

Abstract
Alzheimer's disease is a major cause of dementia for which treatments remain unsatisfactory. Cyclin-dependent kinase 5 (CDK5) is a relevant kinase that has been hypothesized to contribute to the tau pathology. Several classes of chemical inhibitors for CDK5 have been developed, but they generally lack the specificity to distinguish among various ATP-dependent kinases. Therefore, the efficacy of these compounds when tested in animal models cannot definitively be attributed to an effect on CDK5. However, RNA interference (RNAi) targeting of CDK5 is specific and can be used to validate CDK5 as a possible treatment target. We delivered a CDK5 RNAi by lentiviral or adenoassociated viral vectors and analyzed the results in vitro and in vivo. Silencing of CDK5 reduces the phosphorylation of tau in primary neuronal cultures and in the brain of wild-type C57BL/6 mice. Furthermore, the knockdown of CDK5 strongly decreased the number of neurofibrillary tangles in the hippocampi of triple-transgenic mice (3×Tg-AD mice). Our data suggest that this downregulation may be attributable to the reduction of the CDK5 availability in the tissue, without affecting the CDK5 kinase activity. In summary, our findings validate CDK5 as a reasonable therapeutic target for ameliorating tau pathology.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder, considered a major cause of dementia and a public health problem around the world. The main hallmarks of AD are β-amyloid extracellular plaques and intracellular tau pathology that appear progressively. Tau pathology is characterized by excessive tau phosphorylation, reduced binding affinity of tau to microtubules, and the formation of aberrant tau aggregates known as neurofibrillary tangles (NFTs). Because the presence and extent of NFTs has been correlated with the level of dementia (Arriagada et al., 1992), drugs that control the phosphorylation state of tau deserve attention (Iqbal et al., 2005). Several strategies have been tried to inhibit tau phosphorylation but with limited success (Churcher, 2006; Skovronsky et al., 2006; Mazanetz and Fischer, 2007).
Nineteen phosphorylation sites have been identified in tau associated with paired helical filaments (PHFs) (Morishima-Kawashima et al., 1995; Augustinack et al., 2002; Yu et al., 2009). In vitro experiments have demonstrated that tau is a substrate for several kinases (Kobayashi et al., 1993; Paudel et al., 1993; Patrick et al., 1999). Cyclin-dependent kinase 5 (CDK5) has been considered a major tau kinase that contributes to tau pathology (Baumann et al., 1993; Paudel et al., 1993).
CDK5 is a serine threonine kinase with pleiotropic effects in the mammalian CNS (Dhavan and Tsai, 2001). Interaction of CDK5 with either p35 or p39, two proteins abundantly expressed in postmitotic neurons, is necessary for its activation (Lew et al., 1994; Tsai et al., 1994; Tang et al., 1995). Cleavage of p35 to a truncated form, p25, by calpain delocalizes and deregulates CDK5 (Kusakawa et al., 2000). The aberrant activity of CDK5 by p25 has been associated with NFTs (Cruz et al., 2003; Noble et al., 2003).
Several classes of potent chemical inhibitors for CDK5 have been reviewed (Fischer, 2001). Most of them are competitive inhibitors at the ATP binding site, resulting in a lack of specificity among kinases (Garrett and Fattaey, 1999; Gray et al., 1999; Leost et al., 2000; Meijer et al., 2000; Sielecki et al., 2000; Fischer, 2001; Knockaert et al., 2002; Mettey et al., 2003). Discovery of more specific CDK5 inhibitors could be a useful therapeutic (Shiradkar et al., 2007; Gong and Iqbal, 2008). However, before undertaking this onerous task, one would like better evidence that CDK5 is a reasonable treatment target. In the present study, we used RNA interference (RNAi) to target CDK5 with very high specificity and evaluated its effect on the phosphorylation of tau and on the tau pathology, using a triple-transgenic mouse model of Alzheimer's disease.
Materials and Methods
RNAi design.
The RNAi [short hairpin RNAmiR (shRNAmiR)] sequences for silencing of CDK5 (shRNAmiR-CDK5) and a scrambled RNA sequence as control (shRNAmir-SCR) were based on previously published sequences (Chang et al., 2006). These sequences were cloned into human miR-30-based stem loops by polymerase extension of overlapping DNA oligonucleotides. For cloning of lentiviral (LV) RNAi constructs, the following primers were used for polymerase extension: shCDK5miR, forward primer, 5′-CAGAAGGCTCGAGAAGGTATATGCTGTTGACAGTGAGCGACATGACCAAGCTGCCAGACTATAGTGAAGCCACAGATGTA-3′, and shCDK5miR, reverse primer, 5′-CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCACCATGACCAAGCTGCCAGACTATACATCTGTGGCTTCAC-3′, or shSCRmiR, forward primer, 5′-CAGAAGGCTCGAGAAGGTATATGCTGTTGACTAGCACACATCAGGAAGCGCTCGACAGTGATAGTGAAGCCACAGATGTA-3′, and shSCRmiR, reverse primer, 5′-CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCACCTAGCACACATCAGGAAGCGCTCGACAGTGATACATCTGTGGCTTCAC-3′. The extension products were digested with XhoI and EcoRI for directional cloning into the vector pCMV-GIN-ZEO.GFP (Open Biosystems). For cloning of RNAi vectors for adenoassociated virus (AAV) production, the following primers were used for polymerase extension: shCDK5miR, forward primer, 5′-AAAACTCGAGTGAGCGCTGACCAAGCTGCCAGACTATACTGTAAAGCCACAGATGGG-3′, and shCDK5miR, reverse primer, 5′-AAAAACTAGTAGGCGTTGACCAAGCTGCCAGACTATACCCATCTGTGGCTTTACAG-3′, or shSCRmiR, forward primer, 5′-AAAACTCGAGTGAGCGCACCATCGAACCGTCAGAGTTACTGTAAAGCCACAGATGGG-3′, and shSCRmiR, reverse primer, 5′-AAAAACTAGTAGGCGTACCATCGAACCGTCAGAGTTACCCATCTGTGGCTTTACAG-3′. These extension products were digested with XhoI and SpeI for directional cloning into a U6 expression plasmid cut with XhoI and XbaI (Boudreau et al., 2008).
Viral particle production.
To produce lentiviral particles, the plasmid pCMV-GIN-ZEO.GFP (Open Biosystems) (Silva et al., 2005; Stegmeier et al., 2005), which coexpressed green fluorescent protein (GFP) and the designed miRNA was cloned. The lentivirus vector was produced by cotransfection of 10 cm plates of highly confluent human embryonic kidney 293 cells (HEK-293T cells) with 30 μg of vector core plasmid pCMV-GIN-ZEO.GFP, 27 μg of packaging plasmid psPAX2 (Addgene; plasmid 12260), and 3 μg of VSV-G envelope plasmid pMD2G (Addgene; plasmid 12259) using calcium phosphate (Zufferey and Trono 2000). Supernatant of conditioned medium was collected at 48 h after transfection. The supernatant was clarified by filtering through a 0.45 μm filter. The viral particles were concentrated by ultracentrifugation at 30,000 rpm for 2 h at 4°C with a Beckman 50TI rotor on a 20% sucrose cushion. The viral pellet was resuspended in 200 μl of 1× PBS and titered by human immunodeficiency virus (HIV) p24 ELISA, Lentivirus Quantitation kit (Cell Biolabs) and transducing units (TU/ml) in HEK-293T (Naldini et al., 1996).
The protocol to produce AAV particles was for large-scale production of heterologous proteins by Sf9 insect cells culture for coinfecting recombinant baculovirus derived from the Autographa californica nuclear polyhedrosis virus (Urabe et al., 2002). The shRNAmir-CDK5 and shRNAmir-SCR expression cassettes—driven by the mouse U6 promoter—were cloned into pAAV.CMV.hrGFP, which contains AAV serotype 2/5 inverted terminal repeats, and a CMV-humanized Renilla GFP (hrGFP)-simian virus 40 poly(A) reporter cassette (Urabe et al., 2002; Boudreau et al., 2009). AAV titers were determined by using quantitative PCR and/or DNA slot blot analysis. The AAV were dialyzed before use.
Transfections and transductions.
Hippocampal and cortical primary cultures from C57BL/6 mice or Wistar rat embryos [embryonic day 17 (E17) to E18] were dissected, trypsinized, dissociated (Banker and Kimberly, 2002), and cultured at a density of 5 × 105 cells on poly-l-lysine (Sigma-Aldrich)-precoated 24-well or 6-well plates in Neurobasal medium (Invitrogen) containing B-27 supplement (Sigma-Aldrich), and penicillin–streptomycin antibiotic mixture (Invitrogen), at 37°C and 5% CO2 in a humidified atmosphere.
Transient transfection with lentiviral vector plasmid was done in HEK293T cells at day in vitro 3 (DIV3) and neuronal primary cultures at DIV5 using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). At DIV5, neuronal primary cultures plated in six-well plates were transduced for 3 d with 2 μl of LV with a titer of 109 lentiviral particles per milliliter or for 15 d with 2 μl of AAV with a titer of 1012 genomes per milliliter.
Western blotting.
Neuronal primary cultures, HEK-293T cell or hippocampal samples were lysed on 150 mm NaCl, 20 mm Tris, pH 7.4, 10% glycerol, 1 mm EDTA, 1% NP-40, 100 μm phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, and leupeptin (Sigma-Aldrich) and 100 μm orthovanadate (Cardona-Gómez et al., 2004). Proteins were loaded on 10% SDS-PAGE gel and transferred to nitrocellulose membranes (GE Healthcare) at 250 mA for 2 h using an electrophoretic transfer system. The membranes were incubated overnight at 4°C with rabbit anti-CDK5 (C-8) (1:1000; Santa Cruz Biotechnology), PHF-1 monoclonal antibody, which recognizes tau phospho-Ser-396/404 donated by P. Davies (Feinstein Institute for Medical Research, Manhasset, NY), rabbit anti-p35/p25 (C-19) (1:1000; Santa Cruz Biotechnology), total tau (tau-5) (1:1000; Invitrogen), and mouse anti-β-actin (1:2000; Sigma-Aldrich). IRDye 800CW goat anti-mouse or -rabbit (LI-COR; diluted 1:5000) and anti-mouse IgG or anti-rabbit IgG, peroxidase conjugated (Jackson ImmunoResearch; diluted 1:10,000), were used as secondary probes. The blots were developed using the Odyssey Infrared Imaging System or chemiluminesence method (ECL Western blotting system; GE Healthcare) followed by an exposure to a radiographic film (ECL Hyperfilm; GE Healthcare). The films were analyzed using Quantity One, version 4.3.0 (Bio-Rad).
Immunofluorescence.
Cell cultures were fixed 20 min in 4% paraformaldehyde in cytoskeleton buffer (CB) (Gallego-Gómez et al., 2003). Autofluorescence was reduced with 50 mm NH4Cl. Cells were permeabilized and blocked with 0.1% PBS/Triton X-100 and 1% bovine serum for 1 h. Cultures were incubated overnight at 4°C with primary antibodies, rabbit anti-CDK5 (C-8) (1:1000; Santa Cruz Biotechnology), and mouse anti-human-PHF-tau (AT-8; clone that recognizes tau phospho-Ser-202/Thr205, 1:500; Pierce Biotechnology). We used Alexa 488 and Alexa 594 secondary fluorescent antibodies (Invitrogen) during 1 h, and the nuclei were stained with Hoechst (1:5000; Invitrogen) for the last 15 min. The cells were observed by fluorescence microscopy (Olympus IX81). The images were analyzed individually to evaluate GFP-, CDK5-, and AT-8-immunoreactive expression, and the fluorescent intensity per area selected (soma or dendritic processes) was determined in 30 neurons per assay by software Image Scope-Pro (Media Cybernetics).
Mouse brains were cut in 50 μm coronal sections with a vibratome (Leica 1000) and treated with 50 mm NH4Cl for 10 min at room temperature. Slices were preincubated 1 h in 1% BSA with 0.3% Triton X-100 in 0.1 m PB. Primary antibodies were incubated overnight at 4°C. We used rabbit anti-CDK5 (C-8) (1:500; Santa Cruz Biotechnology) and PHF-1 antibody. Alexa 488 and Alexa 594 were used as secondary fluorescent probes. The slices were observed by fluorescence microscopy Olympus IX-81 or Olympus FV-1000 CLS, and analyzed as individual images for GFP, CDK5, and PHF-1 expression. Deconvolution and fluorescence intensity were done using Image Scope-Pro software (Media Cybernetics) and Cell software (Olympus).
Animal procedures.
Neuronal primary cultures were prepared from pregnant C57BL/6 mice or Wistar rats at E17–E18 housed in the vivarium at Sede de Investigación Universitaria–University of Antioquia (Medellin, Colombia). Animals were handled following the Colombian regulations (Law 84 of 1989 and Resolution 8430 of 1993) and the National Institutes of Health animal welfare care guidelines (Public Law 99-158, November 20, 1985, “Animals in Research”). A total of 20 6-month-old C57BL/6 wild-type mice and 42 18- to 23-month-old triple-transgenic Alzheimer mice (3×Tg-AD) was used (Oddo et al., 2003).
Animals were injected with 2 μl of AAV2-shRNAmirSCR (SCRmiR) or AAV2-shRNAmirCDK5 into the right hippocampus (bregma coordinates were −1.7 anteroposterior, −0.7 lateral, and −1.75 depth). For CDK5 kinase activity measurements, the mice were bilaterally injected into the hippocampus with the viral vectors. Injections were performed with a 10 μl Hamilton syringe at a rate of 0.2 μl/min and 5 min elapsed after infusion before withdrawal of the syringe. Animals were perfused transcardially with 4% paraformaldehyde in PBS and processed for the immunodetection. The brains were cryopreserved with 30% sucrose and stored at −20°C. For biochemical measures, the hippocampi were dissected and immediately frozen on dry ice and stored at −80°C before use.
In vitro CDK5 kinase assay. Three weeks after stereotactic injection of AAV2-CDK5miR or AAV2-SCRmiR into the hippocampus of 3×Tg-AD and C57BL/6 wild-type mice, the animals were killed and the hippocampi were dissected, placed on a 1.5 ml microfuge tube containing lysis buffer, and rapidly frozen in liquid nitrogen immersion. Brain tissue was kept at −70°C to preserve enzymatic activity until the assay was performed. Brain tissue was thawed on ice and homogenized, incubated for 15 min on ice, and centrifuged at 13,000 rpm/4°C. Supernatant was recovered in clean microfuge tubes and protein concentration was measured with the bicinchonic acid method (Thermo Fisher Scientific). CDK5 was immunoprecipitated from 250 μg of total protein using 1 μg of IgG rabbit polyclonal anti-CDK5 (C-8) antibody (Santa Cruz). Antibody and protein extract was incubated overnight at 4°C in a rotator; protein G-Sepharose (Sigma-Aldrich) was added and incubated for an additional 1 h at 23°C, and protein G-Sepharose beads were washed five times with immunoprecipitation (IP) buffer (Sigma-Aldrich), keeping the sample at 4°C. After the fifth wash, the protein G-Sepharose beads were resuspended in 200 μl of kinase assay buffer (20 mm Tris-HCl, pH 7.5, 100 μm sodium orthovanadate, 10 mm MgCl2, 50 mm NaCl, 1 mm DTT, and 1 mm NaF), and ATP was added to the resuspended beads at a 10-fold excess (0.5 mm). Histone from calf thymus type III-S (Sigma-Aldrich) was added as a substrate for CDK5 at a final concentration of 6 μm, and then the reaction was gently vortexed, and aliquoted in up to eight fractions 25 μl each, and incubated at 37°C, for times ranging from 15 to 60 min. To stop the reaction, 5 μl of SDS-PAGE loading buffer (250 mm Tris-HCl, 10% SDS, 30% glycerol, 0.5 m DTT, 0.02% bromophenol blue) was added, immediately followed by 5 min incubation at 95°C. The samples were separated electrophoretically at 120 V/2 h and transferred to a nitrocellulose membrane at 200 mA/1.5 h. Ponceau Red in 5% acetic acid was used to stain the transferred proteins. Histone was clearly visible, and its migration on the gel was identified around 21 kDa. Western blotting detection for CDK5 (C-8 antibody) and rabbit polyclonal anti-histone H1 phosphorylated (Millipore; 06-597) were used. Goat anti-rabbit IRDye 800WE (LI-COR) was used and detected using Odyssey Infrared Imaging System (LI-COR). Band intensities for histone were measured with NIH ImageJ software and normalized to IgG heavy chain intensity. To evaluate the kinetic properties of the CDK5 phosphorylation, the reaction was performed with varying amounts of substrate (histone concentrations ranging from 1 to 11.5 μm), and reaction was followed up to 35 min. Vo was calculated as the change in the normalized histone band intensity normalized to the intensity of the CDK5 band and then divided by the time observed in minutes to draw a Lineweaver–Burk projection.
Immunohistochemistry.
Mouse brain sections (50 μm) were treated for 20 min on 0.1 m PB/methanol (1:1), 1% hydrogen peroxide, and incubated 1 h in 0.1 m PB with 1% BSA, 0.3% Triton X-100. Then the slices were incubated with mouse anti-human-PHF-tau (AT-8; 1:500; Pierce Biotechnology), mouse anti-human-PHF-tau (AT-8) primary antibody during overnight at 4°C in 0.1 m PB with 0.3% BSA, 0.3% Triton X-100. Slices were incubated with biotinylated mouse secondary antibody and then incubated with ABC–HRP complex (Pierce Biotechnology) for 2 h. Detection was developed with diaminobenzidine (DAB). The tissue was dehydrated and covered with mounting solution and observed by an Eclipse E200 optical microscope (Nikon).
Quantification of PHF-1-immunoreactive neurons in CA1.
To analyze the effects of CDK5miR on the PHF-1-positive cells in the hippocampus of 3×Tg-AD mice, we used quantitative image analysis techniques using CLSM imaging of CA1. We used Hoechst labeling to automate the detection of cells in this area. First, we acquired three-dimensional images on an Olympus FV-1000 CLS microscope using a 40× objective while imaging two channels: one for the Hoechst signal (350 nm) and the other for immunodetected PHF-1 stain (secondary antibody conjugated to Alexa 594). To analyze the whole CA1 area, we acquired two consecutive three-dimensional images for each sample by manually panning the motorized stage; these three-dimensional images were later automatically stitched together. The ultraviolet channel was used to automatically detect all nuclei present in the paired image, and the red channel was used to classify all cells within the CA1 region expressing PHF-1.
The three-dimensional stack alignment was performed automatically using an automatic registration and seamless mosaicking technique (Fedorov et al., 2002, 2006). We chose the translational transformation model considering deviations in rotation and scale introduced by the motorized stage being negligible. First, candidate locations were extracted as uniformly distributed local maxima over the condition surface (Kenney et al., 2003). Then, for each of these locations, the point descriptors were extracted. Considering small perturbations among input images, our choice sets the descriptors that can be computed efficiently. We use small circular windows whose intensity content was normalized and orientation was aligned with the average gradient orientation (Zuliani et al., 2004). Preliminary matches of the tie points were established identifying the pairs with minimum distance in the descriptor space. Afterward, the inevitable outliers were pruned off using RANSAC-like algorithm producing final tie point matches. The transformation parameters were then estimated using matched tie points by the normalized direct linear transformation algorithm (Hartley and Zisserman, 2004).
To blend stack planes together, we used multiresolution splines (Burt and Adelson, 1983). Images were first decomposed into a multiresolution Laplacian pyramid, and the pyramids were then spliced level by level according to a weighted average over a transition zone. The blended image was obtained by reversely composing the spliced Laplacian pyramid. Therefore, the splice was matched to the scale of features and images were blended gradually without blurring finer image details. The averaged transition zone was identified by minimizing the error surface created by the absolute value of the difference of the gradients of overlapping areas (Ia and Ib). The approximate global solution for the minimization of the problem was given by a computationally efficient graph-cut algorithm (Boykov and Kolmogorov, 2001).
In all fused images, cell nuclei were automatically detected in the ultraviolet channel by a three-dimensional nuclei detection algorithm (Obara et al., 2008). We focused on the pyramidal cells in CA1 and removed nuclei lying outside of this region by manually masking the region of interest, using open-source Digital Notebook software (Kvilekval et al., 2010). The detection algorithm was divided into two steps: (1) candidate location detection and (2) candidate location pruning. The nuclei centroid candidate locations were detected by local maxima of an image convolved with the three-dimensional LoG kernel. The LoG kernel size is controlled by a user-defined average nuclear size parameter. The local maxima detection process continues by detecting locations of higher magnitude until the low-intensity bound, defined by the user, was reached. The candidate locations were then filtered to prune spurious locations because of circle-ellipsoid mismatch (we model nuclei as spheres), image noise, and other issues. During this process, all visible nuclei in the image were detected as indicated by a yellow outline in Figure 6, a and b.
Figure 6.
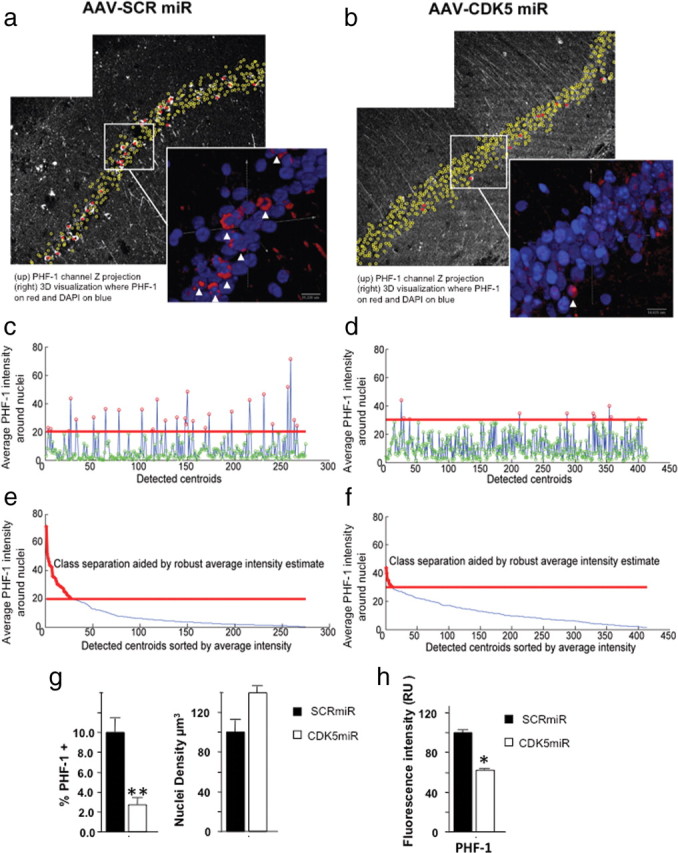
Silencing of CDK5 reduced the number of neurofibrillary tangles in the hippocampus of triple-transgenic Alzheimer mice. In this figure, the left-side images, a, c, and e, correspond to AAV-SCRmiR, and the right-side images, b, d, and f, correspond to AAV-CDK5miR injection. Representative PHF-1 channels of hippocampal CA1 region of 23-month-old 3×Tg-AD mice, imaged 3 weeks after AAV-SCRmiR (a) or AAV-CDK5miR (b) injection. The inset panels show magnified three-dimensional visualizations of CA1 cell nuclei, generated using bioView3D software (Kvilekval et al., 2010), in which PHF-1 channel is shown in red and Hoechst is in blue. The arrows point toward cells automatically classified as high PHF-1. The plots in c and d show average PHF-1 intensities within detected cells in which point color represents the classification result; red indicates high PHF-1, and green indicates low PHF-1. The red line separates the classes. The plots in e and f show the same data sorted according to the average intensity, in which the plot portion colored in red is classified as high PHF-1. Note that the CDK5miR image set was acquired at 1.5 times higher PMT voltage and offset than the SCRmiR. The bar graph in g shows a significant difference in the percentage of CA1 cells classified as high PHF-1 for both conditions (left). The density of the detected nuclei (right) shows a tendency to increase in old mice treated with CDK5miR. h, Intensity fluorescence quantification of PHF-1 also decreased after CDK5miR treatment. Data are presented as mean ± SEM. n = 8 to controls and n = 10 to treated. *p < 0.05; **p < 0.001.
The detected nuclei were classified according to PHF-1 expression in a small volume around the centroid of each nucleus. Thus, each candidate location was described by computing the voxel intensity sum within the sphere constructed at that centroid location with a diameter 1.5 times larger than the nuclei diameter. Because of fluctuations in the intensity of PHF-1 penetration, expression, and acquisition parameters, we estimate the average background intensity of the PHF-1 signal by using robust median absolute deviation estimator (Wang and Suter, 2004). This value is not affected by outliers to the same extent as the average estimation. Finally, the nuclei were classified using a modified K-means approach (Bishop, 1995), in which one of the classes was bound by the robust median estimator. This approach avoids forcefully splitting nuclei into two classes if they lie below or above the PHF-1 background intensity. Cells classified as high PHF-1 after this analysis were indicated by a red outline in Figure 5, a and b. The small insets in these figures zoom into an area of the CA1 nuclei, to emphasize the accuracy of the automated detection. An arrowhead was pointed to cells that belong to the high PHF-1 class, and we could visually confirm that the pointed cells express a high amount of PHF-1 immunoreactivity, noticed in the red pseudocolor of the image.
Figure 5.
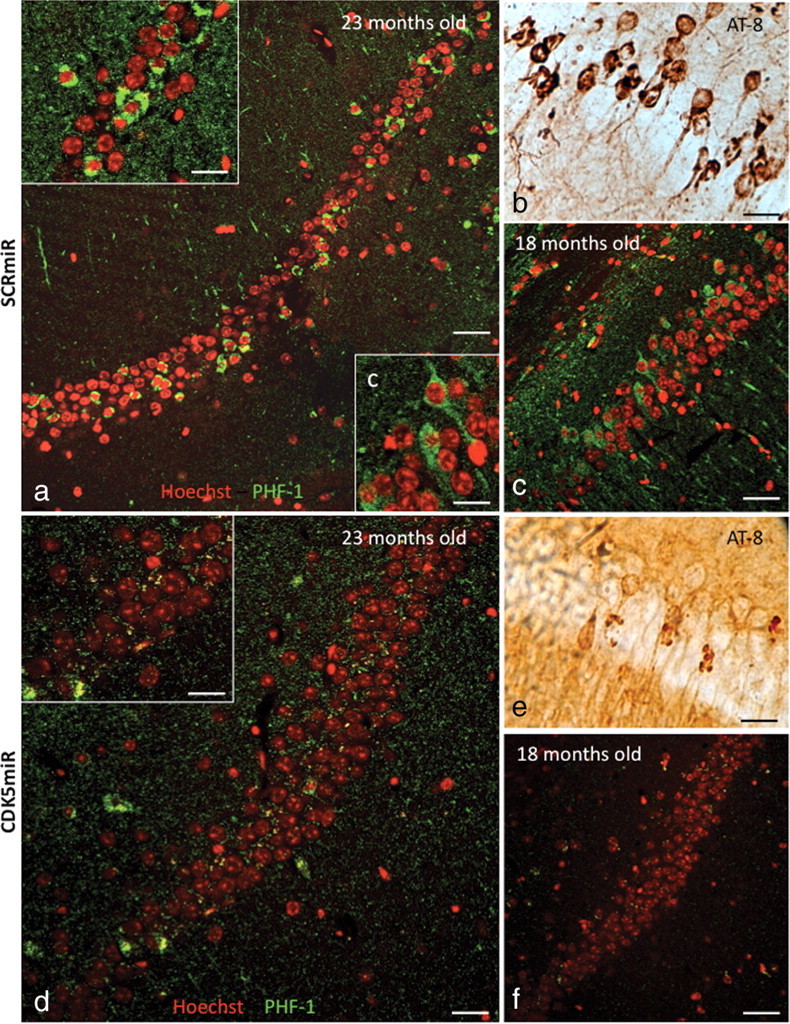
CDK5miR decreases phosphorylated tau and neurofibrillary tangles in 3×Tg-AD mice. a, b, Phospho-tau (PHF-1) immunofluorescence showing NFT-positive cells (a) and phospho-tau (AT-8)-immunoreactive cells (b) present in the CA1 area of hippocampus of 23-month-old 3×Tg-AD mice 3 weeks after injection with AAV-SCRmiR. c, PHF-1 immunofluorescence showing positive cells in CA1 in hippocampus of 18-month-old 3×Tg-AD mice, 3 weeks after hippocampal injection with AAV-SCRmR. d–f, PHF-1 immunofluorescence (d) and AT-8-immunoreactive cells in the CA1 area of 23-month-old (e) and 18-month-old (f) 3×Tg-AD mice 3 weeks after injection with CDK5miR. n = 6. Red pseudocolor: Nucleus staining with Hoechst; green pseudocolor: PHF-1 IF, Alexa 594. Magnification, 60×. Scale bar, 50 μm. Magnification: insets a, c, d, 100×. Scale bar, 20 μm.
Statistical analyses.
The n used for in vitro and in vivo experiments was 3–6. The parametric data were compared using multivariable two-way ANOVA followed by Tukey's post hoc test for comparison between several independent groups. A confidence level of p < 0.05 in a two-tailed test was adopted for statistical significance. The comparison between two groups was done using Student's t test. Data were expressed as mean ± SEM. Analyses were performed with SPSS (IBM) and GraphPad Prism, version 4.00, 2003 (GraphPad Software).
Results
CDK5 RNAi reduced phosphorylated tau in vitro
Following the model proposed by Chang et al. (2006), we designed a microRNA that contains the backbone of the naturally occurring miR30, and the complementary sequence to target the CDK5 mRNA. This construct, referred to as shRNAmirCDK5 (or briefly, CDK5miR), was cloned in pCMV-GIN-ZEO.GFP plasmid (Open Biosystems). To validate the ability of our construct to knock down CDK5 expression in vitro, we transfected it into HEK 293T cells with Lipofectamine 2000 reagent (Invitrogen) and detected transfected cells by GFP expression (Fig. 1a,c). Immunofluorescence (IF) of CDK5 was performed on transfected cultures, and we observed a decrease of the CDK5 IF in transfected cells compared with untransfected cells present in the same optical field (Fig. 1d). Cells transfected with pCMV-GIN-ZEO.GFP plasmid carrying a scrambled version of the CDK5 targeting sequence, expressed the GFP protein, but did not show a change in the intensity of CDK5 fluorescence compared with untransfected cells present in the optical field (Fig. 1a,b). These observations were also evaluated by Western blotting, and quantified by densitometry using Quantity One software, version 4.6.0 (Bio-Rad). We observed a significant decrease of CDK5 immunoreactivity of 76 ± 5.95% in HEK 293T cells (Fig. 1e). Lentiviral particles were produced in HEK293T cells by cotransfecting the pCMV-GIN-ZEO plasmids containing either CDK5miR or SCRmiR, together with pMD2G and psPAX2 using calcium phosphate. Viral supernatants were recovered 2 d later, quantified by HIV p24 ELISA, Lentivirus Quantification kit (Cell Biolabs), and titered by transducing units (TU/ml) in HEK-293T (Naldini et al., 1996). Primary cultures of hippocampal neurons were transduced at DIV5 with these lentiviral particles (LV) at a titer of 109 LV/ml and at a multiplicity of infection of 10. After 3 d, the cultures were fixed with CB-PFA (4%) and immunostained for CDK5 (Fig. 1f,h). In accord with observations made on HEK cells, LV-CDK5miR produced a reduction of CDK5 immunofluorescence in cells that expressed the reporter GFP, compared with untransduced (GFP−) cells (Fig. 1i). Cells transduced with LV carrying the scrambled construct showed no change in the CDK5 immunofluorescence (Fig. 1g). We estimated that the fluorescence intensity of CDK5 in CDK5miR-transduced neurons differed significantly from those transduced with the control SCRmiR, showing a decrease of 52.5 ± 3.38% in CDK5miR-treated cells (Fig. 1j).
Figure 1.

Knockdown of CDK5 in HEK-293T cell line and neuronal primary cultures by CDK5miR constructs. a, GFP expression of HEK-293T cells transfected with pCMV-GIN-ZEO-shRNAmirSCR.GFP (SCRmiR). b, CDK5 immunofluorescence in HEK-293T cells transfected with SCRmiR. c, GFP expression of HEK-293T cells transfected pCMV-GIN-ZEO-shRNAmirCDK5.GFP (CDK5miR). d, CDK5 immunofluorescence in HEK-293T cells transfected with CDK5miR; the arrows point to GFP+ cells in c. Decrease of the CDK5 immunoreactivity is observed in transfected cells. Green, GFP fluorescence; red, Alexa 594. Magnification, 60×. Scale bar, 20 μm. n = 6. e, CDK5 Western blotting in HEK-293T transfected cells with SCRmiR and CDK5miR. A representative plot is shown. βIII-Tubulin was used as loading control. Data are presented as mean ± SEM. n = 6. *p < 0.05. f, GFP expression of neuronal primary cultures transduced with LV- SCRmiR. g, Immunofluorescence of CDK5 in transduced neurons with LV-SCRmiR. h, GFP expression of neuronal primary cultures transduced with LV-CDK5miR. i, CDK5 immunofluorescence in neurons transduced with LV-CDK5miR. The arrows point to GFP+ transduced neurons seen in h. Decrease of CDK5 immunoreactivity in transduced neurons with LV-CDK5miR is observed. Green, GFP fluorescence; red, Alexa 594. Magnification, 60×. Scale bar, 20 μm. n = 6. **p < 0.001. j, Quantification of the fluorescence intensity of CDK5 immunoreactivity of neurons transduced with LV-SCRCDK5miR and LV-SCRmiR, using the software Image Scope-Pro (Media Cybernetics). RU, Relative units. n = 6. **p < 0.001.
CDK5miR and SCRmiR in pCMV-GIN-ZEO.GFP plasmid were also transfected in neuronal cultures at DIV5 for 3 d (Fig. 2a,c) and stained for phosphorylated tau using AT-8 antibody (Fig. 2b,d). As shown in Figure 2, d and inset and e, knocking down of CDK5 by transfection of CDK5miR reduced the AT-8 immunoreactivity in the neuritic processes, compared with that observed in transfected neurons with the SCRmiR (Fig. 2e, inset). Fluorescence intensity, quantified using the software Image Scope-Pro, showed significantly less immunofluorescence in neurons transfected with CDK5miR (33.1 ± 10.3%) compared with the neurons transfected with SCRmiR (Fig. 2f).
Figure 2.

CDK5miR decreased the levels of phosphorylated tau. a, GFP expression of hippocampal neurons transfected with pCMV-GIN-ZEO.shRNAmirSCR.GFP (SCRmiR). b, Immunofluorescence of phosphorylated tau with AT-8 antibody in neurons transfected with SCRmiR expressed in pCMV-GIN-ZEO-GFP plasmid. c, GFP expression of hippocampal neurons transfected with pCMV-GIN-ZEO.shRNAmirCDK5.GFP (CDK5miR). d, Immunofluorescence of phosphorylated tau with AT-8 antibody in neurons transfected with CDK5miR; a decrease in AT-8 immunoreactivity was detected. Green, GFP fluorescence; red, Alexa 594. Magnification, 60×. Scale bar, 20 μm. n = 4. e, Zoom insets from b and d. The arrows indicate neuritic processes of transfected cells. The phospho-tau (AT-8) immunoreactivity decrease in neurites transfected with CDK5miR (GFP-positive cells). f, Quantification of the fluorescence intensity of AT-8 immunoreactivity in neurons transfected with CDK5miR and SCRmiR, using the software Image Scope-Pro (Media Cybernetics). RU, Relative units. n = 4. *p < 0.05. g, CDK5, PHF-1, p35, and p25 were evaluated by Western blotting in neuronal primary cultures transduced with AAV-CDK5miR and AAV-SCRmiR. β-Actin was used as loading control. Representative blots are shown. h, Densitometric quantification from g; p25/p35 ratio was calculated. Data are presented as mean ± SEM. p25 and p35 densitometry values were previously normalized to β-actin. n = 6. *p < 0.05. i, CDK5, PHF-1 tau, pSer199 tau, and tau-5 Western blotting from hippocampal lysates of C57BL/6 mice injected with LV-CDK5miR and LV-SCRmiR. Representative blots are shown. j, Densitometry of Western blots in LV-CDK5miR- and LV-SCRmiR-treated mice was measured and normalized to actin. Data are plotted as the percentage of change between control (SCRmiR) and treated (CDK5miR) groups and presented as mean ± SEM. n = 4. *p < 0.05; **p < 0.001.
CDK5miR and SCRmiR synthetic microRNAs were cloned in the plasmid AAV-CMV.hrGFP, and adenoassociated viral particles serotype 2/5 were produced. Hippocampal neurons were transduced with these adenoassociated particles in DIV5 neurons at a titer of 1012 genomes per milliliter with a high efficiency of transduction. We obtained a monolayer of neurons that homogeneously expressed the viral transgenes, as seen by the GFP reporter (data not shown). After culturing the adenoassociated transduced neurons for 15 d with AAV-CDK5miR or AAV-SCRmiR, protein extracts were immunoblotted for phosphorylated tau with PHF-1 antibody. In this experiment, we confirmed a significant reduction of the PHF-1-immunoreactive band in the neuronal primary cultures expressing the CDK5miR with respect to the SCRmiR as control (Fig. 2g,h).
We then sought to determine the effect of CDK5 knockdown on the subunits p35 and p25 in the same protein extracts of AAV-transduced neurons. We confirmed a significant decrease of the CDK5 immunoreactivity; however, p35 and p25 were strikingly increased in the neuronal cultures expressing CDK5miR compared with SCRmiR cultures (Fig. 2g). CDK5 activity has been linked to the ratio between p25 and p35 (Patrick et al., 1999; Tseng et al., 2002). We determined that this ratio was not modified in the neuronal cultures treated with AAV-CDK5miR (Fig. 2h).
CDK5 RNAi reduced phosphorylated tau in vivo
We next assayed in vivo the silencing of CDK5 on the levels of phosphorylated tau. We stereotactically injected lentiviral particles carrying CDK5miR or SCRmiR in the hippocampus of 6-month-old C57BL/6 wild-type mice. After being killed at 1 or 3 weeks after injection, their hippocampi were dissected, homogenized, and protein extracts were analyzed by Western blotting to determine the levels of CDK5, phosphorylated tau with PHF-1 and pSer199 tau antibodies, and total tau with tau-5 antibody. In the mice injected with LV-CDK5miR for 1 week, CDK5 was significantly reduced (Fig. 2i,j). However, at 3 weeks after injection, there was greater variability in the CDK5 immunoreactivity, and the difference did not achieve significance. When tau immunoreactivity was analyzed at the same two points, the PHF-1 and pSer199 tau signals were significantly decreased at 3 weeks, but not at 1 week. The expression of tau-5 was not affected (Fig. 2i,j). The delay in a decrease of tau phosphorylation relative to CDK5 knockdown may be attributable to the long half-life of the CDK5-induced phosphorylation sites. Together, these results demonstrate that the CDK5miR mediated the knockdown of CDK5 in vivo was associated with a reduction in the phosphorylation of tau.
We investigated the effect of CDK5miR in vivo in the triple-transgenic Alzheimer's disease mouse model (3×Tg-AD mice). At 18 months and older, this murine model (Oddo et al., 2003) displays hippocampal neurofibrillary tangles similar to those in AD patients. We injected either AAV-CDK5miR or AAV-SCRmiR bilaterally into the hippocampi of 18-month-old 3×Tg-AD mice. These animals were killed 3 weeks after injection; one hemisphere was frozen and stored at −80°C, and the other was immersed in 4% PFA for 48 h, sectioned, and analyzed by fluorescence microscopy (IX 81; Olympus). In fixed cerebral tissues, we found that the 3×Tg-AD mice treated with AAV-CDK5miR had a notable decrease of the CDK5 immunofluorescence in dentate gyrus, CA1, CA2, and CA3 areas with AAV-CDK5miR transduction, compared with AAV-SCRmiR (Fig. 3).
Figure 3.
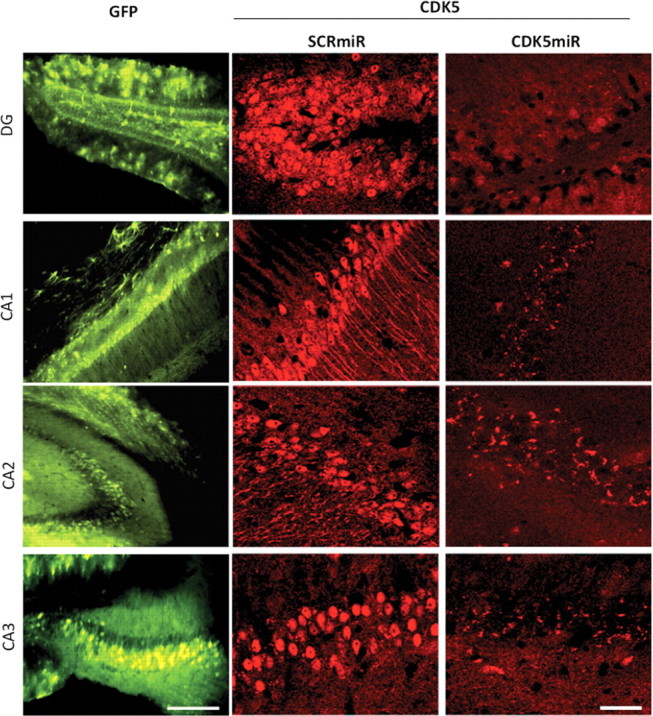
CDK5miR reduces CDK5 expression in the brain of triple-transgenic Alzheimer mice. 3×Tg-AD mice (18–23 months of age) were killed 3 weeks after injection with AAV. Silencing of CDK5 expression by CDK5miR in the dentate gyrus, CA1, CA2, and CA3 regions of 3×Tg-AD mice. GFP represent the transduced areas in the hippocampus. Magnification, 15×. Scale bar, 100 μm. CDK5 immunofluorescence: magnification, 40×. Scale bar, 50 μm. n = 6.
The frozen tissue was slowly thawed on ice, the hippocampus was dissected and homogenized, and protein extracts were analyzed by Western blotting. The AAV-CDK5miR-injected brains had a weak reduction in the CDK5-immunoreactive band. Despite this moderate effect on CDK5 levels in vivo, we confirmed that the phosphorylation of tau was strongly reduced by the CDK5miR treatment (i.e., the PHF-1 band detected in hippocampi of 3×Tg-AD mice injected with CDK5miR resulted in a fivefold reduction compared with the band in mice treated with SCR-miR) (Fig. 4a). In addition, p35 levels were significantly increased in mice treated with CDK5miR compared with the control SCRmiR. However, p25 did not show a comparable increase (Fig. 4a). To analyze the upregulation of p35, CDK5 immunoprecipitation was performed from protein extracts of transduced neuronal cultures. We found that the increased p35 was in a pool that is not associated with CDK5 in the neurons treated with AAV-CDK5miR compared with the AAV-SCRmiR-treated cells (Fig. 4b).
Figure 4.
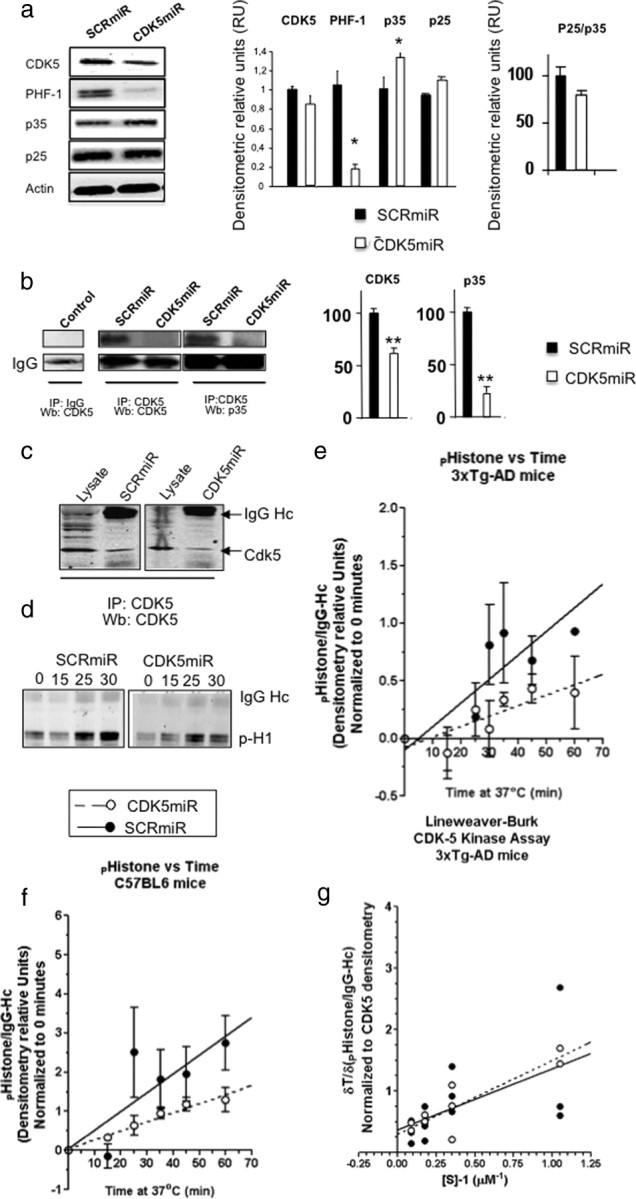
CDK5miR reduces CDK5 levels in the hippocampus of triple-transgenic Alzheimer mice without affecting its kinetic activity. a, CDK5, PHF-1, p35, and p25 Western blotting from hippocampus of 3×Tg-AD mice treated with SCRmiR and CDK5miR. β-Actin was loading control. Representative blots are shown. Densitometric quantification was done. RU, Relative units. n = 3. *p < 0.05. b, CDK5 IP, Western blot (Wb): CDK5, p35 of neuronal primary cultures. A band corresponding to the IgG heavy chain was detected and used as loading control. Negative for IgG immunoprecipitation was used as an internal control. Representative blots are shown. Densitometric quantification of CDK5 and p35 proteins was done. RU, Relative units. n = 3. **p < 0.01. c, CDK5 IP, Wb: CDK5 from hippocampus of 3×Tg-AD mice treated with SCRmiR and CDK5miR. d, Temporal course for CDK5 kinase activity was detected by phosphorylated histone antibody. A band corresponding to the IgG heavy chain was detected. e, f, Product formation using CDK5 IP from hippocampi of transgenic (e) and wild-type (f) mice was measured by densitometry and plotted against the reaction time. Linear regression was used and the fitting line was drawn (GraphPad Software). SCRmiR is shown as closed circles and a straight line, and CDK5miR is shown as open circles and a dotted line. Data are presented as mean ± SEM. n = 3. *p < 0.05. g, Phosphohistone formation was calculated as indicated above in several assays where substrate concentration was varied and normalized to CDK5 densitometry in the immunoprecipitation. Reaction rates were measured as the slope of the curve, and Lineweaver–Burk plot is graphed. Each value is presented in the dot plot and lineal regression was used to fit the model. CDK5miR is shown as closed circles and a straight line, and SCRmiR is shown as open circles and a dotted line.
CDK5 RNAi decreased CDK5 protein availability without affecting kinase activity
To determine whether CDK5 kinase activity was reduced by CDK5miR, we quantified kinase activity in the hippocampi of AAV-CDK5miR-treated 18- to 23-month-old 3×Tg-AD mice. CDK5 was immunoprecipitated and its steady-state kinetics was studied using excess histone as substrate. In agreement with total lysates (Fig. 4a), lower amounts of CDK5 were detected in the immunoprecipitated fractions from CDK5miR-injected mice compared with SCRmiR-injected mice (Fig. 4c). The product formation of phosphorylated histone was observed over a 60 min time course when incubated at 37°C in the presence of CDK5 from the CDK5miR- and SCRmiR-treated tissues, suggesting that the CDK5 remaining in the AAV-CDK5miR-treated fraction was still functionally active (Fig. 4d). We then performed quantitative analysis using data obtained from Western blots, and results were presented in Figure 4d. Data were fit to steady-state linear expression, and the reaction rates were calculated according to the slope. We found a significant 50% product formation rate reduction after CDK5miR treatment of transgenic mice compared with control (Fig. 4e). A significant 45% reduction was also found in replicate experiments done in wild-type mice (Fig. 4f). This result might be attributable to reduced CDK5 availability alone or in combination with partially impaired kinase activity.
To answer this question, we performed the same in vitro assay as in the transgenic mice study, but varied concentrations of substrate at the 30 min time point when product formation was still linear, so the reaction rate was directly correlated with enzyme concentration in the assay. Enzyme concentration was normalized by the density of CDK5 observed in the immunoprecipitated fraction, and results were plotted on a Lineweaver–Burk projection (Lineweaver and Burk, 1934). From three independent experiments, average KM and VMAX were estimated as follows: with CDK5miR treatment, KM = 4.02 μm and VMAX = 2.72 ± 1.76 min−1, and with SCRmiR treatment, KM = 2.71 μm and VMAX = 3.35 ± 4.5 min−1. No apparent changes in the kinetic properties of the enzyme were observed (Fig. 4g). These findings suggest that a reduced amount of CDK5 after CDK5miR treatment led to reduced enzyme availability, whereas CDK5 activity remained unchanged.
Silencing of CDK5 reduced the number of neurofibrillary tangles in the hippocampi of triple-transgenic Alzheimer mice
Triple-transgenic Alzheimer's disease mice (3×Tg-AD) had highly PHF-1-immunoreactive cells in the CA1 area of the hippocampus at 18–23 months of age. Immunohistochemistry performed on 50 μm coronal sections of the mouse brains showed PHF-1+ cells in mice 18 and 23 months old, using both fluorescence (PHF-1+ cells in green/nuclei in red by Hoechst) (Fig. 5a) and DAB (AT-8-positive cells) (Fig. 5b), as previously observed (Oddo et al., 2003). In the 23-month-old animals, PHF-1 immunoreactivity was stronger in the cellular cytoplasm surrounding the nucleus than in the younger 18-month-old mice (Fig. 5a, inset frame). Eighteen-month-old mice had stronger PHF-1 immunoreactivity in neuritic processes compared with the 23-month-old animals (Fig. 5c). When we injected AAV-CDK5miR intrahippocampally and killed the mice 3 weeks later, the extent of the tau pathology appeared to be reduced by immunodetection (Fig. 5d,e) compared with the SCRmiR injection (Fig. 5a,b). This finding was replicated in 18-month-old mice (Fig. 5f). Therefore, silencing of CDK5 is capable of reducing the extent of tau pathology in mice at an advanced age.
To quantify the observed reduction in tau immunoreactivity, we analyzed the sections with CLSM imaging of hippocampal CA1 in 3×Tg-AD mice. Our goal was to quantify the number of cells with high PHF-1 expression in the CA1 area, in mice treated by intrahippocampal injection of CDK5miR or SCRmiR. We acquired images from a total of 8 control mice injected with SCRmiR (6, 18 months; 2, 23 months of age) and a total of 10 experimental mice injected with CDK5miR (6 of the mice were 18 months, and 4 of them were 23 months of age) (Fig. 6a,b). All CA1 neurons in these images were automatically classified according to PHF-1 intensity in two classes: low and high PHF-1 immunoreactivity (Fig. 6c–f), and the percentage of high PHF-1 cells to total cells was calculated. (Fig. 6g). These automated quantitative analyses corroborated the findings observed in the Figure 5. An average of 10.05 ± 1.3% of cells in the CA1 region of 3×Tg-AD mice were classified as high PHF-1 in control mice, whereas the CDK5miR intrahippocampal treatment caused a significant reduction of cells classified as high PHF-1 in mice of the same age, reaching values as low as 2.7 ± 0.67% (Fig. 6g, left bars), which was supported by the single fluorescence quantification (Fig. 6h). Interestingly, the density of cells in this area also differed. We detected more neurons in the CA1 area of 18- to 23-month-old transgenic mice treated with CDK5miR, suggesting that the silencing of CDK5 might prevent the rapid loose of neurons in elder mice (Fig. 6g, right bars).
Discussion
The knockdown of CDK5 reduces the levels of phosphorylated tau, the number of neurofibrillary tangles, and blocks the rapid neuronal loss in the hippocampi of elder triple-transgenic mice. Based on our results, we propose CDK5 RNAi-mediated silencing as a novel gene therapy strategy against the tau pathology associated with Alzheimer's disease.
Several studies report CDK5 inhibition by pharmacological inhibitors such as olomoucine, roscovitine, purvalanol, flavopiridol, butyrolactone, indirubins, hymenialdisine, the paullones, and the aloisines (Garrett and Fattaey, 1999; Gray et al., 1999; Leost et al., 2000; Meijer et al., 2000; Sielecki et al., 2000; Knockaert et al., 2002; Mettey et al., 2003). Interestingly, hymenialdisine inhibits in vivo phosphorylation of tau at AD-specific sites in cultured insect cells (Meijer et al., 2000). Alsterpaullone, an inhibitor for CDK5 and GSK-3β, inhibits the in vivo phosphorylation of tau at AD-specific sites in slices from mouse striatum (Leost et al., 2000). A notable disadvantage to all these inhibitors is that these are competitive inhibitors at the ATP binding site, resulting in a lack of specificity among the many ATP-dependent kinases. However, the shRNAmiCDK5 (CDK5miR) offers specific inhibition of CDK5. This study shows that CDK5 is a valid therapeutic target. The previous chemical inhibitors lacked the specificity as compared with RNAi. Therefore this is a proof-of-concept study, which suggests that RNAi is a valid therapeutic approach but also suggests that developing more specific inhibitors of CDK5 is a worthy goal.
We observed that 3 weeks of CDK5miR treatment decreased the phosphorylated tau in adult brain of wild-type mice. However, the CDK5 protein levels displayed tendency to recover at 3 weeks of treatment compared with 1 week. This may indicate an endogenous feedback operating to maintain the CDK5 necessary for its normal function in the nervous tissue. In vivo data of CDK5 kinetic showed normal activity after silencing of CDK5. We calculated a CDK5 KM of 4 μm with histone as substrate. This value is comparable with the previously described KM of 4 μm in vivo tau (Ahn et al., 2004) and KM of 10 μm in vitro for histone (Liu et al., 2008). A 45–50% reduction in the histone phosphorylation rate with CDK5 obtained from cerebral tissue of CDK5miR-treated mice was associated with a comparable reduction of CDK5 in the same tissues. Based on these data, we suggest that diminution of the amount of CDK5 available in the tissue regulates or normalizes the activity of CDK5 in the transgenic mice, resulting in amelioration of the tau pathology.
We also observed increased p35 levels in the brain of 3×Tg-AD mice treated with CDK5miR. Mice that overexpress human p35, CDK5, and tau display increased CDK5 activity without an evident increase in tau phosphorylation (Van den Haute et al., 2001). These findings are consistent with our results and indicate that p35/CDK5 does not efficiently phosphorylate tau. However, recombinant p25/CDK5 phosphorylates tau with higher efficiency than p35/CDK5 in vitro (Hashiguchi et al., 2002). The 3×Tg-AD mice treated with CDK5miR in our study had increased p35 and reduced CDK5. These circumstances may reduce p25-mediated CDK5 phosphorylation of tau. As a consequence, a lower number of tangle-bearing neurons were seen in treated mice, despite their advanced age.
Recent studies show that p25/CDK5 deregulates HCDAC1 (histone deacetylase 1) activity inducing aberrant cell cycle and double-strand DNA breaks before neuronal death in CK-p25 mice (Kim et al., 2008). Other reports showed that conditional knock-out of CDK5 in the adult mouse brain improved performance in spatial learning tasks, enhanced hippocampal long-term potentiation, and decreased CDK5-induced NR2B (NMDA subunit) degradation by calpain (Hawasli et al., 2007). Moreover, partial inhibition of CDK5 has been a proposed as therapeutic option (Zheng et al., 2005; Camins et al., 2006).
Decreasing the availability of CDK5 could be a useful therapeutic approach. Excessive upregulation of CDK5 by the truncated activators may contribute to neurodegeneration and alter the phosphorylation state of cytosolic and cytoskeletal proteins. Increased CDK5 activity has been implicated not only in AD but also in amyotrophic lateral sclerosis, Parkinson's disease, Niemann–Pick type C disease, ischemia, and animal models with some of these conditions (Lee et al., 1999; Patrick et al., 1999; Nguyen et al., 2001; Bu et al., 2002; Lu et al., 2003; Nguyen and Julien, 2003; Smith et al., 2003; Wang et al., 2003). In summary, silencing CDK5 opens a novel gene therapy strategy to control the amount of CDK5 to reduce the tau pathology associated with Alzheimer's disease and other tauopathies.
Footnotes
This work was supported by National Institutes of Health (NIH)–Fogarty Grant R21 AG024024063 (K.S.K., G.P.C.-G.) and National Institute on Aging–NIH Grant R01 AG029802-01 (K.S.K., G.P.C.-G.); Colciencias Grants 11150418078 and 111545921503 (G.P.C.-G.); NIH Grant NS 50210 (B.D., K.S.K.), Advanced Microscopy Unit and Viral Vector Core and Gene Therapy, Group of Neuroscience of Antioquia. University of Antioquia was also supported by Colciencias Grant 111540820511 and 111545921525 (J.C.G.-G.); Committee for Research Development, University of Antioquia, Colombia (G.P.C.-G., J.C.G.-G.). Confocal imaging was done under the aegis of the Center for Bio-Image Informatics using an Olympus FV1000 CLSM microscope. The Center for Bio-Image Informatics is partially supported by National Science Foundation (NSF) Information Technology Research Grant 0331697, NSF Grant EIA-0080134, and NSF Grant III-0808772. D.P. was the recipient of a Doctoral Fellowship from Colciencias. We thank Dr. P. Davies (Feinstein Institute for Medical Research, Manhasset, NY) for the donation of PHF-1 antibody. We extend our appreciation to Dr. Xuemei Zhang (Neuroscience Research Institute, University of California, Santa Barbara, Santa Barbara, CA) for her expert advice on the CDK5 enzyme assays. We thank Tania Marquez for assistance in maintaining the mouse and rat colonies in the SPF vivarium of the University of Antioquia.
References
- Ahn JS, Musacchio A, Mapelli M, Ni J, Scinto L, Stein R, Kosik KS, Yeh LA. Development of an assay to screen for inhibitors of tau phosphorylation by Cdk5. J Biomol Screen. 2004;9:122–131. doi: 10.1177/1087057103260594. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Banker G, Kimberly G. Ed 2. London: MIT; 2002. Culturing nerve cells. [Google Scholar]
- Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993;336:417–424. doi: 10.1016/0014-5793(93)80849-p. [DOI] [PubMed] [Google Scholar]
- Bishop CM. Oxford: Oxford UP; 1995. Neural networks for pattern recognition. [Google Scholar]
- Boudreau RL, Monteys AM, Davidson BL. Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. RNA. 2008;14:1834–1844. doi: 10.1261/rna.1062908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RL, McBride JL, Martins I, Shen S, Xing Y, Carter BJ, Davidson BL. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice. Mol Ther. 2009;17:1053–1063. doi: 10.1038/mt.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boykov Y, Kolmogorov V. Energy minimization methods in computer vision and pattern recognition. Berlin, Heidelberg: Springer-Verlag; 2001. An experimental comparison of min-cut/max-flow algorithms for energy minimization in vision; pp. 359–374. [Google Scholar]
- Bu B, Li J, Davies P, Vincent I. Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann–Pick type C murine model. J Neurosci. 2002;22:6515–6525. doi: 10.1523/JNEUROSCI.22-15-06515.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt PJ, Adelson EH. A multiresolution spline with application to image mosaics. ACM Trans Graph. 1983;2:217–236. [Google Scholar]
- Camins A, Verdaguer E, Folch J, Canudas AM, Pallàs M. The role of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect. 2006;19:453–460. doi: 10.1358/dnp.2006.19.8.1043961. [DOI] [PubMed] [Google Scholar]
- Cardona-Gómez P, Perez M, Avila J, Garcia-Segura LM, Wandosell F. Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol Cell Neurosci. 2004;25:363–373. doi: 10.1016/j.mcn.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Chang K, Elledge SJ, Hannon GJ. Lessons from nature: microRNA-based shRNA libraries. Nat Methods. 2006;3:707–714. doi: 10.1038/nmeth923. [DOI] [PubMed] [Google Scholar]
- Churcher I. Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr Top Med Chem. 2006;6:579–595. doi: 10.2174/156802606776743057. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–483. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Fedorov D, Fonseca LM, Kenney C, Manjunath BS. Automatic registration and mosaicking system for remotely sensed imagery. Paper presented at SPIE 9th International Symposium on Remote Sensing; September; Agia Pelagia, Crete, Greece. 2002. [Google Scholar]
- Fedorov D, Sumengen B, Manjunath BS. Multi-focus imaging using local focus estimation and mosaicking. Paper presented at IEEE International Conference on Image Processing 2006 (ICIP06); October; Atlanta, GA. 2006. [Google Scholar]
- Fischer PM. Recent advances and new directions in the discovery and development of cyclin-dependent kinase inhibitors. Curr Opin Drug Discov Devel. 2001;4:623–634. [PubMed] [Google Scholar]
- Gallego-Gómez JC, Risco C, Rodríguez D, Cabezas P, Guerra S, Carrascosa JL, Esteban M. Differences in virus-induced cell morphology and in virus maturation between MVA and other strains (WR, Ankara, and NYCBH) of vaccinia virus in infected human cells. J Virol. 2003;77:10606–10622. doi: 10.1128/JVI.77.19.10606-10622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett MD, Fattaey A. CDK inhibition and cancer therapy. Curr Opin Genet Dev. 1999;9:104–111. doi: 10.1016/s0959-437x(99)80015-x. [DOI] [PubMed] [Google Scholar]
- Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N, Détivaud L, Doerig C, Meijer L. ATP-site directed inhibitors of cyclin-dependent kinases. Curr Med Chem. 1999;6:859–875. [PubMed] [Google Scholar]
- Hartley RI, Zisserman A. Ed 2. Cambridge, UK: Cambridge UP; 2004. Multiple view geometry in computer vision. [Google Scholar]
- Hashiguchi M, Saito T, Hisanaga S, Hashiguchi T. Truncation of CDK5 activator p35 induces intensive phosphorylation of Ser202/Thr205 of human tau. J Biol Chem. 2002;277:44525–44530. doi: 10.1074/jbc.M207426200. [DOI] [PubMed] [Google Scholar]
- Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10:880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Kenney CS, Manjunath BS, Zuliani M, Hewer MGA, Nevel AV. A condition number for point matching with application to registration and postregistration error estimation. IEEE Trans Pattern Anal Mach Intell. 2003;25:1437–1454. [Google Scholar]
- Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, Broodie N, Mazitschek R, Delalle I, Haggarty SJ, Neve RL, Lu Y, Tsai LH. Deregulatin of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–817. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23:417–425. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Ishiguro K, Omori A, Takamatsu M, Arioka M, Imahori K, Uchida T. A cdc2-related kinase PSSALRE/cdk5 is homologous with the 30 kDa subunit of tau protein kinase II, a proline-directed protein kinase associated with microtubule. FEBS Lett. 1993;335:171–175. doi: 10.1016/0014-5793(93)80723-8. [DOI] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Kvilekval K, Fedorov D, Obara B, Singh A, Manjunath BS. Bisque: a platform for bioimage analysis and management. Bioinformatics. 2010;26:544–552. doi: 10.1093/bioinformatics/btp699. [DOI] [PubMed] [Google Scholar]
- Lee KY, Clark AW, Rosales JL, Chapman K, Fung T, Johnston RN. Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci Res. 1999;34:21–29. doi: 10.1016/s0168-0102(99)00026-7. [DOI] [PubMed] [Google Scholar]
- Leost M, Schultz C, Link A, Wu YZ, Biernat J, Mandelkow EM, Bibb JA, Snyder GL, Greengard P, Zaharevitz DW, Gussio R, Senderowicz AM, Sausville EA, Kunick C, Meijer L. Paullones are potent inhibitors of glycogen synthase kinase-3beta and cyclin-dependent kinase 5/p25. Eur J Biochem. 2000;267:5983–5994. doi: 10.1046/j.1432-1327.2000.01673.x. [DOI] [PubMed] [Google Scholar]
- Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc. 1934;56:658–666. [Google Scholar]
- Liu M, Choi S, Cuny GD, Ding K, Dobson BC, Glicksman MA, Auerbach K, Stein RL. Kinetic studies of Cdk5/p25 kinase: phosphorylation of tau and complex inhibition by two prototype inhibitors. Biochemistry. 2008;47:8367–8377. doi: 10.1021/bi800732v. [DOI] [PubMed] [Google Scholar]
- Lu L, Grimm JW, Shaham Y, Hope BT. Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J Neurochem. 2003;85:1604–1613. doi: 10.1046/j.1471-4159.2003.01824.x. [DOI] [PubMed] [Google Scholar]
- Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- Meijer L, Thunnissen AM, White AW, Garnier M, Nikolic M, Tsai LH, Walter J, Cleverley KE, Salinas PC, Wu YZ, Biernat J, Mandelkow EM, Kim SH, Pettit GR. Inhibition of cyclin-dependent kinases, GSK-3beta and CK1 by hymenialdisine, a marine sponge constituent. Chem Biol. 2000;7:51–63. doi: 10.1016/s1074-5521(00)00063-6. [DOI] [PubMed] [Google Scholar]
- Mettey Y, Gompel M, Thomas V, Garnier M, Leost M, Ceballos-Picot I, Noble M, Endicott J, Vierfond JM, Meijer L. Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects. J Med Chem. 2003;46:222–236. doi: 10.1021/jm020319p. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–829. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP. Cyclin-dependent kinase 5 in amyotrophic lateral sclerosis. Neurosignals. 2003;12:215–220. doi: 10.1159/000074623. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Larivière RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Obara B, Byun J, Fedorov D, Manjunath BS. Automatic nuclei detection and data flow in Bisquik system. Paper presented at Workshop on Bio-Image Informatics: Biological Imaging, Computer Vision and Data Mining; January; Santa Barbara, CA. 2008. [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Paudel HK, Lew J, Ali Z, Wang JH. Brain proline-directed protein kinase phosphorylates tau on sites that are abnormally phosphorylated in tau associated with Alzheimer's paired helical filaments. J Biol Chem. 1993;268:23512–23518. [PubMed] [Google Scholar]
- Shiradkar MR, Padhalingappa MB, Bhetalabhotala S, Akula KC, Tupe DA, Pinninti RR, Thummanagoti S. A novel approach to cyclin-dependent kinase 5/p25 inhibitors: a potential treatment for Alzheimer's disease. Bioorg Med Chem. 2007;15:6397–6406. doi: 10.1016/j.bmc.2007.06.053. [DOI] [PubMed] [Google Scholar]
- Sielecki TM, Boylan JF, Benfield PA, Trainor GL. Cyclin-dependent kinase inhibitors: useful targets in cell cycle regulation. J Med Chem. 2000;43:1–18. [PubMed] [Google Scholar]
- Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, Sheth N, Bradshaw J, Burchard J, Kulkarni A, Cavet G, Sachidanandam R, McCombie WR, Cleary MA, Elledge SJ, Hannon GJ. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet. 2005;37:1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Lee VM, Trojanowski JQ. Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu Rev Pathol. 2006;1:151–170. doi: 10.1146/annurev.pathol.1.110304.100113. [DOI] [PubMed] [Google Scholar]
- Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S, Slack RS, Przedborski S, Anisman H, Park DS. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:13650–13655. doi: 10.1073/pnas.2232515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:13212–13217. doi: 10.1073/pnas.0506306102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- Tseng HC, Zhou Y, Shen Y, Tsai LH. A survey of Cdk5 activator p35 and p25 levels in Alzheimer's disease brains. FEBS Lett. 2002;523:58–62. doi: 10.1016/s0014-5793(02)02934-4. [DOI] [PubMed] [Google Scholar]
- Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- Van den Haute C, Spittaels K, Van Dorpe J, Lasrado R, Vandezande K, Laenen I, Geerts H, Van Leuven F. Coexpression of human cdk5 and its activator p35 with human protein tau in neurons in brain of triple transgenic mice. Neurobiol Dis. 2001;8:32–44. doi: 10.1006/nbdi.2000.0333. [DOI] [PubMed] [Google Scholar]
- Wang H, Suter D. Robust adaptive-scale parametric model estimation for computer vision. IEEE Trans Pattern Anal Mach Intell. 2004;26:1459–1474. doi: 10.1109/TPAMI.2004.109. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y. Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci. 2003;6:1039–1047. doi: 10.1038/nn1119. [DOI] [PubMed] [Google Scholar]
- Yu Y, Run X, Liang Z, Li Y, Liu F, Liu Y, Iqbal K, Grundke-Iqbal I, Gong CX. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem. 2009;108:1480–1494. doi: 10.1111/j.1471-4159.2009.05882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC. A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J. 2005;24:209–220. doi: 10.1038/sj.emboj.7600441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Trono D. Current protocols in neuroscience, Unit 4.21. New York: Wiley; 2000. Production of high-titer lentiviral vectors. [Google Scholar]
- Zuliani M, Kenney C, Manjunath BS. A mathematical comparison of point detectors. Paper presented at Second IEEE Image and Video Registration Workshop; July; Washington, DC. 2004. [Google Scholar]