synchronized L4 animals were grown on 8P/NA22 plates containing 25μM MeHg for 8 h and then incubated with DCF-DA for 60 min. Change in fluorescence was normalized to protein concentration. Shown are mean values of ± SE of four individual replicates. Mean ROS levels between MeHg and control groups were analyzed using a paired t-test, and the difference was statistically significant (*p = 0.02). MeHg exposure induces the expression of GST mRNAs (B). Caenorhabditis elegans were exposed to 25μM for 2 h (black bars) or 8 h (gray bars), mRNA extracted, and reverse-transcribed to cDNA. Relative gene expression changes of the GSTs were quantitated using real-time PCR. The fold change in gene expression relative to GAPDH was calculated for each gene following the ΔΔCT method. Shown are mean values for ± SE of at least three individual replicates. The level of significance between the groups was calculated using paired t-test. MeHg induced (p < 0.03) gst-4, gst-12, and gst-21 gene expression at 2- and at 8-h time point, gene expression of gst-4, gst-5, gst-12, gst-21, and gst-38 were increased (indicated by “*,” p < 0.05). To determine gene expression changes between 2- and 8-h time point, log fold-change values were analyzed using one-way ANOVA followed by unpaired t-test. Transcript levels of gst-5, gst-12, and gst-38 were increased (indicated by #, p ≤ 0.05) in worms exposed to MeHg for 8 h compared with worms exposed for 2 h. Exposure to MeHg did not change GAPDH gene expression (p = 0.76) in worms at either time point.
MeHg Induces GST Expression
An increase in cellular ROS levels and oxidative stress can induce GST expression in an attempt to maintain normal cellular and metabolic processes. In order to determine if C. elegans GSTs are induced following exposure to MeHg, we utilized qPCR and evaluated mRNA levels of a number of GSTs, some of which have been associated with toxicant-induced gene expression changes, following sublethal exposures to MeHg. L4 animals exposed to 25μM MeHg for 2 h display a significant increase in a number of GSTs expression including gst-4, gst-5, gst-12, gst-21, and gst-38 (Fig. 3B). Animals exposed under these conditions and then incubated on non-MeHg media do not demonstrate reduced viability relative to nonexposed animals, indicating that these growth conditions are not lethal (data not shown). mRNA levels for gst-5 and gst-38 also increased dramatically, up to 10-fold (over 50-fold relative to nonexposed animals), when exposed for an additional 6 h (Fig. 3B). Taken together, these results indicate that C. elegans GSTs are induced following MeHg exposure.
MeHg-Associated Induction of gst-4 and gst-38 Is Dependent on skn-1
Caenorhabditis elegans hyperbaric stress microarray studies indicate that gst-4 and gst-38 mRNA inductions are largely dependent on the transcription factor skn-1 (Park et al., 2009). In order to determine whether MeHg-induced gst-4 and gst-38 expression is skn-1 dependent, mRNA was isolated from RNAi-sensitive nematodes incubated on media plates containing bacteria expressing skn-1 double-stranded RNA (dsRNA) following exposure to 25μM MeHg, and gst-4 and gst-38 expression levels were determined by RT-PCR. A brief 4-h incubation in the presence of MeHg results in an approximate 15-fold and 55-fold decrease in gst-4 and gst-38 mRNA levels relative to RNA levels from worms grown on control bacteria, respectively (Fig. 4A). These results indicate that MeHg-associated induction of gst-4 and gst-38 mRNAs is largely dependent on the expression of skn-1 and suggests that the corresponding proteins levels may also be skn-1 dependent. To determine if GST-38 protein expression is also dependent on skn-1, we generated antibodies to GST-38 and determined protein levels following exposure to 25μM MeHg for 4 h. Consistent with the induction of gst-38 mRNA expression, exposure to the toxicant results in an approximate 12-fold increase in GST-38 protein levels (Fig. 4B; data not shown). Furthermore, MeHg-associated induction of the GST-38 protein appears to be highly dependent on skn-1 as genetic knockdown of the transcription factor does not result in an increase in GST-38 following exposure to MeHg (Fig. 4B). Taken together, these results indicate that MeHg-associated induction of gst-4 and gst-38 is largely skn-1 dependent and suggests that skn-1 may inhibit cellular toxicity following exposure to the toxicant.
FIG. 4.
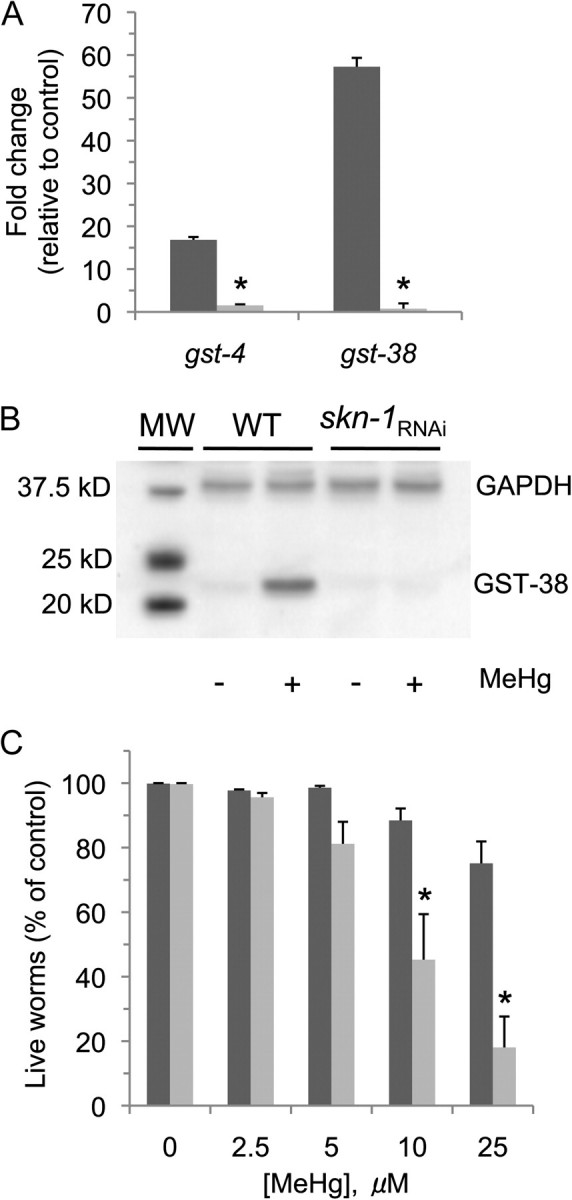
SKN-1 induces MeHg-associated gst-4 and gst-38 expression and decreases whole-animal toxicity. MeHg induces skn-1–dependent mRNA expression (A). RNAi-sensitive nematodes (NL2099) were grown on bacteria on NGM plates containing either an empty vector (black bars) or a vector expressing dsRNA to skn-1 (gray bars) and were exposed to 25μM MeHg for 4 h, mRNA extracted, and reverse transcribed to cDNA. Relative gene expression changes of the gst-4 and gst-38 were quantitated utilizing RT-PCR. Shown are mean values for ± SE of three individual replicates. The fold change in gene expression relative to GAPDH was calculated for each gene following the ΔΔCT method. Paired t-test was performed on log-transformed fold-change values to determine changes in gene expression between worms exposed to MeHg versus controls. Genetic knockdown of skn-1 decreased (p ≤ 0.01) gst-4 and gst-38 gene expression following MeHg exposure. MeHg-induced expression of GST-38 protein is dependent on skn-1 (B). Animals were exposed as in (A), and protein lysates were prepared and isolated as described in the “Materials and Methods” section; 50 μg of the cell lysate protein was resolved by SDS-PAGE, transferred to PVDF membranes, and probed with antibodies specific for GST-38. Western blots were performed in triplicate, and a representative image is shown. skn-1 mRNA knockdown increases whole-animal vulnerability to MeHg (C). RNAi-sensitive L4 nematodes (NL2099) (≥ 50 worms/condition) were grown on bacteria on media plates containing either an empty vector (black bars) or a vector expressing dsRNA to skn-1 (gray bars) for 48 h and then exposed to various concentrations of MeHg for 24 h, and the number of live animals was determined as described in the “Materials and Methods” section. Data were analyzed using one-way ANOVA followed by Dunnett's multiple comparison test. MeHg significantly induced animal death at 25μM in the HT115-fed animals (p < 0.05) and at 10 and 25μM in the skn-1 RNAi-fed animals (p < 0.01). At 10 and 25μM MeHg, animal death was significantly increased in skn-1 RNAi worms compared with HT115 worms as determined by Tukey's test following one-way ANOVA (*p < 0.01).
skn-1 Inhibits MeHg-Induced Animal Death
Nrf2/SKN-1 has been shown to play a significant role in inhibiting oxidative stress and cellular toxicity following MeHg exposure in vertebrates. Considering that at least two of the skn-1–dependent C. elegans GSTs are highly induced following exposure to MeHg, we asked whether a genetic knockdown of skn-1 may affect toxicant-induced animal vulnerability. To determine whether a reduction in skn-1 may increase sensitivity to MeHg, we grew animals on media plates on bacteria containing either an empty vector or a vector expressing skn-1 dsRNA to reduce skn-1 mRNA expression. A reduction in skn-1 mRNA levels was confirmed by qPCR and likely translates to lower skn-1 protein levels because growth on bacteria expressing the dsRNA also results in a skn-1 deletion phenotype of eggs containing dead embryos (Simmer et al., 2003). Consistent with prior skn-1 studies, genetic knockdown of the transcription factor at this animal's age does not reduce animal viability under nonoxidative stress conditions (An and Blackwell, 2003) (Fig. 4C). Conversely, skn-1 animals exposed to MeHg are up to 5× more sensitive to the toxicant relative to the controls (Fig. 4C). These results indicate that skn-1 expression inhibits MeHg-induced toxicity in C. elegans and further suggests that the signaling cascades involved in MeHg-induced cytotoxicity are conserved between nematodes and humans.
SKN-1 Is Expressed in DA Neurons
The mammalian SKN-1 orthologue NRF2 is expressed in DA neurons, and a decrease in gene expression in vitro and in vivo renders the cells vulnerable to DA neurotoxins (Jakel et al., 2007; Siebert et al., 2009). Detailed analysis of endogenous SKN-1 expression and localization has not been performed in the whole worm, but transgenic animals overexpressing the three isoforms of SKN-1 fused to the green fluorescent protein (SKN-1::GFP) display protein expression in the intestines and the chemosensory ASI neurons (Bishop and Guarente, 2007; Tullet et al., 2008). However, translational reporter fusions may not give a complete representation of endogenous protein expression levels and cellular localization (Boulin et al., 2006). To determine whether the C. elegans DA neurons express SKN-1, we generated C. elegans primary cultures from RJ928 animals that have robust expression of GFP in DA neurons both in vivo and in vitro (Carvelli et al., 2004; Nass et al., 2002). We used affinity-purified anti-SKN-1 to evaluate cellular SKN-1 expression levels. SKN-1 immunoreactivity is observed in all DA neurons (Figs. 5A–D). No specific staining was observed in primary DA neurons or other cells from animals in which skn-1 mRNA levels were reduced using RNAi (Figs. 5E–H; data not shown). Furthermore, SKN-1–associated DA neuron immunoreactivity was not observed following coincubation of the cultures with the SKN-1 antibody and the complementary antigenic peptide, providing further evidence that the antibody is binding to the SKN-1 protein in the DA neurons (Supplementary fig. 2). Taken together, these results indicate that SKN-1 is expressed in DA neurons and that a reduction in the transcription factor mRNA levels by RNAi results in a significant loss of SKN-1–immunoreactive protein expression in the DAergic cells.
FIG. 5.

SKN-1 is expressed in DA neurons. Primary Caenorhabditis elegans cultures expressing GFP in the DA neurons were generated with WT (A–D) or skn-1 knockdown animals (E–H), and fixation was performed as described under the “Materials and Methods” section. Primary cultures were incubated with SKN-1 primary antibody followed by incubation with Texas Red–conjugated donkey anti-goat secondary antibodies. DIC images of (A) and (E) of WT and skn-1RNAi cultures, respectively. DA neurons from WT and skn-1 knockdown animals expressing GFP driven by the dat-1 promotor (B) and (F), respectively. SKN-1 expressed in DA neurons in WT animals (C) but not in skn-1 knockdown animals (G). (D) Overlay of (B–C) and (H) overlay of (F–G). Images were observed under a Zeiss confocal microscope (Zeiss LSM 510). Scale bar represents 45 μm.
SKN-1 Inhibits MeHg-Induced DA Neurodegeneration
The activation of Nrf2-dependent stress response pathways have been shown to inhibit MeHg- and PD-associated toxicant pathologies (Jakel et al., 2007; Taylor et al., 2008). Considering SKN-1 inhibits MeHg-induced animal death and is expressed in DA neurons, we asked whether the transcription factor may also mitigate toxicant-associated DA neuron vulnerability. Nematodes were grown on RNAi bacteria to reduce expression of skn-1; reduction in mRNA levels of skn-1 was confirmed by quantitative PCR (data not shown). Animals were then exposed to various concentrations of toxicant ranging from 0 to 2μM MeHg for 96 h, and DA neuron integrity was assessed in vivo as previously described (Settivari et al., 2009). We found that low chronic exposures to MeHg caused a significant loss of DA neurons in animals (up to 30% of the animals exposed to 1μM MeHg) with a reduction of skn-1 mRNA within 96 h at all concentrations tested (Figs. 6A and 6B). The DA neuron degeneration appear similar to our prior PD-associated toxicant studies in which we characterized the cellular pathology by loss of DA neuron GFP expression in the CEP dendrites (since our prior studies have correlated a loss of GFP in the dendrites with DA neuronal death), DA levels, and loss of DA neuron integrity by electron microscopy (Nass et al., 2002; Settivari et al., 2009). Animals exposed at these concentrations for up to 120 h did not display a decrease in animal viability, and approximately a 10% decrease in viability following exposure for 144 h (consistent with the decrease in longevity of the skn-1 mutants [Oliveira et al., 2009]), suggesting that there is not large-scale MeHg-associated cellular death following the 96 h exposure (Supplementary fig. 3). Taken together, these results indicate that the DA neurons are vulnerable to MeHg, and the expression of SKN-1 inhibits the toxicant-induced DA neuron degeneration.
FIG. 6.

SKN-1 inhibits MeHg-induced DA neuron degeneration. Synchronized L1 nematodes (RJ928) (≥ 50 worms/condition) were grown on 1.0μM MeHg media plates containing either control (empty vector) or skn-1 RNAi bacteria (vector expressing dsRNA for skn-1) for 96 h, and worms were scored for DA neuron degeneration as described in the “Materials and Methods” section. (A) GFP-expressing DA neurons (CEPs) within the head of a control worm exposed to MeHg. (B) DIC image of the corresponding control animal in (A). (C) GFP-expressing DA neurons within the head of an skn-1 knockdown animal exposed to MeHg; image chosen emphasizes significant loss of CEP cell bodies and dendrites. (D) DIC image of the corresponding control animal (C). (E) Quantification of DA neuron integrity in control animals or skn-1 knockdown animals exposed to various concentrations MeHg. Shown are mean values of ± SE of three individual replicates. Data were analyzed using two-way ANOVA. All comparisons between control and skn-1 knockdown animals were significant (p ≤ 0.01) at 0.5, 1, and 2μM MeHg concentrations. Within the skn-1 RNAi group, all the MeHg concentration exposure groups were significantly different (p < 0.03) from the 0μM MeHg group.
DISCUSSION
MeHg is a potent xenobiotic that is associated with human developmental abnormalities, neurological dysfunction, and embryonic defects (Budtz-Jorgensen et al., 2007; Grandjean et al., 1997; Murata et al., 2004; Rodier, 1995). Vertebrate studies indicate that low chronic exposure to MeHg confers animal and cellular developmental delays and embryonic defects, impairs cellular proliferation and migration, and toxic exposures decrease the number of offspring and animal death (Glover et al., 2009; Limke et al., 2004). Recent Drosophila studies involving relatively low MeHg exposures also show toxicant-induced inhibition of embryonic development and delays, suggesting conservation of the molecular targets across phyla (Rand et al., 2009). Our studies recapitulate key hallmarks of MeHg-induced mammalian toxicity as low and chronic exposure of the toxicant cause embryonic defects, postembryonic developmental delays, a reduction in brood size, and concentration-dependent animal death (Figs. 1 and 2).
In vertebrate systems, glutathione (GSH) and the phase II detoxification enzymes play significant roles in inhibiting MeHg-induced cellular pathology. MeHg exposure has been shown to induce gene expression of the rate-limiting enzyme for GSH synthesis, γ-glutamylcysteine synthetase, which results in an increase in cellular GSH levels and cytoprotection (Thompson et al., 2000). The toxicant also induces the expression of GSTs that conjugate the compound to GSH, allowing for cellular excretion through phase III detoxification proteins. Caenorhabditis elegans contains over 40 putative GSTs, and previous studies demonstrate that there are significant gene expression changes for several of the GSTs following exposure to neurotoxicant and oxidative stress (Hasegawa et al., 2008; Liao and Freedman, 1998; Oliveira et al., 2009; Park et al., 2009). Consistent with vertebrate studies, exposure of C. elegans to MeHg results in a significant and robust increase in mRNA levels of a number of GSTs, with the highest induction in gst-4, gst-5, gst-12, and gst-38 (Fig. 3B). Among the GSTs examined, gst-38, a GST that is expressed in the intestines and nervous system, shows the largest change in gene expression relative to non–toxicant-exposed animals, with a 50-fold increase in mRNA levels, as well as a 12-fold increase in GST-38 protein levels (Fig. 3B) (Hasegawa et al., 2008). GST-38 expression levels have previously been shown to increase fivefold or less following exposure to cadmium, acrylamide, arsenite, or hyperoxia, suggesting that gst-38 expression is likely a sensitive indicator of tissue-associated oxidative stress (Hasegawa et al., 2008; Liao and Freedman, 1998; Oliveira et al., 2009; Park et al., 2009). Furthermore, although care must be taken in comparing mRNA expression changes across analysis platforms (microarray vs. RT-PCR), as well as growth conditions (i.e., different medias and bacteria), age and length of toxicant exposures, the greater than 10-fold induction of gst-38 relative to those found in previously reported toxicant exposure studies suggests that the C. elegans neurons and/or intestinal cells may be particularly sensitive to MeHg.
Numerous phase II detoxification enzymes, including the GSTs in mammals and C. elegans, are regulated by the transcription factor Nrf2/SKN-1 (Oliveira et al., 2009; Yu et al., 2010). In vertebrates and nematodes, oxidative stress can trigger Nrf2/SKN-1 translocation from the cytoplasm to the nucleus that results in the induction of cytoprotective proteins (Sykiotis and Bohmann, 2010). In vertebrates, Nrf2-deficient animals are also sensitive to MeHg and oxidative stress, and exposure to MeHg confers an increase in Nrf2 expression (Toyama et al., 2007). The most highly induced GSTs in our C. elegans studies (gst-4, gst-5, gst-12, and gst-38) have at least three canonical SKN-1–binding sites within 1 kb 5′ of the start ATG, and gene expression for each of these GSTs has previously been shown to be at least partially dependent on skn-1 (An and Blackwell, 2003; Park et al., 2009). Consistent with MeHg-induced skn-1–dependent gene expression, knockdown of skn-1 results in decreases in GST mRNA and protein levels (Figs. 4A and 4B). Furthermore, a decrease in skn-1 mRNA levels in the nematode results in a dramatic increase in MeHg sensitivity (Fig. 4C). Taken together, these results indicate that SKN-1 and its downstream targets play an essential role in mediating MeHg-induced cellular toxicity.
Developing and adult DA neurons are particularly sensitive to MeHg, and recent studies suggest that the toxicant may be a contributing factor in the development of PD (Dreiem et al., 2009; Newland et al., 2008; Petersen et al., 2008). Here we show that SKN-1 is expressed in C. elegans DA neurons, and genetic knockdown of skn-1 renders the DA neurons highly vulnerable to MeHg in vivo, as 1μM of the toxicant causes DA neuron degeneration in approximately 30% of the animals examined (Figs. 5 and 6). These results indicate that SKN-1 plays a vital role in limiting MeHg-induced DA neuron toxicity. It is interesting to speculate that in individuals who have reduced levels of Nrf2, MeHg exposure could be an environmental risk factor for the development of PD. Indeed, Nrf2 haplotypes have been correlated with the propensity to develop PD (von Otter et al., 2010). Furthermore, DNA polymorphisms in Nrf2 that result in reduced cellular levels have been associated with a number of human diseases that could be affected by environmental toxicant exposures, consistent with Nrf2 expression levels potentially mediating DA neuron vulnerability and the propensity to develop PD (Sykiotis and Bohmann, 2010).
SKN-1 likely contributes to limiting MeHg-induced DA neuron degeneration through the upregulation of phase II enzymes, although a reduction in SKN-1 may also adversely affect the regulation of DA synthesis that could contribute to DA neuron vulnerability. For example, Blackwell and colleagues have recently shown through microarray expression profiles that a reduction in skn-1 mRNA levels reduces GTP cyclohydrolase I (GTPCH) and 6-pyruvoyl tetrahydrobiopterin synthase (PTPS) gene expression (Oliveira et al., 2009). Both proteins are involved in the synthesis of tetrahydrobiopterin (BH4) that is required for the generation of tyrosine hydroxylase, the rate-limiting step in DA synthesis, and reduced GTPCH and PTPS expression could theoretically increase toxic metabolite levels that contribute to DA neuron pathogenesis (Blau et al., 2001). Consistent with a reduction in GTPCH or PTPS gene expression and a concomitant increase in DA neuron pathogenesis, a reduction in gene expression of both proteins is observed prior to PD-associated behavioral changes and neurodegeneration in a Drosophila PD model (Scherzer et al., 2003). Furthermore, autosomal dominant human mutations in GTPCH have been correlated with the development of parkinsonism in children and adults, and cerebrospinal fluid from PD patients have been shown to have reduced BH4 levels relative to control subjects (Blau et al., 2001; Lovenberg et al., 1979; Nygaard et al., 1992). Taken together, these studies suggest that the SKN-1–dependent MeHg-induced DA neuron vulnerability may also be because of dysregulation of enzymes involved in DA synthesis or metabolism.
The concentrations of MeHg that C. elegans were exposed in this study are environmentally relevant and similar to those incorporated in vertebrate studies (Wang et al., 2009; Yu et al., 2010). Considering low and chronic exposures of 10μM MeHg reduces whole-animal viability, brood size, and normal embryonic development within 2–3 days, our studies suggest that C. elegans is highly sensitive to the toxicant. This is in sharp contrast to a number of other toxicity studies in C. elegans (acute and chronic) that often require a 10- to a 100-fold or greater toxicant concentration exposure relative to vertebrates to facilitate toxicant transport through the worm's orifices or across the moderately impermeable cuticle; toxicant concentrations within the cytoplasm though are often similar to vertebrates (Nass and Hamza, 2007; Rand and Johnson, 1995). It is likely that MeHg's lipophilic properties facilitate its ease in crossing the cuticle and cellular membranes and its intracellular bioaccumulation (Lakowicz and Anderson, 1980; Nakada and Imura, 1982; Parran et al., 2001). A recent phenomenological study that describes effects of significantly higher concentrations of MeHg on C. elegans (100–1000μM) also reports a decrease in nematode viability and larvae growth (Helmcke et al., 2009); embryo or brood size defects were not apparent possibly because of the length of the exposure period or the severity of the insult that may reduce embryo viability. The rapid and robust induction of GST mRNA (within 2 h of MeHg exposure) in this study also further emphasizes the acute sensitivity of the nematode to the toxicant. The induction of these phase II enzymes likely increase animal survival until other Nrf2/SKN-1–regulated events (i.e., GSH generation, induction of multidrug resistance efflux proteins, etc.), or other stress-associated pathways can reverse the cellular redox imbalance to normal cellular homeostasis.
In summary, our findings describe a novel C. elegans model for MeHg toxicity that demonstrates that low and chronic exposure to MeHg confers embryonic defects, developmental delays, decreases in brood size, and concentration-dependent whole-animal death. We also show for the first time that the stress-associated transcription factor SKN-1 is expressed in DA neurons, specific SKN-1–dependent GSTs are induced following exposure to MeHg, and a reduction in SKN-1 gene expression increases MeHg-induced animal vulnerability and confers DA neuron degeneration. The development of this genetic model should facilitate the identification of genes and molecular pathways in vivo involved in MeHg-induced developmental defects and cellular dysfunction.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institute of Environmental Health (R011ES014459) (to R.N.); Vanderbilt University Toxicology Program Pilot Grant (to R.N.); Saastamoinen Foundation (to G.W.); a PhRMA Foundation Predoctoral Award (to N.V.).
Supplementary Material
Acknowledgments
We gratefully appreciate the technical assistance of Ken Grimes and Randy Hunter. We thank Lihsia Chen for her invaluable assistance with the embryonic development assays. We also thank Bill Atchison for valuable discussions. Some of the strains were also provided by the Caenorhabditis Genetics Center, which is supported by the National Institutes of Health Center for Research Resources.
References
- An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003;17:1882–1893. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Aschner JL. Mercury neurotoxicity: mechanisms of blood-brain barrier transport. Neurosci. Biobehav. Rev. 1990;14:169–176. doi: 10.1016/s0149-7634(05)80217-9. [DOI] [PubMed] [Google Scholar]
- Audhya A, Hyndman F, McLeod IX, Maddox AS, Yates JR, III, Desai A, Oegema K. A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J. Cell. Biol. 2005;171:267–279. doi: 10.1083/jcb.200506124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A, al-Rawi NY, Tikriti S, Dahahir HI, Clarkson TW, Smith JC, et al. Methylmercury poisoning in Iraq. Science. 1973;181:230–241. doi: 10.1126/science.181.4096.230. [DOI] [PubMed] [Google Scholar]
- Beyrouty P, Stamler CJ, Liu JN, Loua KM, Kubow S, Chan HM. Effects of prenatal methylmercury exposure on brain monoamine oxidase activity and neurobehaviour of rats. Neurotoxicol. Teratol. 2006;28:251–259. doi: 10.1016/j.ntt.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Driscoll M. Culture of embryonic C. elegans cells for electrophysiological and pharmacological analyses (September 30, 2006) WormBook. 2006 doi: 10.1895/wormbook.1.122.1. (The C. elegans Research Community, WormBook Ed.). doi/10.1895/wormbook.1.122.1. Available at: http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Guarente L. Genetic links between diet and lifespan: shared mechanisms from yeast to humans. Nat. Rev. Genet. 2007;8:835–844. doi: 10.1038/nrg2188. [DOI] [PubMed] [Google Scholar]
- Blau N, Bonafe L, Thony B. Tetrahydrobiopterin deficiencies without hyperphenylalaninemia: diagnosis and genetics of dopa-responsive dystonia and sepiapterin reductase deficiency. Mol. Genet. Metab. 2001;74:172–185. doi: 10.1006/mgme.2001.3213. [DOI] [PubMed] [Google Scholar]
- Boulin T, Etchberger J, Hobert O. Reporter gene fusions (April 5, 2006) WormBook. 2006 doi: 10.1895/wormbook.1.106.1. (The C. elegans Research Community, WormBook Ed.). doi/10.1895/wormbook.1.122.1. Available at: http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges CC, Zalups RK. Molecular and ionic mimicry and the transport of toxic metals. Toxicol. Appl. Pharmacol. 2005;204:274–308. doi: 10.1016/j.taap.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budtz-Jorgensen E, Grandjean P, Weihe P. Separation of risks and benefits of seafood intake. Environ. Health Perspect. 2007;115:323–327. doi: 10.1289/ehp.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho MC, Nazari EM, Farina M, Muller YM. Behavioral, morphological, and biochemical changes after in ovo exposure to methylmercury in chicks. Toxicol. Sci. 2008;106:180–185. doi: 10.1093/toxsci/kfn158. [DOI] [PubMed] [Google Scholar]
- Carvelli L, McDonald PW, Blakely RD, Defelice LJ. Dopamine transporters depolarize neurons by a channel mechanism. Proc. Natl. Acad. Sci. U.S.A. 2004;101:16046–16051. doi: 10.1073/pnas.0403299101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol. 2006;36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- Curle DC, Ray M, Persaud TV. In vivo evaluation of teratogenesis and cytogenetic changes following methylmercuric chloride treatment. Anat. Rec. 1987;219:286–295. doi: 10.1002/ar.1092190309. [DOI] [PubMed] [Google Scholar]
- Di Simplicio P, Gorelli M, Vignani R, Leonzio C. The differential modulation of the enzymes of glutathione metabolism. Indication of overlapping effects of toxicity and repair in mouse liver and kidney after dietary treatment with methylmercury and sodium selenite. Biol. Trace Elem. Res. 1993;36:167–181. doi: 10.1007/BF02783176. [DOI] [PubMed] [Google Scholar]
- Dreiem A, Shan M, Okoniewski RJ, Sanchez-Morrissey S, Seegal RF. Methylmercury inhibits dopaminergic function in rat pup synaptosomes in an age-dependent manner. Neurotoxicol. Teratol. 2009;31:312–317. doi: 10.1016/j.ntt.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Friberg L, Mottet NK. Accumulation of methylmercury and inorganic mercury in the brain. Biol. Trace Elem. Res. 1989;21:201–206. doi: 10.1007/BF02917253. [DOI] [PubMed] [Google Scholar]
- Glover CN, Zheng D, Jayashankar S, Sales GD, Hogstrand C, Lundebye AK. Methylmercury speciation influences brain gene expression and behavior in gestationally-exposed mice pups. Toxicol. Sci. 2009;110:389–400. doi: 10.1093/toxsci/kfp105. [DOI] [PubMed] [Google Scholar]
- Goering PL, Fisher BR, Noren BT, Papaconstantinou A, Rojko JL, Marler RJ. Mercury induces regional and cell-specific stress protein expression in rat kidney. Toxicol. Sci. 2000;53:447–457. doi: 10.1093/toxsci/53.2.447. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, Murata K, Sorensen N, Dahl R, Jorgensen PJ. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol. Teratol. 1997;19:417–428. doi: 10.1016/s0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Miwa S, Isomura K, Tsutsumiuchi K, Taniguchi H, Miwa J. Acrylamide-responsive genes in the nematode Caenorhabditis elegans. Toxicol. Sci. 2008;101:215–225. doi: 10.1093/toxsci/kfm276. [DOI] [PubMed] [Google Scholar]
- Helmcke KJ, Syversen T, Miller DM, III, Aschner M. Characterization of the effects of methylmercury on Caenorhabditis elegans. Toxicol. Appl. Pharmacol. 2009;240:265–272. doi: 10.1016/j.taap.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope I, editor. C. elegans: A Practical Approach. Oxford University Press: New York; 1999. [Google Scholar]
- Hughes JA, Annau Z. Postnatal behavioral effects in mice after prenatal exposure to methylmercury. Pharmacol. Biochem. Behav. 1976;4:385–391. doi: 10.1016/0091-3057(76)90052-6. [DOI] [PubMed] [Google Scholar]
- InSug O, Datar S, Koch CJ, Shapiro IM, Shenker BJ. Mercuric compounds inhibit human monocyte function by inducing apoptosis: evidence for formation of reactive oxygen species, development of mitochondrial membrane permeability transition and loss of reductive reserve. Toxicology. 1997;124:211–224. doi: 10.1016/s0300-483x(97)00153-4. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kampkotter A, Pielarski T, Rohrig R, Timpel C, Chovolou Y, Watjen W, Kahl R. The Ginkgo biloba extract EGb761 reduces stress sensitivity, ROS accumulation and expression of catalase and glutathione S-transferase 4 in Caenorhabditis elegans. Pharmacol. Res. 2007;55:139–147. doi: 10.1016/j.phrs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Kunimoto M, Suzuki T. Migration of granule neurons in cerebellar organotypic cultures is impaired by methylmercury. Neurosci. Lett. 1997;226:183–186. doi: 10.1016/s0304-3940(97)00273-5. [DOI] [PubMed] [Google Scholar]
- Lakowicz JR, Anderson CJ. Permeability of lipid bilayers to methylmercuric chloride: quantification by fluorescence quenching of a carbazole-labeled phospholipid. Chem. Biol. Interact. 1980;30:309–323. doi: 10.1016/0009-2797(80)90054-x. [DOI] [PubMed] [Google Scholar]
- Lee JM, Shih AY, Murphy TH, Johnson JA. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J. Biol. Chem. 2003;278:37948–37956. doi: 10.1074/jbc.M305204200. [DOI] [PubMed] [Google Scholar]
- Liao VH, Freedman JH. Cadmium-regulated genes from the nematode Caenorhabditis elegans. Identification and cloning of new cadmium-responsive genes by differential display. J. Biol. Chem. 1998;273:31962–31970. doi: 10.1074/jbc.273.48.31962. [DOI] [PubMed] [Google Scholar]
- Limke TL, Heidemann SR, Atchison WD. Disruption of intraneuronal divalent cation regulation by methylmercury: are specific targets involved in altered neuronal development and cytotoxicity in methylmercury poisoning? Neurotoxicology. 2004;25:741–760. doi: 10.1016/j.neuro.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Lovenberg W, Levine RA, Robinson DS, Ebert M, Williams AC, Calne DB. Hydroxylase cofactor activity in cerebrospinal fluid of normal subjects and patients with Parkinson's disease. Science. 1979;204:624–626. doi: 10.1126/science.432666. [DOI] [PubMed] [Google Scholar]
- Murata K, Weihe P, Budtz-Jorgensen E, Jorgensen PJ, Grandjean P. Delayed brainstem auditory evoked potential latencies in 14-year-old children exposed to methylmercury. J. Pediatr. 2004;144:177–183. doi: 10.1016/j.jpeds.2003.10.059. [DOI] [PubMed] [Google Scholar]
- Myers GJ, Davidson PW. Prenatal methylmercury exposure and children: neurologic, developmental, and behavioral research. Environ. Health Perspec.t. 1998;106(Suppl. 3):841–847. doi: 10.1289/ehp.98106841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada S, Imura N. Uptake of methylmercury and inorganic mercury by mouse glioma and mouse neuroblastoma cells. Neurotoxicology. 1982;3:249–258. [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM, III, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3264–3269. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass R, Hamza I. The nematode Caenorhabditis elegans as a model to explore toxicology in vivo: solid and axenic growth culture conditions and compound exposure parameters. Curr. Protoc. Toxicol. 2007:1.9.1–1.9.18. doi: 10.1002/0471140856.tx0109s31. [DOI] [PubMed] [Google Scholar]
- Nass R, Merchant KM, Ryan T. Caenohabditis elegans in Parkinson's disease drug discovery: addressing an unmet medical need. Mol. Interv. 2008;8:284–293. doi: 10.1124/mi.8.6.6. [DOI] [PubMed] [Google Scholar]
- Nass R, Przedborski S, editors. Parkinson's Disease: Molecular and Therapeutic Insights from Model Systems. 1st ed. Elsevier Academic Press; 2008. [Google Scholar]
- Newland MC, Paletz EM, Reed MN. Methylmercury and nutrition: adult effects of fetal exposure in experimental models. Neurotoxicology. 2008;29:783–801. doi: 10.1016/j.neuro.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novillo A, Won S-J, Li C, Callard IP. Changes in nuclear receptor and vitellogenin gene expression in response to steroids and heavy metal in Caenorhabditis elegans. Int. Comp. Biol. 2005;45:61–71. doi: 10.1093/icb/45.1.61. [DOI] [PubMed] [Google Scholar]
- Nygaard TG, Takahashi H, Heiman GA, Snow BJ, Fahn S, Calne DB. Long-term treatment response and fluorodopa positron emission tomographic scanning of parkinsonism in a family with dopa-responsive dystonia. Annu. Neurol. 1992;32:603–608. doi: 10.1002/ana.410320502. [DOI] [PubMed] [Google Scholar]
- Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell. 2009;8:524–541. doi: 10.1111/j.1474-9726.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Tedesco PM, Johnson TE. Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell. 2009;8:258–269. doi: 10.1111/j.1474-9726.2009.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parran DK, Mundy WR, Barone S., Jr Effects of methylmercury and mercuric chloride on differentiation and cell viability in PC12 cells. Toxicol. Sci. 2001;59:278–290. doi: 10.1093/toxsci/59.2.278. [DOI] [PubMed] [Google Scholar]
- Petersen MS, Halling J, Bech S, Wermuth L, Weihe P, Nielsen F, Jorgensen PJ, Budtz-Jorgensen E, Grandjean P. Impact of dietary exposure to food contaminants on the risk of Parkinson's disease. Neurotoxicology. 2008;29:584–590. doi: 10.1016/j.neuro.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Rand JB, Johnson CD. Genetic pharmacology: interactions between drugs and gene products in Caenorhabditis elegans. Methods Cell. Biol. 1995;48:187–204. doi: 10.1016/s0091-679x(08)61388-6. [DOI] [PubMed] [Google Scholar]
- Rand MD, Dao JC, Clason TA. Methylmercury disruption of embryonic neural development in Drosophila. Neurotoxicology. 2009;30:794–802. doi: 10.1016/j.neuro.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier PM. Developing brain as a target of toxicity. Environ. Health Perspect. 1995;103(Suppl. 6):73–76. doi: 10.1289/ehp.95103s673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samali A, Orrenius S. Heat shock proteins: regulators of stress response and apoptosis. Cell Stress Chaperones. 1998;3:228–236. doi: 10.1379/1466-1268(1998)003<0228:hspros>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer CR, Jensen RV, Gullans SR, Feany MB. Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson's disease. Hum. Mol. Genet. 2003;12:2457–2466. doi: 10.1093/hmg/ddg265. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Settivari R, Levora J, Nass R. The divalent metal transporter homologues SMF-1/2 mediate dopamine neuron sensitivity in Caenorhabditis elegans models of manganism and parkinson disease. J. Biol. Chem. 2009;284:35758–35768. doi: 10.1074/jbc.M109.051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert A, Desai V, Chandrasekaran K, Fiskum G, Jafri MS. Nrf2 activators provide neuroprotection against 6-hydroxydopamine toxicity in rat organotypic nigrostriatal cocultures. J. Neurosci. Res. 2009;87:1659–1669. doi: 10.1002/jnr.21975. [DOI] [PubMed] [Google Scholar]
- Simmer F, Moorman C, van der Linden AM, Kuijk E, van den Berghe PV, Kamath RS, Fraser AG, Ahringer J, Plasterk RH. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 2003;1:E12. doi: 10.1371/journal.pbio.0000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D. Stress-activated cap′n′collar transcription factors in aging and human disease. Sci. Signal. 2010;3:re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T. Pathology of Minamata disease. With special reference to its pathogenesis. Acta Pathol. Jpn. 1982;32(Suppl. 1):73–99. [PubMed] [Google Scholar]
- Taylor RC, Acquaah-Mensah G, Singhal M, Malhotra D, Biswal S. Network inference algorithms elucidate Nrf2 regulation of mouse lung oxidative stress. PLoS Comput. Biol. 2008;4:e1000166. doi: 10.1371/journal.pcbi.1000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, White CC, Krejsa CM, Eaton DL, Kavanagh TJ. Modulation of glutathione and glutamate-L-cysteine ligase by methylmercury during mouse development. Toxicol. Sci. 2000;57:141–146. doi: 10.1093/toxsci/57.1.141. [DOI] [PubMed] [Google Scholar]
- Timmons L, Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- Toyama T, Sumi D, Shinkai Y, Yasutake A, Taguchi K, Tong KI, Yamamoto M, Kumagai Y. Cytoprotective role of Nrf2/Keap1 system in methylmercury toxicity. Biochem. Biophys. Res. Commun. 2007;363:645–650. doi: 10.1016/j.bbrc.2007.09.017. [DOI] [PubMed] [Google Scholar]
- Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–1038. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Otter M, Landgren S, Nilsson S, Celojevic D, Bergstrom P, Hakansson A, Nissbrandt H, Drozdzik M, Bialecka M, Kurzawski M, et al. Association of Nrf2-encoding NFE2L2 haplotypes with Parkinson's disease. BMC Med. Genet. 2010;11:36. doi: 10.1186/1471-2350-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jiang H, Yin Z, Aschner M, Cai J. Methylmercury toxicity and Nrf2-dependent detoxification in astrocytes. Toxicol. Sci. 2009;107:135–143. doi: 10.1093/toxsci/kfn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis JS. Reproductive, developmental, and neurobehavioral effects of methylmercury in fishes. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009;27:212–225. doi: 10.1080/10590500903310088. [DOI] [PubMed] [Google Scholar]
- Wilce MCJ, Parker MW. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta. 1994;1205:1–18. doi: 10.1016/0167-4838(94)90086-8. [DOI] [PubMed] [Google Scholar]
- Yu X, Robinson JF, Sidhu JS, Hong S, Faustman EM. A system-based comparison of gene expression reveals alterations in oxidative stress, disruption of ubiquitin-proteasome system and altered cell cycle regulation after exposure to cadmium and methylmercury in mouse embryonic fibroblast. Toxicol. Sci. 2010;114:356–377. doi: 10.1093/toxsci/kfq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.