

Abstract
The NMDA receptor coagonist D-serine is important in a number of different processes in the central nervous system, ranging from synaptic plasticity to disease states, including schizophrenia. D-serine appears to be the major coagonist acting on retinal ganglion cell NMDA receptors, but the cell type from which it originates and whether its release can be modulated by activity are unknown. In this study, we utilized a mutant mouse line with elevated D-serine to investigate this question. Direct measurements of extracellular D-serine using capillary electrophoresis demonstrate that D-serine can be released from the intact mouse retina through an AMPA receptor dependent mechanism. AMPA-evoked D-serine release persisted in the presence of a cocktail of neural inhibitors but was abolished after administration of a glial toxin. These findings provide the first evidence that extracellular D-serine levels in the retina can be modulated, and that such modulation is contingent upon glial cell activity.
Keywords: Retina, D-serine, NMDA receptor, AMPA receptor, capillary electrophoresis, glia
Introduction
Activation of N-methyl-D-aspartate receptors (NMDARs) requires glutamate bound to the NR2 subunit as well as a separate coagonist bound to the NR1 subunit (Johnson and Ascher 1987;Kleckner and Dingledine 1988). Glycine was originally thought to be the endogenous coagonist, but in recent years D-serine has emerged as the more likely coagonist at several CNS sites, including the retina. D-serine is now known to be abundant in the brain (Hashimoto et al. 1992), especially in regions rich in NMDARs (Schell et al. 1995). The prospects of D-serine as an endogenous coagonist were further strengthened when Wolosker discovered serine racemase, the enzyme that synthesizes D-serine from L-serine (Wolosker et al. 1999). D-serine has since been implicated in numerous mechanisms ascribed to NMDARs, including neuroplasticity (Basu et al. 2009;Panatier et al. 2006) and learning and memory (Duffy et al. 2008). Furthermore, abnormalities in D-serine regulation have been implicated in the NMDAR hypofunction theory of schizophrenia (Verrall et al. 2010;Labrie et al. 2009).
Early immunohistochemical and ultrastructural studies localized D-serine to astrocytes adjacent to neurons expressing NMDARs (Schell et al. 1997). A similar result was later confirmed in the retina, where D-serine was found in astrocytes and Müller glia (Stevens et al. 2003). However, the recent discovery of D-serine in neurons throughout the brain (Kartvelishvily et al. 2006) and serine racemase mRNA in retinal neurons (Takayasu et al. 2008) brings into question the idea that D-serine originates solely from glia in the CNS.
While the cellular origins of D-serine are presently unclear, it serves as an endogenous coagonist of retinal ganglion cell (RGC) NMDARs. In the intact retina, applying D-serine deaminase (DsDa), a highly selective D-serine degrading enzyme, has the same effect on light-evoked RGC NMDAR currents as completely blocking NMDARs with conventional antagonist. Adding exogenous coagonist augments RGC NMDAR currents (Gustafson et al. 2007;Stevens et al. 2003) demonstrating that the coagonist site is not saturated and additional NMDARs would be recruited by D-serine release.
Studies on cultured cortical astrocytes suggest that α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor (AMPAR) activation stimulates the synthesis (Kim et al. 2005) and release of D-serine (Mothet et al. 2005). However, cultured cortical neurons (Kartvelishvily et al. 2006) and, more recently, neurons from cortical slice have displayed similar AMPA-evoked D-serine release (Rosenberg et al. 2010). It remains unclear which cell type contributes to the endogenous pool of D-serine in intact CNS tissue and whether the same glutamatergic release mechanisms are employed.
Previous attempts at measuring extracullar D-serine in the retina have proven difficult because of the relatively low levels of D-serine (O’Brien et al. 2004), despite the fact that D-serine modulates retinal ganglion cell NMDARs. To aid us in our study of D-serine release, we utilized a mutant mouse with a point mutation in the only known mammalian D-serine degrading enzyme, D-amino acid oxidase (DAO-), rendering the enzyme inactive (Konno and Yasumura 1983). Direct measurements of extracellular D-serine in the intact retina via capillary electrophoresis demonstrate glutamate-evoked D-serine release through an AMPAR-dependent pathway. This D-serine release persisted in the presence of neural inhibitors but was abolished by a glial toxin.
Materials and Methods
Materials
AMPA, cyclothiazide, TFB-TBOA, GYKI 52466 hydrochloride, TTX, and suramin were purchased from Tocris Bioscience (Ellisville, MO). NBD-F and Fluo-4 AM were purchased from Invitrogen (Eugene, OR). All other chemicals were purchased from Sigma (St Louis, MO).
Isolation and pharmacological treatment of retinas
Experiments were performed in strict accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Minnesota. Adult ddY mice were euthanized by an overdose of Nembutal (0.1ml of 50mg/ml, i.p.) and pneumo-thoraxed. Eyes were enucleated and placed in bicarbonate Ringer’s solution (111 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 32 mM NaHCO3, 0.5 mM NaH2PO4, and 15 mM Dextrose; bubbled with 95% O2, 5% CO2) for the surgical isolation of the retina. For each data point, two isolated retinas were incubated in 100 μL of Ringer’s solution and oxygenated in a humidified chamber containing 95% O2 and 5% CO2 gas with shaking for 50 minutes, unless indicated otherwise. Following incubation in the drug solution or control, the bathing media from the retinas was extracted and partitioned into three equal volumes. These samples were used for the identification of D-serine via capillary electrophoresis.
Capillary electrophoresis
Capillary electrophoresis (CE) with a commercial LIF detection system (MDQ, Beckman-Coulter, Fullerton, CA, USA), was used to separate and measure D-serine as described previously (O’Brien et al. 2005). 25 μL of the sample was treated with 2.5 μL of the internal standard α-aminoadipic acid (5 μM final concentration). D-glutamate was used as an internal standard in experiments using α-aminoadipic acid as a glial toxin. The amino acids from extracellular retinal samples were fluorescently derivitized by adding 2.5 μL of 4-fluro-7-nitrobenz-2oxa-1,3-diazole (NBD-F) dissolved in acetonitrile (3.6 mM final concentration; 30 μL final volume) and reacting at 60°C for 15 minutes.
Samples were pressure injected for 5 s at 0.5 psi into a fused cilica capillary and run at -15 kV (70 μA) for 30 minutes. Before each run, the capillary was rinsed with 1M NaOH and loaded with 34 mM hydroxypropyl-β-cyclodextrin in 165 mM borate pH 10.2 to separate D-serine from its enantiomeric partner L-serine. A 4 mW argon laser (488 nm excitation) was used to detect fluorescence. A beam splitter combined with two separate photo multiplier tubes allowed simultaneous recording of samples at different gain settings. Amino acids were quantified by integrating the generated electropherogram using 32 karat analysis software.
Retinal protein determination
Total protein measurement was used to normalize all CE amino measurements (expressed as nmol/g protein). Retinas were homogenized by sonication and centrifuged at 11,000 g for 5 minutes. The pellets from each sample were re-suspended in 120 μL of 2M NaOH and diluted (1:20) in water. Protein concentration in the diluted samples was quantified using a Pierce (Rockford, IL) bicinchoninic acid assay.
Identification of D-serine
The D-serine peak was identified by spiking a fraction of the extracellular sample with an additional 5 μM D-serine. To be sure that another molecule with a similar migration time was not interfering with our D-serine measurements, D-serine was also measured after treating the sample with DsDa (50 μg/ml, 20 minutes). DsDa specifically eliminated D-serine from a series of amino acid standards (Figure 1A) and also completely removed the peak matching the D-serine migration time in samples (Figure 1B). The mass of D-serine in each sample was computed from a standardized curve.
Figure 1.
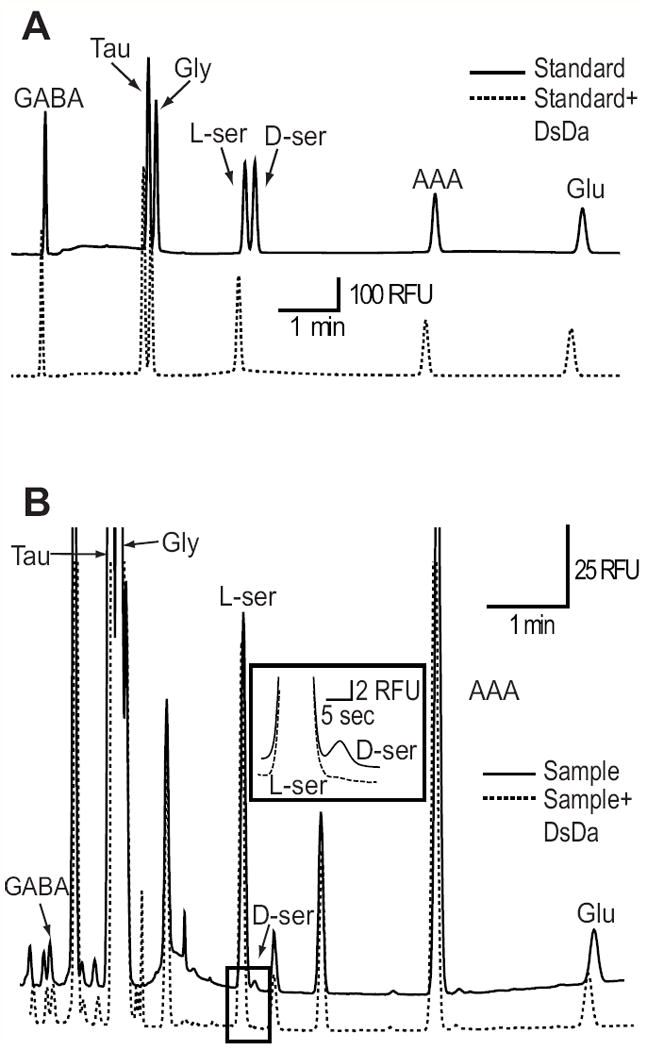
Identification of D-serine via capillary electrophoresis. (A) Electropherogram showing the separation of D-serine (D-ser), from a series of amino acid standards; Gamma-aminobutyric acid (GABA), Taurine (Tau), Glycine (Gly), L-serine (L-ser), α-aminoadipic acid (AAA), and Glutamate (Glu). Treatment with D-serine deaminase (DsDa) (50 ug/ml, 20 min) specifically eliminated D-serine (dotted trace). (B) Separation of amino acids from mouse retina extracellular media, with L-serine and D-serine expanded. D-serine was completely eliminated following DsDa incubation.
Glial toxin α-aminoadipic acid
Retinas were preincubated in Ringer’s solution containing 10 mM α-aminoadipic acid (AAA) for 3 hours, while control retinas were incubated for the same period without the toxin. The treated retinas were then incubated for 50 minutes in presence of 1mM AAA (10 mM AAA added too much noise to the CE trace) and the extracellular media was sampled.
Ca2+ imaging
Retinas were incubated in Ringer’s solution containing 230 μM Fluo-4 AM for 45 minutes to label glial cells (Newman and Zahs 1998). Whole-mount retinas were flattened on nitrocellulose paper, with the ganglion cell side up, and perfused in bicarbonate Ringer’s solution. Whole field fluorescence in the ganglion cell layer revealed Müller cell end feet surrounding dark unlabeled ganglion cell bodies. Images were acquired every 2 seconds with a multiphoton laser (λ=820 nm) (Prairie Technologies). Data were expressed as ΔF/F, where F is the average baseline fluorescence measured over the first 30 seconds. Drugs blocking neural signaling were applied 1 minute prior to imaging. Figure 4A (bottom) was reconstructed from a Z-series of images taken at 1um intervals.
Figure 4.
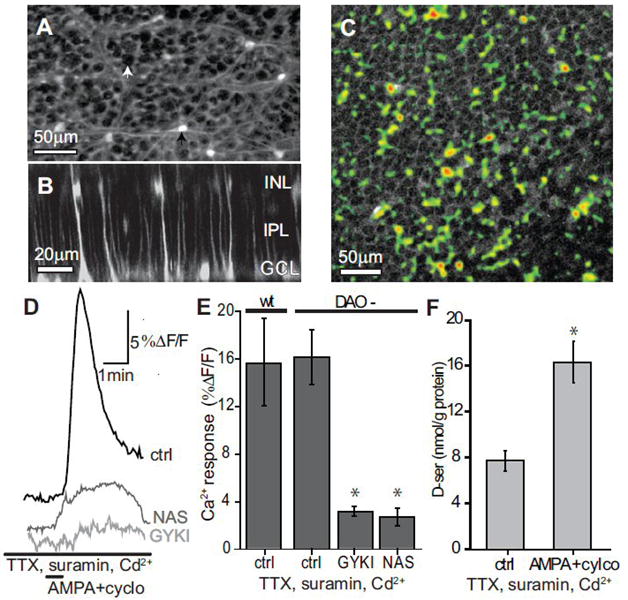
AMPA+cyclothiazide D-serine release is not blocked by inhibition of neural activity. (A) Fluo-4 AM loaded retina showing labeling of Astrocytes (black arrow) and Müller cell endfeet surrounding unlabeled ganglion cell bodies (white arrow). (B) Cross-section of retina in A reconstructed from a Z-series showing Müller cell labeling of endfeet near the ganglion cell layer (GCL), with stalks spanning the inner plexiform layer (IPL), and cell bodies in the inner nuclear layer (INL). (C) Ca2+ response of Müller cell endfeet to AMPA+cylco. Retinas were bulk loaded with Fluo-4 AM then treated with 1μM TTX, 100 μM suramin, and 200 μM Cd2+ 2 minutes prior to a 30 second bath application of AMPA+cylco. Image shows the change in Ca2+ in response to AMPA+cylco as a thresholded maximal projection (pseudocolor) overlaid on the average baseline image. Ca2+ elevations excluded ganglion cell bodies. (D) Change in Ca2+ over time from the region in C. (E) No significant difference was observed in the Ca2+ increase between wt (n=4) and DAO- (n=3) retinas following AMPA+cylco application. GYKI (n=5) and NAS (n=6) significantly reduced the AMPA+cylco induced Ca2+ response in DAO- retinas (*, p<0.05, compared to ctrl). (F) DAO- retinas exposed to AMPA+cyclo still released D-serine in the combined presence of TTX, suramin, and Cd2+. (*, statistically different from control, p<0.05, n=4-5).
Statistical analysis
Each data point collected (n) for CE data was derived from an incubation of two retinas, while those for Ca2+ imaging denotes the response of a single retina. Student’s one-tailed t-test was used to calculate significance. All data are expressed as mean ± SE. Significance defined as p<0.05.
Results
AMPA induced D-serine release
DAO- mice have significantly elevated levels of D-serine in brain regions rich in DAO expression, including the brainstem and cerebellum (Hamase et al. 2005). In this study, we found that DAO- retinas incubated for 50 min in oxygenated Ringer’s solution had significantly greater extracellular D-serine levels compared to wild-type (wt) controls (Table 1, Figure 2B). In contrast, no significant difference was observed in any of the other amino acids measured, including L-serine, glycine, taurine, GABA, or glutamate (Table 1).
Table 1.
Extracellular amino acid comparison between wt and DAO- mice. Data expressed as the mean extracellular amino acid measurement in nmol/g protein ± S.E.
| D-serine | L-serine | Glycine | Glutamate | GABA | Taurine | |
|---|---|---|---|---|---|---|
| wt | 0.69 ± 0.12 | 492.68 ± 57.61 | 336.36 ± 70.37 | 86.02 ± 56.58 | 31.17 ± 16.59 | 4579.72 ± 883.25 |
| DAO- | 4.66* ± 1.13 | 715.40 ± 148.08 | 395.11 ± 127.38 | 118.29 ± 48.61 | 42.14 ± 18.23 | 5667.64 ± 1252.42 |
statistically different from wt at p<0.05. n=5.
Figure 2.

AMPA+cyclothiazide evokes D-serine release in wt and DAO- retinas. (A) Extracellular D-serine in DAO- retinas incubated for 50 minutes (Ctrl) or with 50 μM AMPA plus 50 μM cyclothiazide (AMPA+cylco). Both peaks were eliminated by DsDa. (B) Comparison of extracellular D-serine between wild-type (wt) and DAO- mice, ctrl or with AMPA+cyclo. *, statistically different from control at p<0.05. #, statistically different from wt under same drug condition at p<0.05. (C) D-serine release measured in DAO- retinas every 10 minutes, for 5 times total. (D) sum of D-serine release at each 10 minute time point in C. C-D, AMPA alone failed to elevate D-serine (*, significantly different from control, p<0.05) and AMPA+cyclo release was significantly attenuated by 1-naphthyl acetyl spermine (NAS) (#, comparison of two groups, p<0.05). Data reported as the mean ± S.E. n=3-6.
We examined the possibility that D-serine is released through glutamatergic mechanisms. To carry out this objective, we exposed retinas to AMPA+cyclothiazide (which prevents AMPAR desensitization (Zhang et al. 2006)) and sampled the extracellular media. AMPA+cylcothiazide treatment resulted in an approximately twofold increase in extracullar D-serine in both wt and DAO- retinas (figure 2B), implying that there is no difference in the D-serine release mechanism between the genotypes. DAO- mice were used in all subsequent pharmacological experiments to more readily detect changes in D-serine.
We tested whether the D-serine release induced by AMPA+cyclothiazide was detectable over shorter exposures. Retinas were moved to a new well containing fresh Ringer’s solution every 10 minutes for five consecutive incubations, and the incubation sample measured for D-serine at each point (50 minutes total). Over this time period, the AMPA+cyclothiazide-induced D-serine release was steady and inexhaustible (Figure 2C). This finding suggests that the source of releasable D-serine was continuously replenished, presumably by serine racemase. Coapplication of 1-naphthyl acetyl spermine (NAS), a Ca2+ permeable AMPAR antagonist (Tsubokawa et al. 1995), significantly reduced the AMPA+cyclothiazide-induced D-serine release observed over time (Figure 2C, 2D). AMPA alone, in the absence of cyclothiazide, was incapable of increasing extracellular D-serine, suggesting that this mode of D-serine release is prone to AMPAR desensitization (Figure 2C, 2D). Cyclothiazide alone had no effect on D-serine levels (data not shown).
Blocking Glutamate uptake elevates extracellular D-serine by acting through AMPA receptors
Our findings raised the possibility that endogenous glutamate is capable of evoking D-serine release. In healthy retinas, extracellular glutamate concentrations are tightly regulated by excitatory amino acid transporters (EAAT1-EAAT5) and removal of EAAT1 or EAAT2 significantly elevates extracellular glutamate (Vorwerk et al. 2000). We treated the retinas with TFB-TBOA, a high affinity EAAT1 and EAAT2 blocker (Shimamoto et al. 2004), which dramatically elevated extracellular glutamate over the course of 50 minutes (Figure 3A, 3B).
Figure 3.
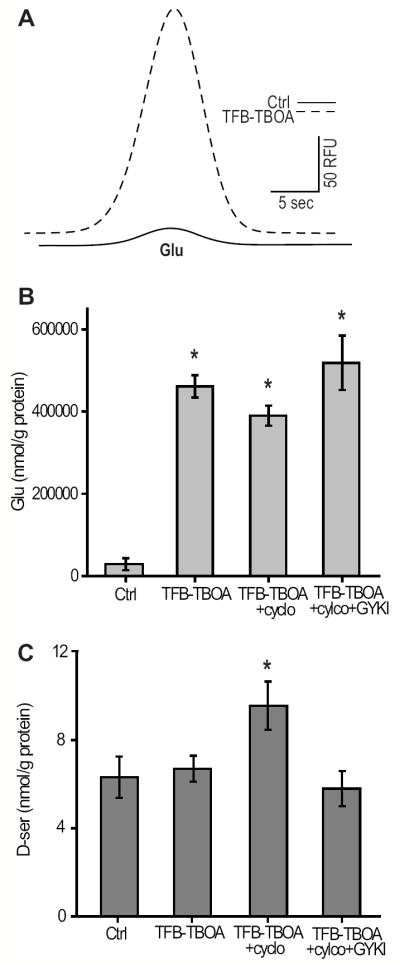
Blocking Glutamate uptake elevates extracellular D-serine by acting through AMPA receptors. (A) Raw trace showing glutamate (Glu) increase following a 50 minute incubation in 10 μM TFB-TBOA. (B) TFB-TBOA significantly elevated Glu in all conditions tested. There was no difference in Glu between experiments where TFB-TBOA was added. (C) TFB-TBOA elevated D-serine in the presence of cyclothiazide which was blocked by the application of 50 μM GYKI. A, B, C DAO- retinas (*, statistically different from control, p<0.05, n=6-7).
We found that elevating glutamate alone was insufficient to measurably increase D-serine, consistent with our findings that AMPA alone did not elevate extracellular D-serine (Figure 2D). However, using cyclothiazide, in addition to TFB-TBOA, resulted in a significant elevation of D-serine. This increase was blocked by the selective non-competitive AMPAR antagonist GYKI 52466, confirming that glutamate was acting through an AMPAR-dependent pathway to elevate D-serine. GYKI did not bring D-serine levels significantly below baseline (Figure 3C), which implies that the baseline levels of D-serine measured in these experiments were set by a non-AMPAR mechanism, possibly by the alanine-serine-cysteine transporter (ASCT2) as described previously (O’Brien et al. 2005;Dun et al. 2007).
AMPA induced D-serine release is independent of neural activity
We next wanted to evaluate the cell type responsible for AMPAR-dependent D-serine release. Given that D-serine could be present in both neurons and glia, there are three basic possibilities: (1) AMPA could act directly on neurons stimulating them to release D-serine; (2) AMPA could act on neurons which then signal to glial cells to release D-serine; or (3) AMPA could act directly on glial cells to release D-serine. We addressed possibilities (1) and (2) by adding pharmacological inhibitors of neuronal activity and neurotransmitter release, combined with an inhibitor of neuron to glia signaling.
For possibility (3) to hold true, AMPA must have some direct effect on glia. Müller cells appear to express AMPA receptors (Vitanova 2007) and are responsive to AMPA in isolation (Wakakura and Yamamoto 1994). To test this in the intact retina, we looked at intracellular Ca2+ in retinal glia in response to AMPA+cyclothiazide treatment in the presence of a cocktail of neural signaling inhibitors. TTX was used to prevent neural impulses, and cadmium chloride (Cd2+) was also added to prevent synaptic transmission in retinal neurons with graded potentials by blocking voltage gated Ca2+ channels. To inhibit neuron-to-glia and glia-to-glia communication, which is primarily carried out by ATP in the retina (Newman 2005), the purenurgic (P2) antagonist suramin was added. Whole-mount retinas were incubated in Fluo-4 AM over time periods favoring glial loading (Newman and Zahs 1998). We observed that Fluo-4 AM loaded astrocytes and Müller cells but not neurons (Figure 4A-B). Imaging with a multiphoton laser we found that retinal Müller cell endfeet in the ganglion cell layer of the retina show significant increases in intracellular Ca2+ in response to AMPA+cylothiazide in the absence of neural signaling which was blocked by the AMPAR antagonists GYKI and NAS (Figure 4C-E), suggesting that AMPA is capable of directly acting on glia, perhaps through Ca2+ permeable AMPARs. There was no difference in the Ca2+ response between wt and DAO- retinas (Figure 4E), implying that the DAO mutation did not indirectly influence D-serine levels by altering glial sensitivity to agonist. AMPA+cyclothiazide application still evoked D-serine release in the presence of suramin and the neural signaling inhibitors (Figure 4F), which implies that neurons are not contributing to the AMPAR-dependent D-serine release in these studies.
Glial cells regulate baseline as well as evoked D-serine release
To address whether or not AMPA+cyclothiazide can act directly on glia to evoke D-serine release, we utilized the glial toxin α-aminoadipic acid (AAA). AAA toxicity is thought to originate by causing free radical buildup in glial cells (Kato et al. 1993), and eliminates retinal Müller cell function, while retaining the retina’s responsiveness to light (Wang et al. 2009;Wang and Kefalov 2009). Preincubating retinas in 10 mM AAA significantly reduced extracellular glutamine (Gln) concentrations compared to control retinas (Figure 5A), indicating that the glial cells, the major source of Gln (Bringmann et al. 2009), were impaired. In the same preparations, D-serine release evoked by AMPA+cyclothiaizide was dramatically reduced (Figure 5B). Glial impairment also significantly lowered baseline D-serine (Figure 5B), implying that glia also contribute to basal concentration of D-serine in the extracellular environment.
Figure 5.
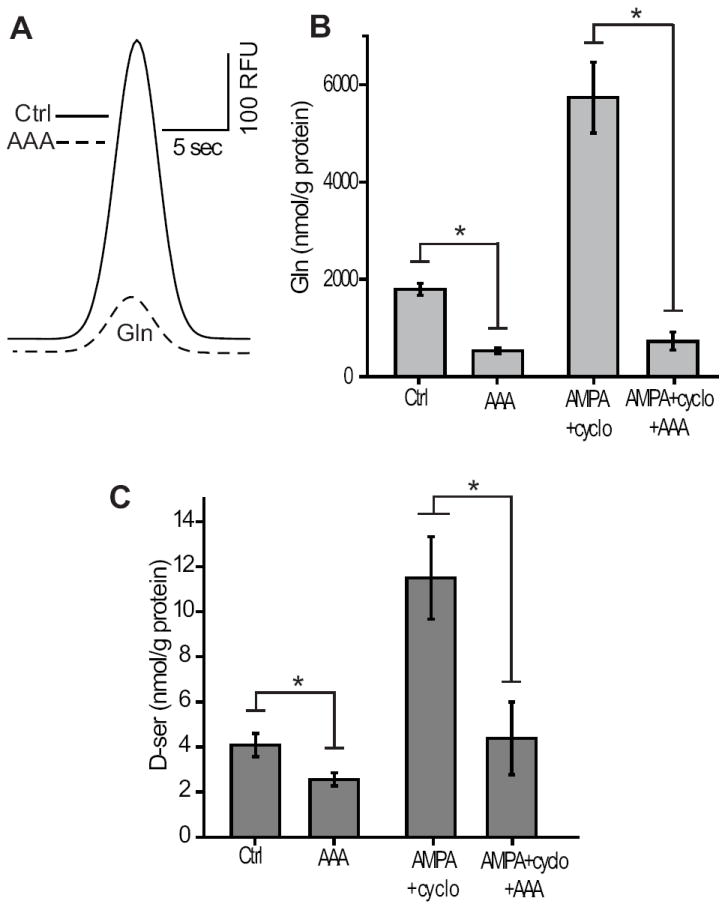
Glial toxin prevents AMPA receptor dependent D-serine release. (A) Electropherogram showing the reduction of Glutamine (Gln) following a 3 hour incubation in 10 mM AAA. (B) Gln was significantly reduced by AAA irrespective of AMPA+cyclo application. (C) Baseline D-serine and AMPA+cyclo induced D-ser release was reduced by AAA. A, B, C DAO- retinas (*, p<0.05, n=5).
Discussion
We have provided the first direct measurements of dynamic D-serine release from the isolated retina. This pathway for D-serine release involved the activation of AMPARs, a model consistent with previous findings in cortical astrocytes (Mothet et al. 2005). Because the coagonist site is not saturated on RGC NMDARs (Gustafson et al. 2007;Kalbaugh et al. 2009;Daniels and Baldridge 2009;Stevens et al. 2003), stimulated D-serine release would influence the output of the retina to the brain.
Our results reveal that DAO- retinas have significantly greater D-serine than wt, while a number of other amino acids are unaffected. The similar increase in D-serine evoked by AMPA+cylcothiaizide in DAO- and wt suggests that the mechanism of D-serine release is unaltered in the mutants, making them ideal for studying D-serine release in the retina. In other areas of the nervous system, DAO expression appears to correlate negatively with D-serine levels. Brain regions that typically show the lowest levels of D-serine, such as the cerebellum, display the greatest relative increase in DAO- mice, whereas brain regions with high levels of D-serine appear to be unaffected by the mutation (Hamase et al. 2005). Based on our results, we would predict the utilization of DAO in the regulation of retinal D-serine to most likely fall somewhere between the extremes of cortex and cerebellum.
We showed that raising extracellular glutamate by blocking glutamate transport can induce D-serine release via an AMPAR-dependent pathway. Many retinal stressors are known to increase extracellular glutamate, including: hypoglycemia, anoxia (Zeevalk and Nicklas 1990), optic nerve crush (Vorwerk et al. 2004;Schuettauf et al. 2000), and models of glaucoma (Schuettauf et al. 2002). A leading theory as to how these stressors pathologically develop is that the excess glutamate becomes excitotoxic through its action on NMDARs. Our studies suggest that glutamate elevations could also cause retinal glia to release D-serine, which would recruit more NMDARs for the same level of glutamate to act on, thus exacerbating excitotoxicity. Congruently, oxygen-glucose deprivation in the cerebrum leads to efflux of glutamate and D-serine (Kirschner et al. 2009) and serine racemase knockout mice are protected against ischemia (Mustafa et al. 2010), while the addition of coagonist exaggerates NMDA excitotoxicity in the retina and D-serine removal attenuates it (Hama et al. 2006).
The fact that the glial toxin AAA reduced but did not completely eliminate baseline extracellular D-serine might mean that AAA only partially impaired the glial metabolic machinery required for D-serine synthesis. Alternatively, neurons might also contribute to baseline extracellular D-serine levels. However, retinal D-serine uptake is thought to occur solely through ASCT2 (O’Brien et al. 2005), expressed primarily by glial cells (Dun et al. 2007). If there was a neural source of D-serine, disrupting the main mechanism of D-serine uptake by functionally eliminating glia would be expected to elevate the levels of extracellular D-serine, but the opposite was observed. Although a potential role of neurons in the regulation of D-serine cannot be ruled out, the fact that there was no apparent D-serine release observed when glia were functionally eliminated, and that inhibitors of neurons and neuron-to-glia signaling had no effect on D-serine release, suggests that the AMPAR-dependent D-serine release observed in our studies was exclusively from glia.
Electrophysiological observations in our lab have shown that increasing the contrast of light stimuli leads to a greater occupancy of the NMDAR coagonist site on RGCs (unpublished findings). Another group has shown, in rat retinal slice, that a pharmacologically mimicked light stimulus is still capable of recruiting additional RGC NMDA current in the presence of saturating NMDA concentrations, again implying dynamic coagonist release (Kalbaugh et al. 2009). In this report, stimulated coagonist release was unaffected by the addition of DAO, leading the authors to conclude that glycine was enhancing NMDAR currents. However, previous studies in the salamander retina showed that using an enzyme with greater specific activity against D-serine, DsDa, abolished light-evoked RGC NMDAR (Gustafson et al. 2007), implying that released glycine was not reaching the coagonist site.
Glycine serves as a major inhibitory neurotransmitter in the retina (Miller et al. 1977). It is noteworthy that our CE measurement of the extracellular retinal environment showed a significantly higher baseline concentration for glycine than D-serine. It is plausible that the high affinity glycine transporter GlyT1 expressed in amacrine (Pow and Hendrickson 2000) and Müller (Lee et al. 2005) cells keeps glycine levels low at the local micro-environment of RGC NMDARs. Coexpression of both GlyT1 and NMDARs in Xenopus oocytes dramatically reduces the level of glycine capable of reaching the coagonist site (Supplisson and Bergman 1997), despite the fact that glycine and D-serine have comparable potencies in expression systems void of amino acid transporters (McBain et al. 1989). Similarly, mutant mice with reduced expression of the GlyT1 transporter display greater coagonist site occupancy in the retina (Reed et al. 2009). On the other hand, transport of D-serine in the retina is thought to occur almost exclusively through the relatively slow neutral amino acid transporter ASCT2 (O’Brien et al. 2005;Dun et al. 2007), unlike the cortex where the high affinity transport ASC-1 is also expressed (Rutter et al. 2007). It is possible that the absence of a high affinity transport system for D-serine, combined with spatially precise release, allows such small extracellular levels of D-serine to serve as the major coagonist in the retina.
The mechanism of D-serine release in response to light stimulation remains to be elucidated. Our findings provide a model for D-serine release whereby glutamate released from bipolar cells during light stimulation could act on Müller cell processes in the IPL, which in turn release D-serine onto RGC NMDARs. The added NMDAR currents recruited by releasable D-serine could serve to increase the dynamic range of light-evoked RGC responses, or may lead to more long-term changes in ganglion cell activity by the activation of intracellular signaling pathways.
Acknowledgments
We thank Ryuichi Konno for providing us with the DAO- mice and Herman Wolosker for providing us with DsDa. We would also like to thank Eric Gustafson and Manny Esguerra for discussions and advice and Derek Miller for manuscript corrections. Sources of funding: NIH EY03014, T32GM008471, T32EY07133.
Abbreviations
- AMPAR
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor
- AAA
α-aminoadipic acid
- ASCT2
alanine-serine-cysteine transporter
- cyclo
cylothiazide
- DAO-
D-amino acid oxidase mutant
- DsDa
D-serine deaminase
- EAAT
excitatory amino acid transporter
- GlyT1
Glycine transporter 1
- GYKI 52466
4-(8-Methyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-5-yl)-benzenamine hydrochloride
- NAS
1-naphthyl acetyl spermine
- NBDF
4-fluoro-7-nitrobenz-2-oxa-1,3-diazole
- NMDAR
N-methyl-D-aspartate receptor
- TFB-TBOA
(3S)-3-[[3-[[4-(Trifluoromethyl)benzoyl]amino]phenyl]methoxy]-L-aspartic acid
- TTX
tetrodotoxin
- wt
wild type
References
- Basu AC, Tsai GE, Ma CL, Ehmsen JT, Mustafa AK, Han L, Jiang ZI, Benneyworth MA, Froimowitz MP, Lange N, Snyder SH, Bergeron R, Coyle JT. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry. 2009;14:719–727. doi: 10.1038/mp.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Biedermann B, Francke M, Iandiev I, Grosche J, Wiedemann P, Albrecht J, Reichenbach A. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem Int. 2009;54:143–160. doi: 10.1016/j.neuint.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Daniels BA, Baldridge WH. D-Serine enhancement of NMDA receptor-mediated calcium increases in rat retinal ganglion cells. J Neurochem. 2009 doi: 10.1111/j.1471-4159.2009.06532.x. [DOI] [PubMed] [Google Scholar]
- Duffy S, Labrie V, Roder JC. D-serine augments NMDA-NR2B receptor-dependent hippocampal long-term depression and spatial reversal learning. Neuropsychopharmacology. 2008;33:1004–1018. doi: 10.1038/sj.npp.1301486. [DOI] [PubMed] [Google Scholar]
- Dun Y, Mysona B, Itagaki S, Martin-Studdard A, Ganapathy V, Smith SB. Functional and molecular analysis of D-serine transport in retinal Muller cells. Exp Eye Res. 2007;84:191–199. doi: 10.1016/j.exer.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson EC, Stevens ER, Wolosker H, Miller RF. Endogenous D-serine contributes to NMDA-receptor-mediated light-evoked responses in the vertebrate retina. J Neurophysiol. 2007;98:122–130. doi: 10.1152/jn.00057.2006. [DOI] [PubMed] [Google Scholar]
- Hama Y, Katsuki H, Tochikawa Y, Suminaka C, Kume T, Akaike A. Contribution of endogenous glycine site NMDA agonists to excitotoxic retinal damage in vivo. Neurosci Res. 2006;56:279–285. doi: 10.1016/j.neures.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Hamase K, Konno R, Morikawa A, Zaitsu K. Sensitive determination of D-amino acids in mammals and the effect of D-amino-acid oxidase activity on their amounts. Biol Pharm Bull. 2005;28:1578–1584. doi: 10.1248/bpb.28.1578. [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Nishikawa T, Hayashi T, Fujii N, Harada K, Oka T, Takahashi K. The presence of free D-serine in rat brain. FEBS Lett. 1992;296:33–36. doi: 10.1016/0014-5793(92)80397-y. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Kalbaugh TL, Zhang J, Diamond JS. Coagonist release modulates NMDA receptor subtype contributions at synaptic inputs to retinal ganglion cells. J Neurosci. 2009;29:1469–1479. doi: 10.1523/JNEUROSCI.4240-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. J Biol Chem. 2006;281:14151–14162. doi: 10.1074/jbc.M512927200. [DOI] [PubMed] [Google Scholar]
- Kato S, Ishita S, Sugawara K, Mawatari K. Cystine/glutamate antiporter expression in retinal Muller glial cells: implications for DL-alpha-aminoadipate toxicity. Neuroscience. 1993;57:473–482. doi: 10.1016/0306-4522(93)90080-y. [DOI] [PubMed] [Google Scholar]
- Kim PM, Aizawa H, Kim PS, Huang AS, Wickramasinghe SR, Kashani AH, Barrow RK, Huganir RL, Ghosh A, Snyder SH. Serine racemase: activation by glutamate neurotransmission via glutamate receptor interacting protein and mediation of neuronal migration. Proc Natl Acad Sci U S A. 2005;102:2105–2110. doi: 10.1073/pnas.0409723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner DL, Wilson AL, Drew KL, Green TK. Simultaneous efflux of endogenous D-ser and L-glu from single acute hippocampus slices during oxygen glucose deprivation. J Neurosci Res. 2009;87:2812–2820. doi: 10.1002/jnr.22092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science. 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Konno R, Yasumura Y. Mouse mutant deficient in D-amino acid oxidase activity. Genetics. 1983;103:277–285. doi: 10.1093/genetics/103.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie V, Fukumura R, Rastogi A, Fick LJ, Wang W, Boutros PC, Kennedy JL, Semeralul MO, Lee FH, Baker GB, Belsham DD, Barger SW, Gondo Y, Wong AH, Roder JC. Serine racemase is associated with schizophrenia susceptibility in humans and in a mouse model. Hum Mol Genet. 2009;18:3227–3243. doi: 10.1093/hmg/ddp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Zhong YM, Yang XL. Expression of glycine receptor and transporter on bullfrog retinal Muller cells. Neurosci Lett. 2005;387:75–79. doi: 10.1016/j.neulet.2005.06.023. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Kleckner NW, Wyrick S, Dingledine R. Structural requirements for activation of the glycine coagonist site of N-methyl-D-aspartate receptors expressed in Xenopus oocytes. Mol Pharmacol. 1989;36:556–565. [PubMed] [Google Scholar]
- Miller RF, Dacheux RF, Frumkes TE. Amacrine cells in Necturus retina: evidence for independent gamma-aminobutyric acid- and glycine-releasing neurons. Science. 1977;198:748–750. doi: 10.1126/science.910159. [DOI] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci U S A. 2005;102:5606–5611. doi: 10.1073/pnas.0408483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Ahmad AS, Zeynalov E, Gazi SK, Sikka G, Ehmsen JT, Barrow RK, Coyle JT, Snyder SH, Dore S. Serine racemase deletion protects against cerebral ischemia and excitotoxicity. J Neurosci. 2010;30:1413–1416. doi: 10.1523/JNEUROSCI.4297-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J Neurosci. 2005;25:5502–5510. doi: 10.1523/JNEUROSCI.1354-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien KB, Esguerra M, Miller RF, Bowser MT. Monitoring neurotransmitter release from isolated retinas using online microdialysis-capillary electrophoresis. Anal Chem. 2004;76:5069–5074. doi: 10.1021/ac049822v. [DOI] [PubMed] [Google Scholar]
- O’Brien KB, Miller RF, Bowser MT. D-Serine uptake by isolated retinas is consistent with ASCT-mediated transport. Neurosci Lett. 2005;385:58–63. doi: 10.1016/j.neulet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Pow DV, Hendrickson AE. Expression of glycine and the glycine transporter Glyt-1 in the developing rat retina. Vis Neurosci. 2000;17:1R–9R. [PubMed] [Google Scholar]
- Reed BT, Sullivan SJ, Tsai G, Coyle JT, Esguerra M, Miller RF. The glycine transporter GlyT1 controls N-methyl-D-aspartic acid receptor coagonist occupancy in the mouse retina. Eur J Neurosci. 2009;30:2308–2317. doi: 10.1111/j.1460-9568.2009.07020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg D, Kartvelishvily E, Shleper M, Klinker CM, Bowser MT, Wolosker H. Neuronal release of D-serine: a physiological pathway controlling extracellular D-serine concentration. FASEB J. 2010 doi: 10.1096/fj.09-147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter AR, Fradley RL, Garrett EM, Chapman KL, Lawrence JM, Rosahl TW, Patel S. Evidence from gene knockout studies implicates Asc-1 as the primary transporter mediating d-serine reuptake in the mouse CNS. Eur J Neurosci. 2007;25:1757–1766. doi: 10.1111/j.1460-9568.2007.05446.x. [DOI] [PubMed] [Google Scholar]
- Schell MJ, Brady RO, Jr, Molliver ME, Snyder SH. D-serine as a neuromodulator: regional and developmental localizations in rat brain glia resemble NMDA receptors. J Neurosci. 1997;17:1604–1615. doi: 10.1523/JNEUROSCI.17-05-01604.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci U S A. 1995;92:3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettauf F, Naskar R, Vorwerk CK, Zurakowski D, Dreyer EB. Ganglion cell loss after optic nerve crush mediated through AMPA-kainate and NMDA receptors. Invest Ophthalmol Vis Sci. 2000;41:4313–4316. [PubMed] [Google Scholar]
- Schuettauf F, Quinto K, Naskar R, Zurakowski D. Effects of anti-glaucoma medications on ganglion cell survival: the DBA/2J mouse model. Vision Res. 2002;42:2333–2337. doi: 10.1016/s0042-6989(02)00188-8. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Sakai R, Takaoka K, Yumoto N, Nakajima T, Amara SG, Shigeri Y. Characterization of novel L-threo-beta-benzyloxyaspartate derivatives, potent blockers of the glutamate transporters. Mol Pharmacol. 2004;65:1008–1015. doi: 10.1124/mol.65.4.1008. [DOI] [PubMed] [Google Scholar]
- Stevens ER, Esguerra M, Kim PM, Newman EA, Snyder SH, Zahs KR, Miller RF. D-serine and serine racemase are present in the vertebrate retina and contribute to the physiological activation of NMDA receptors. Proc Natl Acad Sci U S A. 2003;100:6789–6794. doi: 10.1073/pnas.1237052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supplisson S, Bergman C. Control of NMDA receptor activation by a glycine transporter co-expressed in Xenopus oocytes. J Neurosci. 1997;17:4580–4590. doi: 10.1523/JNEUROSCI.17-12-04580.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayasu N, Yoshikawa M, Watanabe M, Tsukamoto H, Suzuki T, Kobayashi H, Noda S. The serine racemase mRNA is expressed in both neurons and glial cells of the rat retina. Arch Histol Cytol. 2008;71:123–129. doi: 10.1679/aohc.71.123. [DOI] [PubMed] [Google Scholar]
- Tsubokawa H, Oguro K, Masuzawa T, Nakaima T, Kawai N. Effects of a spider toxin and its analogue on glutamate-activated currents in the hippocampal CA1 neuron after ischemia. J Neurophysiol. 1995;74:218–225. doi: 10.1152/jn.1995.74.1.218. [DOI] [PubMed] [Google Scholar]
- Verrall L, Burnet PW, Betts JF, Harrison PJ. The neurobiology of D-amino acid oxidase and its involvement in schizophrenia. Mol Psychiatry. 2010;15:122–137. doi: 10.1038/mp.2009.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitanova L. Non-NMDA receptors in frog retina: an immunocytochemical study. Acta Histochem. 2007;109:154–163. doi: 10.1016/j.acthis.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Vorwerk CK, Naskar R, Schuettauf F, Quinto K, Zurakowski D, Gochenauer G, Robinson MB, Mackler SA, Dreyer EB. Depression of retinal glutamate transporter function leads to elevated intravitreal glutamate levels and ganglion cell death. Invest Ophthalmol Vis Sci. 2000;41:3615–3621. [PubMed] [Google Scholar]
- Vorwerk CK, Zurakowski D, McDermott LM, Mawrin C, Dreyer EB. Effects of axonal injury on ganglion cell survival and glutamate homeostasis. Brain Res Bull. 2004;62:485–490. doi: 10.1016/S0361-9230(03)00075-3. [DOI] [PubMed] [Google Scholar]
- Wakakura M, Yamamoto N. Cytosolic calcium transient increase through the AMPA/kainate receptor in cultured Muller cells. Vision Res. 1994;34:1105–1109. doi: 10.1016/0042-6989(94)90293-3. [DOI] [PubMed] [Google Scholar]
- Wang JS, Estevez ME, Cornwall MC, Kefalov VJ. Intra-retinal visual cycle required for rapid and complete cone dark adaptation. Nat Neurosci. 2009;12:295–302. doi: 10.1038/nn.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JS, Kefalov VJ. An alternative pathway mediates the mouse and human cone visual cycle. Curr Biol. 2009;19:1665–1669. doi: 10.1016/j.cub.2009.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolosker H, Sheth KN, Takahashi M, Mothet JP, Brady RO, Jr, Ferris CD, Snyder SH. Purification of serine racemase: biosynthesis of the neuromodulator D-serine. Proc Natl Acad Sci U S A. 1999;96:721–725. doi: 10.1073/pnas.96.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevalk GD, Nicklas WJ. Chemically induced hypoglycemia and anoxia: relationship to glutamate receptor-mediated toxicity in retina. J Pharmacol Exp Ther. 1990;253:1285–1292. [PubMed] [Google Scholar]
- Zhang W, Robert A, Vogensen SB, Howe JR. The relationship between agonist potency and AMPA receptor kinetics. Biophys J. 2006;91:1336–1346. doi: 10.1529/biophysj.106.084426. [DOI] [PMC free article] [PubMed] [Google Scholar]