

Abstract
Background
Completed genome projects have revealed an astonishing diversity of transposable genetic elements, implying the existence of novel element families yet to be discovered from diverse life forms. Concurrently, several better understood transposon systems have been exploited as efficient tools in molecular biology and genomics applications. Characterization of new mobile elements and improvement of the existing transposition technology platforms warrant easy-to-use assays for the quantitative analysis of DNA transposition.
Results
Here we developed a universal in vivo platform for the analysis of transposition frequency with class II mobile elements, i.e., DNA transposons. For each particular transposon system, cloning of the transposon ends and the cognate transposase gene, in three consecutive steps, generates a multifunctional plasmid, which drives inducible expression of the transposase gene and includes a mobilisable lacZ-containing reporter transposon. The assay scores transposition events as blue microcolonies, papillae, growing within otherwise whitish Escherichia coli colonies on indicator plates. We developed the assay using phage Mu transposition as a test model and validated the platform using various MuA transposase mutants. For further validation and to illustrate universality, we introduced IS903 transposition system components into the assay. The developed assay is adjustable to a desired level of initial transposition via the control of a plasmid-borne E. coli arabinose promoter. In practice, the transposition frequency is modulated by varying the concentration of arabinose or glucose in the growth medium. We show that variable levels of transpositional activity can be analysed, thus enabling straightforward screens for hyper- or hypoactive transposase mutants, regardless of the original wild-type activity level.
Conclusions
The established universal papillation assay platform should be widely applicable to a variety of mobile elements. It can be used for mechanistic studies to dissect transposition and provides a means to screen or scrutinise transposase mutants and genes encoding host factors. In succession, improved versions of transposition systems should yield better tools for molecular biology and offer versatile genome modification vehicles for many types of studies, including gene therapy and stem cell research.
Background
Transposable DNA elements constitute a class of discrete genome segments capable of moving from one genomic location to another [1]. These common genome residents are present in all kingdoms of life, being particularly abundant in eukaryotes [1,2], where they cover a sizeable fraction of the genome space (e.g., ~45% in human [3] and nearly 85% in maize [4]). Completed genome sequences demonstrate a wide diversity among mobile DNA and imply the existence of novel transposable element families yet to be discovered from a multitude of diverse life forms [2]. Mobile DNA elements produce genetic variation, supply material for genome innovations (e.g., new genes) and provoke genome instability [5], evidently earning them a highly significant role in the course of evolution.
Transposable elements can be exploited in many types of advanced genetic studies. Typical applications include insertional mutagenesis [6], genome manipulation [7], transgenesis [8], functional genomics studies [9,10], gene therapy [11,12] and generation of induced pluripotent stem cells [12]. Such methodologies are currently under strong development, and it is expected that novel transposition-based applications and new strategies will emerge in the near future.
Transposable elements which move via a DNA intermediate (i.e., class II elements or DNA transposons [1]) are widespread, both in prokaryotes and in eukaryotes. For transposition, they share a common overall reaction mechanism, albeit with some variation in details among different element families [13]. Characteristically, DNA transposons are mobilised by a machinery typically encoded by the elements themselves, and the most critical component is the catalytic protein, a transposase. Initiating transposition, the transposase binds sequence-specifically the transposon ends and, by synapsing the ends as a multimer, assembles a protein-DNA complex called a transpososome. Within the transpososome, transposase catalyses two chemical reactions, donor DNA cleavage and DNA strand transfer, ultimately attaching the transposon DNA to the target DNA. Because of the unity in their reaction mechanisms [13], similar research approaches and analytical methods can be used to study both prokaryotic and eukaryotic DNA transposons.
Bacteriophage Mu uses DNA transposition for propagation and encodes one of the most thoroughly characterised transposition machineries [14]. Despite the complexity of Mu transposition in natural contexts (e.g., certain auxiliary factors involved; see Discussion), a substantially simpler reaction can be performed in vitro. This minimal component reaction requires only a simple reaction buffer and three purified macromolecular components: MuA transposase, mini-Mu transposon DNA and target DNA [15]. The minimal system yields transposition products highly efficiently and with low target site selectivity [15,16]. In general, these properties make the Mu reaction ideal for a variety of advanced molecular biology [15,17-19], protein engineering [20-22] and genomics [9,23] applications. With an addition of an in vivo step, the minimal system can also be used for efficient gene delivery in bacteria, yeast and mammalian cells [24-26].
Various methods have been developed in the past for the in vivo analysis of transposition frequency, with typical examples including mating-out and phage assays [27,28], but these methods are not ideal for large-scale studies. Currently, the most widely used methods to study transposition in vivo exploit coloured microcolonies (papillae), growing within otherwise colourless bacterial colonies. Initially, such papillation assays were designed to score events involving transposon excision from a specific gene locus [29]. More recently, most papillation assays have exploited a reporter transposon, typically including lacZ as a marker gene. Insertion of the reporter transposon into a genomic locus under the control of an active promoter would generate a gene fusion, facilitating the expression of the lacZ gene, detectable as coloured Lac+ papillae on colourless Lac- colonies. The number of papillae per colony is proportional to the frequency of transposition events catalysed by the transposase. Useable papillation assays are currently available for a number of transposons, and typical examples include the assays for Tn5 [30], Tn7 [31], Tn10 [32], IS903 [33] and Mu [34]. All present-day papillation assays are characteristically element-specific, many of them lack adjustability and some are complicated by the use of several plasmids. Clearly, there exists a need for further methodology development, and a papillation assay that could eliminate the above limitations would be beneficial.
An increasing number of transposon systems have recently been adapted for advanced molecular biology and genomics applications, and their modification for better performance is under development. Concurrently, owing to genome projects, new transposons are being discovered essentially on a daily basis. Both improvement of the existing transposition applications and characterization of new transposition systems require robust quantitative assays. In this report, we describe a general in vivo platform for the quantitative analysis of transposition that should be applicable to a variety of mobile DNA elements.
Results
A universal in vivo assay that could be used for the quantitative analysis of mobile DNA activity in various transposition systems would be a valuable tool for future transposon studies and application development. With this goal, we set up a platform that is based on the transformation of E. coli with a single plasmid driving transposon mobilization (Figure 1). The plasmid is engineered to enable straightforward construction of a reporter transposon for any specific DNA transposon system, and it contains a controllable unit for the inducible expression of the cognate transposase protein. The platform provides a visual read-out with blue microcolonies (papillae) growing within otherwise whitish bacterial colonies and allows the adjustment of the transposition frequency to a quantifiable range. As a test system for the platform development, we utilized bacteriophage Mu transposition. For further assessment of the assay and for the demonstration of its universality, we additionally employed IS903 transposition.
Figure 1.
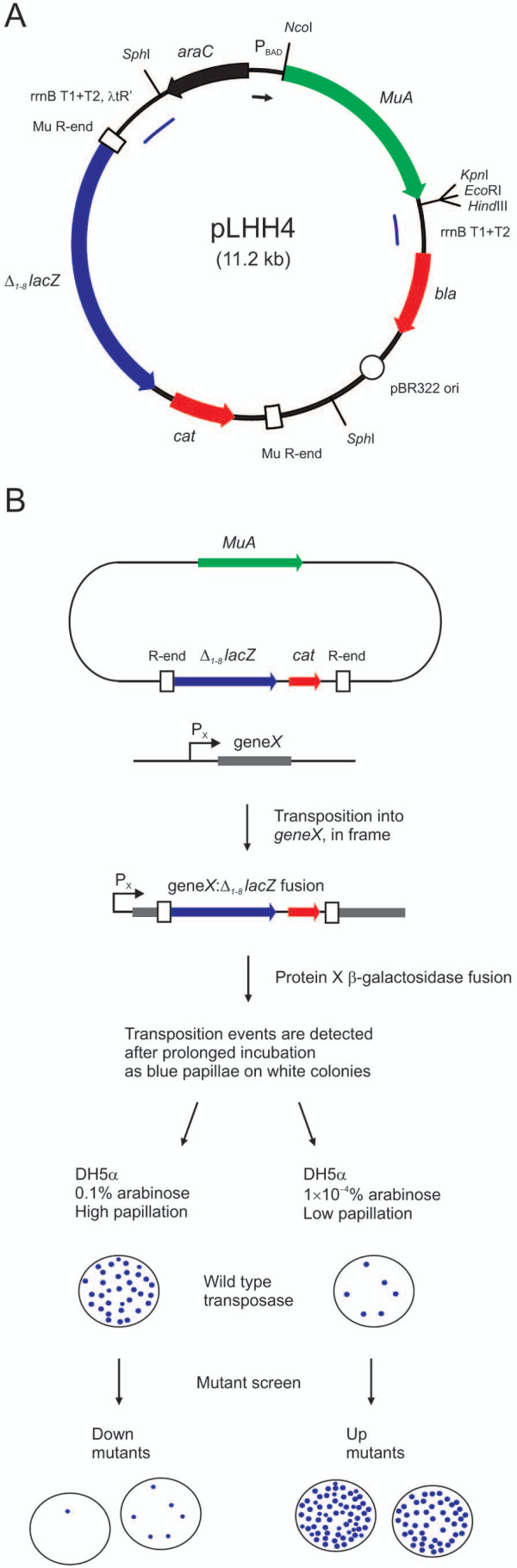
Adjustable in vivo papillation assay. (a) Papillation assay plasmid pLHH4. This plasmid contains the MuA transposase-encoding gene under the arabinose-inducible E. coli PBAD promoter, and the mobilisable mini-Mu reporter transposon Δ1-8LacZ-Cat-Mu. (b) Generation of papillae. The papillation assay plasmid is transferred into a phenotypically Lac- E. coli strain. Expression of the transposase protein is induced by arabinose, mobilising the reporter transposon. Transposition of the element into an expressed gene (geneX) in the correct orientation and reading frame will form a geneX::Δ1-8lacZ gene fusion, and a β-galactosidase fusion protein will be expressed. Such transposition events can be detected on indicator plates after prolonged incubation as blue microcolonies on otherwise whitish colonies. The level of transposition can be adjusted by varying the transposase expression via changes in arabinose or glucose concentration.
Papillation assay
Initially, we constructed the papillation assay plasmid pLHH4 (Figure 1a), which encodes the phage Mu transposase and, in addition, contains a mobilisable mini-Mu reporter transposon. This pBR322-derived plasmid exhibits medium copy number and encodes β-lactamase as a standard selectable marker. The plasmid also includes the arabinose/glucose-controllable PBAD promoter of E. coli, driving the inducible expression of MuA transposase. The expression unit is bordered by strong transcription terminators to prevent surplus gene expression from sequences flanking the unit. The mini-Mu transposon (Δ1-8LacZ-Cat-Mu) contains, as an inverted repeat, a 50-bp segment of Mu R-end DNA in each of the transposon ends. As a reporter gene, the transposon contains a promoterless, 5'-terminally truncated E. coli lacZ gene (Δ1-8lacZ), lacking the codons for amino acids 1-8. The transposon also includes the cat gene for chloramphenicol selection.
The papillation assay for Mu transposition takes advantage of the introduction of the plasmid pLHH4 into a phenotypically Lac- E. coli strain, and the plasmid facilitates the reporter transposon mobilisation upon sufficient transposase expression. Successful transposition into an expressed chromosomal gene in the appropriate orientation and reading frame gives rise to lacZ gene fusions. These events can be detected as blue papillae on a colony grown on an indicator plate containing X-gal, with each papilla representing an independent transposition event (Figure 1b).
Colony phenotypes and papillation characteristics of different E. coli strains
We evaluated three E. coli strains: DH10B, DH5α and JM109 (Additional file 1) with regard to their suitability for the papillation assay. These standard laboratory strains are phenotypically Lac-, and they grow as whitish colonies on X-gal indicator plates. As a low-papillation-frequency control, we also included strain HT321 in the evaluation. This strain harbours a mutation in the pcnB locus, resulting in a reduced plasmid copy number [35], and has previously been used to identify host mutations that increase transposition frequency in IS903, Tn10 and Tn552 systems [33]. Plasmid pLHH4 was transformed into the cells, and colonies were grown for a 115-h reference time period on a standard papillation medium with varying arabinose concentrations (Table 1). The critical characteristics evaluated were: (1) colony size, because large colonies would potentiate a broad dynamic range for the assay; (2) the number of papillae generated should be adjustable by arabinose over a wide range of concentrations; (3) a reasonable maximum transposition frequency should be attained; (4) clearly defined papillae should be evenly distributed on individual colonies; and (5) the number of the papillae per colony should not be overly influenced by the number of growing bacterial colonies on a given plate.
Table 1.
Colony phenotypes and papillation characteristics in different E. coli strains
| Strain | Colony sizea | Adjustabilitywith arabinose | Maximum frequencyof transpositionb | Distribution ofpapillae on colonies | Papillae number dependency on the number of growing colonies per platec |
|---|---|---|---|---|---|
| DH10B | Large | Limited | High (up to ~100 papillae) | Centered in the middle | Somewhat dependent |
| DH5α | Large | Good | Highest (up to ~300 papillae) | Even | Largely independent |
| JM109 | Medium | Good | Intermediate (up to ~40 papillae) | Even | NDd |
| HT321 | Large | Moderate | Low (up to ~10 papillae) | Even | ND |
aLarge colony size: up to ~5 mm of diameter. Medium colony size: up to ~3 mm of diameter.
bThe maximum frequency of transposition is specified in this table as the number of papillae per colony grown at 30°C for 115 h on a standard papillation medium containing 0.1% arabinose.
cSee Additional file 2.
dND, not determined.
Strain DH10B formed large colonies with a convex center and generated a relatively high maximum number of papillae. The number of papillae generated responded to the increase in the arabinose concentration, but not linearly, and two maxima were observed (at 0.001 and 1% arabinose concentrations; data not shown). The reason for this somewhat unexpected behaviour is not known but might possibly point to unknown alterations in the arabinose transporter systems (see Discussion). Most papillae were distributed in the center of the colony, and the number of papillae on even-sized colonies was somewhat influenced by the number of growing colonies per plate (Additional file 2).
Strain DH5α formed large flat colonies, and the number of papillae correlated well with the arabinose concentration in the growth medium. A high maximum number of clearly defined papillae formed throughout a colony. The number of growing colonies per plate did not overly influence the frequency of transposition. Particularly within the range of 40 to 200 colonies per plate, papillae formed with a very similar rate (Additional file 2).
Strain JM109 generated medium-size, irregularly shaped colonies. The number of papillae on a colony was proportional to the arabinose concentration used, reaching the level of ~40 papillae per colony upon 115-h incubation. Up to ~120 papillae per colony formed with a prolonged (164 h) incubation time (data not shown). Clearly defined papillae formed throughout a colony, but curiously, ~7% of the colonies appeared completely white, thus exhibiting Lac- phenotype; the same clones were also proline auxotrophs (data not shown). These data point to the loss of the resident F'-episome, which in this strain encodes the critical lactose utilization and proline biosynthesis proteins.
The control strain HT321 generated large colonies and with the 115-h reference incubation time exhibited a low transposition frequency (up to ~10 papillae per colony), too low to unambiguously evaluate arabinose responsiveness. However, with prolonged incubation time (164 h), up to ~30 papillae per colony could be observed, and arabinose dependency was apparent (data not shown).
The effect of temperature on transposition frequency
Strain DH5α appeared ideal for the further characterization of the papillation assay. We next examined the effect of growth temperature on papillation. DH5α cells carrying the plasmid pLHH4 were grown on papillation plates in standard growth conditions (0.1% arabinose) at four different temperatures: 22°C (room temperature), 25°C, 30°C and 37°C. Emerging papillae were enumerated as a function of incubation time (Figure 2). At each temperature, visible papillae began to appear on colonies after a prolonged incubation period, and the growth rate of the bacteria apparently dictated the time course of the appearance. The two lower temperatures produced a modest number of papillae with a plateau in their appearance. In contrast, the two higher temperatures generated papillae in increasing numbers. Yielding a measure for the maximum dynamic range of the assay, we were able to enumerate papillae up to a time point where ~700 discernible blue spots per colony were visible; thereafter the papillae fused, and the entire colony turned blue. At 30°C and 37°C, papillae formation appeared linear between the time points ~100-150 h and ~50-80 h, respectively. For further studies, we chose to use incubation at 30°C, as at this temperature papillae accumulated more slowly, allowing a more convenient and accurate papillae enumeration.
Figure 2.
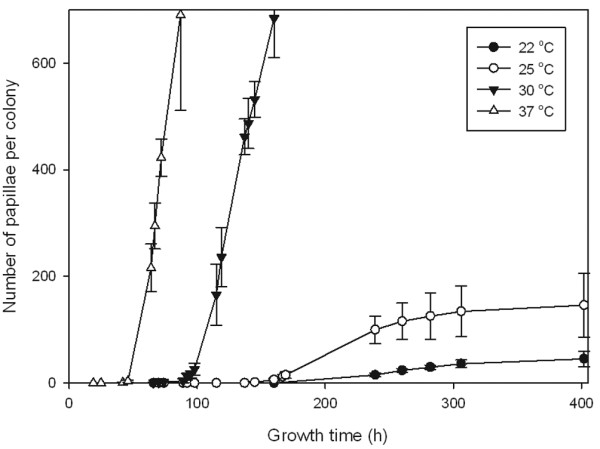
The effect of temperature on transposition frequency depending on temperature. The arabinose content in the growth medium was 0.1%. The transposition frequency at 37°C is shown with an open triangle, at 30°C with a filled triangle, at 25°C with an open circle and at room temperature (22°C) with a filled circle, respectively. Papillae were enumerated from six colonies for each data point. The error bars indicate SD above and below the average value for each data point.
MuA expression and transposition frequency
The expression level of MuA transposase should influence the transposition frequency measurable by the papillation assay. We therefore studied the MuA expression level and transposition frequency under variable inducer concentrations. The former was studied using liquid cultures and the latter, in a separate experiment, using papillation analysis on plate cultures.
The initial MuA expression analysis was done with whole-cell lysates of liquid cultures grown under variable arabinose concentrations. SDS-PAGE and Western blotting were used to reveal population-average protein expression levels of MuA (Figure 3a). The data indicated that MuA expression was increased by the increase in the arabinose concentration up to 1 × 10-2%, and the induction was detectable already at 5 × 10-4%. Altogether, the results demonstrated that the inducible expression system was functional and adjustable over a large range of arabinose concentrations. The apparent reduction in MuA expression at greater than 1 × 10-2% arabinose concentrations in this experiment is difficult to explain, but it may conceivably relate to MuA overexpression possibly causing harmful effects on various cellular functions and ultimately resulting in differences in the growth rate.
Figure 3.

The effect of growth conditions on transposition frequency. (a) Western blot analysis of MuA expression levels at varying arabinose concentrations. Whole cell lysates from E. coli DH5α cells containing plasmid pLHH4 were analysed for MuA content using polyclonal rabbit anti-MuA antibody. The asterisk indicates MuA. (b) The effect of arabinose concentration on colony size and papillation. Wild-type MuA-expressing plasmid (pLHH4) was used for papillation analysis with E. coli DH5α as a plasmid recipient strain. Open and filled circles indicate the colony size and the number of papillae per colony, respectively. Papillae were enumerated from six colonies for each data point. The error bars indicate SD above and below the average value for each data point.
Transposition frequency was subsequently analysed under variable arabinose concentrations by the papillation assay (Figure 3b). Papillae began to appear at ~1 × 10-4% arabinose concentration, and the highest number of discernible papillae was obtained at 1 × 10-1% concentration. Higher arabinose concentrations apparently affected cell survival significantly, as colonies were smaller (Figure 3b) and ragged (data not shown). Furthermore, at these concentrations, the papillae were fuzzy with diffuse colour and impossible to enumerate. These data indicated that measurable papillation could be induced over a wide range of arabinose concentrations.
In general, the MuA expression levels observed from liquid cultures correlated well with the papillation data obtained from plate cultures. However, some discrepancy was seen at 1 × 10-1% arabinose concentration (Figure 3a and 3b). This latter result likely reflects key differences between the analyses, liquid versus plate cultures. In liquid cultures, the ambient arabinose concentration should be equal for all cells, and this is not necessarily the case for cells growing as colonies on agar plates. With regard to the arabinose concentration, the microenvironment within a colony on a plate may vary for each individual cell. Furthermore, the actual arabinose concentration experienced by the cells within a colony conceivably is, in general, somewhat lower than that in the medium because of diffusion characteristics and colony morphology.
The effect of MuA activity on transposition frequency
The suitability of the papillation assay for the quantitative comparison of variable transposition frequencies was examined using several MuA variants (Figure 4a). We used an N-terminally deleted MuA77-663 as well as C-terminally deleted variant MuA1-615, and in addition, a variant missing both termini MuA77-615. We also used a MuA variant with a substitution in the catalytically important active site DDE-triad (MuAE392Q), a protein that is proficient for the transpososome assembly but catalytically defective [36]. Standard papillation conditions with 115-h incubation time were used in these experiments.
Figure 4.
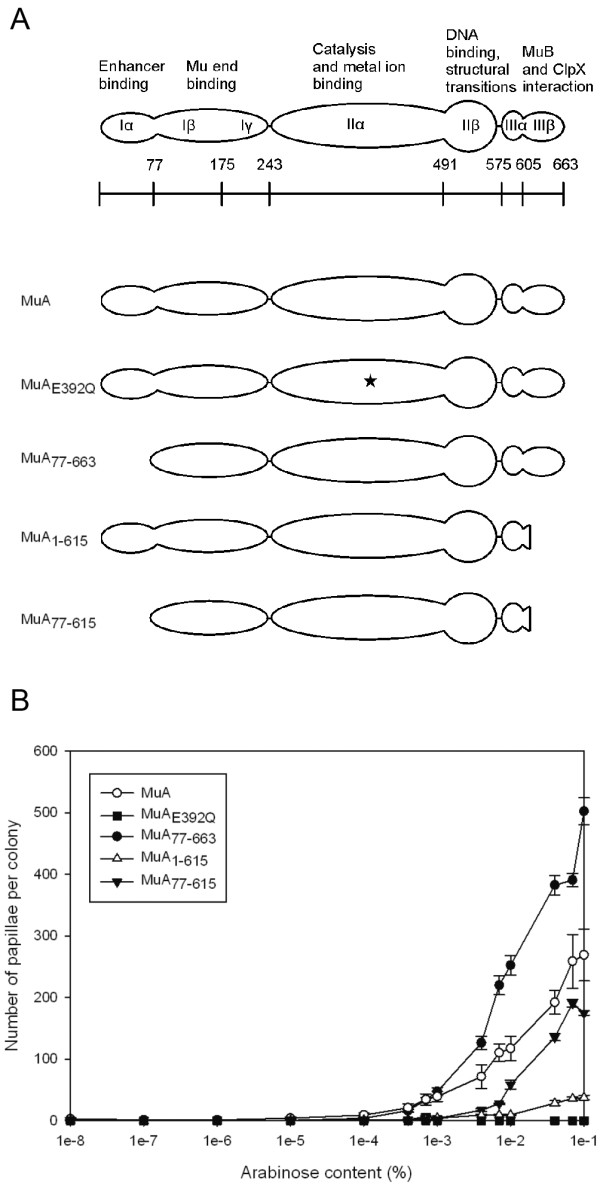
Papillation analysis of different MuA variants at varying arabinose concentrations. (a) MuA proteins used in papillation analysis. Structural organisation of MuA with different functions assigned to various domains [14]. Amino acid numbers corresponding to the amino terminus of each subdomain are shown beneath the structure. (b) In vivo transposition frequency induced by MuA variants. The transposition frequency of MuA77-663 is shown with a filled circle, wild-type MuA with an open circle, MuA77-615 with a filled triangle, MuA1-615 with an open triangle and MuAE392Q with a filled square, respectively. Papillae were enumerated from a total of three to six colonies for each data point. The error bars indicate SD/2 above and below the average value for each data point.
The wild-type MuA and all of the deletion variants studied produced papillae and responded to the increase in the arabinose concentration (Figure 4b and Figure 5). The wild-type protein produced ~300 papillae per colony at the highest arabinose concentration tested (0.1%). Both MuA1-615 and MuA77-615 produced significantly less papillae with maximum yields of ~50 and ~200 papillae, respectively, indicating hypoactivity. MuA77-663 maximally gave rise to ~500 papillae and thus appeared hyperactive. The active site mutant MuAE392Q did not produce papillae in the assay, most probably directly reflecting its reported deficiency in catalysis. Similarly, no papillae were generated when a pLHH4 derivative (pTLH1), missing the MuA gene but retaining the Mu ends, was assayed as a control (data not shown), highlighting the specificity of the system. These results indicate that variable transposition activity levels can be quantified reliably, demonstrating the suitability of the papillation assay to screen transposase mutants with altered activities. The data also show that the transposition frequency can be fine-tuned to a desired initial level for screening purposes by the adjustment of the arabinose concentration in the medium.
Figure 5.

Colonies from papillation assay. Papillation of MuA variants in DH5α at varying arabinose concentrations.
We include a time-lapsed video of growing colonies from a papillation analysis (Additional file 3), which highlights graphically the characteristics the developed assay. The video was recorded using papillation conditions where wild-type MuA produced only a few papillae (1 × 10-4% arabinose). Using these assay conditions, we have recently identified a number of hyperactive MuA variants, and a comprehensive analysis of critical amino acid changes involved in their hyperactivity phenotype will be described in a future report.
Universal plasmid for papillation analysis
We next modified the papillation assay plasmid pLHH4 to be useful with other DNA transposons. In this modified plasmid, pSKT1 (Additional file 4), the Mu R-ends have been replaced by polylinkers, allowing a straightforward directional cloning of heterologous transposon ends. Similarly, the MuA transposase gene is also replaced by a polylinker, facilitating directional cloning of a heterologous transposase gene. The functionality of the universal system was then confirmed as follows: (1) Mu R-ends were recloned into the appropriate newly made linker sites and tested in the context of wild-type MuA; a similar number of papillae were generated, as was observed with the original papillation assay plasmid pLHH4 (data not shown). (2) When the Mu R-ends were omitted, no papillae were produced, further illustrating the specificity of the assay system (similar to the omission of the MuA transposase gene discussed above). (3) Finally, the components of the IS903 transposition system were introduced into pSKT1, and the ensuing plasmid pSKT4 (Additional file 5) was analysed for transpositional activity (results described in the next subsection).
Analysis of IS903 transposition
Plasmid pSKT4, containing the critical components of the IS903 transposition system, was introduced into DH5α cells and analysed for papillation with variable concentrations of arabinose in the growth medium. All tested concentrations yielded papillae, and somewhat surprisingly, even the plates which did not contain arabinose produced ~200 papillae per colony (data not shown), indicating that IS903 transposase was expressed at levels sufficient for transposition. We next tested whether glucose, conceivably via catabolite repression, could be used to tighten the control of transposase expression, which in turn would result in reduced levels of papillation. A constant number of ~200 papillae were produced with glucose concentrations up to 0.15% (data not shown). However, higher concentrations of glucose reduced the number of papillae essentially linearly, and upon 0.25% concentration, no papillae were produced (Figure 6). Thus, in cases where uninduced transposase expression results in transposition frequency that is too high for screening purposes, glucose can be used to fine-tune papillae formation to a desirable level.
Figure 6.
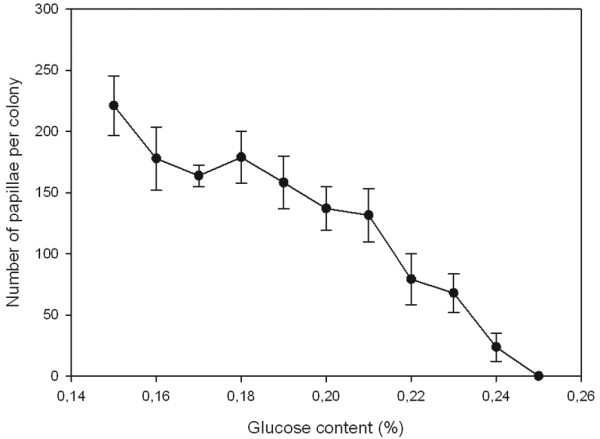
Papillation analysis of IS903 transposition. Plasmid pSKT4 was introduced DH5α cells for papillation at varying glucose concentrations. Papillae were enumerated from six colonies for each data point. The error bars indicate SD above and below the average value for each data point.
Discussion
Papillation assay systems have provided straightforward means to dissect transposition events in vivo. For example, they have enabled simultaneous activity analyses of tens of thousands of randomly generated transposase mutants [31,37-40]. Furthermore, such assays have successfully been used to study the effects of mutations in transposon end sequences [30,34,41-43] and to examine the influence of host factors [33]. Although each particular assay system has performed adequately in the context in which it has been used, in many cases the adjustability has been limited. Furthermore, many of the assays have included a reporter transposon and the cognate transposase gene separately in different plasmids, which is not an ideal arrangement for many applications.
The system we describe provides certain advantages. First, the assay is based on an easy-to-use single plasmid, and it offers a straightforward means to visually screen mutant versions of transposases as well as potentiates the discovery of host factors affecting transposition. Second, the inclusion of two antibiotic marker genes in the assay plasmid, bla and cat, enables double antibiotic selection, which effectively prevents the growth of satellite colonies on plates during the long incubation period needed for papillation. Note that the cat marker gene within the reporter transposon can also be used for the selection of transposition events, a useful feature in prospective downstream applications such as mating-out assays. Third, the transposition frequency is easily adjustable, which potentiates many types of studies under a desired optimal rate of transposition.
The expression system used in our assay, the arabinose-controllable induction via PBAD promoter, has been studied in detail previously [44]. The study revealed that with standard E. coli strains harbouring native arabinose transporter systems araE and araFGH, the arabinose-inducible PBAD promoter is subject to essentially all-or-none induction via a regulatory feedback loop, in which limiting arabinose concentrations give rise to subpopulations of cells that are either highly induced or uninduced [44]. Raising the arabinose concentration increases the fraction of highly induced cells until a saturating concentration is reached, under which concentration essentially all of the cells in the population become highly induced [44]. Nevertheless, even under these saturating arabinose concentrations, a degree of variation exists among the expression levels of individual cells [44]. Considering the above features, it was expected that in our assay system, only a fraction of cells would express the transposase within any given colony under subsaturating arabinose concentrations, and those cells expressing the transposase would be the ones which ultimately produced the observed papillae. Further considering the above features, although all-or-none induction should generate two subpopulations of cells, a degree of transposase expression variation should still be present within these subpopulations in both subsaturating and saturating arabinose concentrations. Overall, our results have shown that good control of population level average transposase expression can be achieved using the PBAD promoter, at least under subsaturating arabinose concentrations. Under these conditions, the transposase expression levels correlate very well with the formation of papillae, forming a basis for a dependable quantitative assay.
The omission of arabinose on selection plates effectively eliminated transposition in the Mu system, but identical conditions resulted in a reasonable level of transposition in the IS903 system. These data indicate that the PBAD control unit of transposase expression in the papillation assay plasmid allows some basal protein expression. As it is likely that similar expression levels would be achieved independently of the expressed protein, the observed differences between Mu and IS903 most probably reflect differences in the inherent activities of the two systems (in the conditions tested) rather than differences in the transposase expression levels. The observed activity differences are necessarily not surprising, because the analysed Mu system is somewhat aberrant (unnatural transposon end configuration, lack of transposition enhancer, lack of MuB protein), whereas the analysed IS903 transposition represents a wild-type system.
We examined several E. coli strains for their suitability in the papillation assay. From these studies, it became evident that a number of critical attributes affecting the assay performance were strain-dependent, evidently reflecting known (plus possibly unknown) genotypic differences, and by all criteria DH5α appeared optimal for the assay. First, the large colony size and the even distribution pattern of papillae within a colony allowed scoring of more than 500 discernible papillae. Second, the adjustability of the assay spans a wide range of arabinose concentrations. Together, these attributes generated a large dynamic range for the assay, and the use of glucose extended the adjustability even further. Third, the colony density on a plate did not influence papillation appreciably, in practice meaning that reliable results could be obtained from standard plates (diameter 9 cm) containing ~50-200 growing colonies.
Several MuA variants were scrutinised for their transpositional activity to test the assay under different levels of transposase activity. Overall, the results showed that activities above and below a certain (in this case, wild-type MuA) activity level could reliably be analysed, potentiating the screen for hyperactive and hypoactive mutants (Figure 4b and 5). MuA77-663, missing the Iα domain [45], has been shown to be hyperactive in certain in vitro assays [46]. Similar hyperactivity was also seen in its proficiency to produce papillae. MuA1-615, lacking the C-terminal 48 amino acids [47], retains a wild-type level of activity in certain in vitro assays [47,48]. However, in our in vivo analysis, it was clearly hypoactive. The details behind the apparent discrepancy are unknown but may reflect the deficiency in the disassembly of the transpososome via ClpX-mediated interaction, a property confined to the C-terminus of MuA [14]. MuA77-615, having both the Iα domain and the C-terminal residues deleted, retained substantial activity. Thus, the hyperactivity-causing property of the domain Iα deletion appeared to compensate for the activity reduction caused by the deletion in the C-terminus.
Mu transposition can be reproduced in vitro using as sole components MuA transposase, transposons that contain 50 bp of Mu R-end in each transposon terminus, and target DNA. This minimal reaction forms the basis of the Mu in vitro transposition technology [15]. With an overall aim to improve the technology, we designed our assay system to mimic this minimal in vitro transposition reaction as closely as possible. Accordingly, we used a reporter transposon which contains two R-ends and did not include in the system the known auxiliary factors of Mu transposition, such as the transposition enhancer (IAS) and activator/targeting protein MuB [14]. Our results demonstrate that Mu transposition in vivo can proceed efficiently independent of the natural L/R (left/right) end configuration and in the absence of the above auxiliary factors. While a massive number of Mu studies have employed the natural L/R end combination for studies in vivo, significantly less in vivo data are available with regard the L/L or R/R end configurations. Nevertheless, these configurations have been shown to promote transposition in vivo, albeit with reduced efficiency, and to generate correct junctions between the Mu DNA and host DNA (unpublished data in [49]). Falling outside the scope of the current study, we did not analyse the effects on transposition of the known E. coli host factors, HU and IHF. Nevertheless, it would be interesting to see whether these two proteins were dispensable under the described in vivo conditions as well.
The papillation assay platform should be applicable to a number of transposition systems. For cut-and-paste type transposons, papillation assays generate gene fusions by the initial detachment of the transposon from the donor site, followed by the integration of the transposon into another locus. Thus, the mechanistic basis of the assay for cut-and-paste transposons is relatively simple. However, Mu and IS903 primarily use the replicative mode of transposition, a somewhat more complicated mechanism. During replicative transposition, a branched transposition intermediate (a/k/a Shapiro intermediate) is formed. This intermediate can be processed either by DNA replication or by DNA repair, and it is known that Mu uses both of these pathways for processing [50]. However, in this work, we have not studied the mechanistic details of the assay. Recent findings indicate that several pathways may be involved in the processing of the Shapiro intermediate [50], which necessarily would complicate such studies. As retroviruses and LTR retrotransposons catalyse a mechanistically related (transposition) reaction called integration [1], it might be possible to apply the assay platform also for them. However, in these cases, the interplay between the integrase protein and other proteins of the integration machinery may complicate the development of a useable strategy.
Conclusions
We have established a papillation assay platform which is easily modifiable for a variety of transposon systems. It provides an efficient tool for the studies of yet unknown transposons and yields means to screen and scrutinise transposase mutants as well as genes encoding critical host factors involved in transposition. Although in this paper we used the papillation assay with two prokaryotic transposons, the system may equally be applied to eukaryotic DNA transposons as well. It is hoped that the use of the developed platform will generate better versions of transposition systems, ultimately producing better tools for molecular biology and offering versatile genome modification vehicles for a variety of future studies. These investigations are expected to involve disciplines such as cancer biology, gene therapy, stem cell research and studies on transgenesis.
Methods
Bacteria, plasmids and oligonucleotides
Escherichia coli strains are listed in Additional file 1. Plasmids and oligonucleotides are described in Additional file 6 and Additional file 7, respectively.
Reagents, enzymes and DNA techniques
Ampicillin, chloramphenicol, arabinose and glucose were from Sigma (St. Louis, MO, USA). Lactose was from BDH/VWR International (West Chester, PA, USA) and 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) was from AppliChem GmbH (Darmstadt, Germany). Restriction endonucleases and T4 DNA ligase were from New England BioLabs (Ipswich, MA, USA). Calf intestinal phosphatase (CIP) was from Finnzymes (Espoo, Finland). Standard DNA techniques were performed as described previously [51]. Plasmids were propagated in E. coli DH5α and were isolated using appropriate Qiagen (Hilden, Germany) kits. Electrocompetent DH5α cells were prepared as described previously [24] and used for standard cloning procedures.
Construction of the papillation assay plasmid expressing wild-type MuA transposase
The papillation assay plasmid pLHH4 (Figure 1a), expressing wild-type MuA transposase (GenBank accession number P07636) was generated as follows: (1) A truncated version of the lacZ gene, encoding E. coli β-galactosidase (EC 3.2.1.23) but missing the nucleotides specifying the amino acids 1-8 (Δ1-8lacZ), was amplified by polymerase chain reaction (PCR) from plasmid pNT105 using the primer pair HSP173/HSP408. The generated PCR fragment was trimmed with BamHI and subsequently cloned into plasmid pSupF-Mu between the two BamHI sites to replace the supF gene, yielding plasmid pLHH1. (2) The cat gene was amplified by PCR from plasmid pBC SK(+) using the primer pair HSP360/HSP361. The generated PCR fragment was trimmed with BamHI and cloned downstream of Δ1-8lacZ into BamHI-linearized pLHH1 (partial digestion), yielding plasmid pLHH2. Prior to cloning, the cat gene was modified by introducing, via overlap PCR with appropriate primers, a silent mutation (codon 177, Thr, ACC→ACT) to eliminate a critical NcoI site. (3) An SphI-fragment containing the tandem E. coli rrnB terminators T1+T2 [52] and phage λ tR' terminator [53] as well as a critical BamHI site for cloning was generated by standard DNA techniques including PCR and annealing of oligonucleotides (see Additional file 7). (4) The above terminator-containing SphI-fragment was then cloned into SphI-digested pALH6 to generate pLHH3. (5) Finally, the mini-Mu transposon of pLHH2 was released with BglII and cloned into BamHI-linearised pLHH3 (partial digestion) to generate pLHH4.
MuA variant-expressing papillation assay plasmids
Initially, plasmid pTLH1 was generated by cloning the 5.2-kb SphI reporter-containing fragment of pLHH4 into SphI-digested pBADHisA, which substituted the MuA gene with the polylinker of pBADHisA. PCR-generated MuA deletion variants (pMPH14, pMPH17, pMPH18; see Additional files 6 and 7) were subsequently cloned between the NcoI and XhoI sites of pTLH1. The plasmid expressing MuAE392Q (pLHH12) was generated by cloning the NcoI-BamHI fragment of pMK616 into pTLH1 digested with NcoI and BglII.
Construction of universal papillation assay plasmid
Initially, the reporter region of pLHH2 containing the Δ1-8lacZ and cat genes was amplified by PCR using the linker-sequence-tailed primers HSP685 and HSP689 (see Additional file 7). The generated PCR fragment was trimmed with BglII and cloned into BamHI-linearized pLHH3 (partial digestion), yielding plasmid pMPH23B. Finally, the universal papillation assay plasmid pSKT1 (Additional file 4) was obtained by replacing the MuA-containing NcoI-ScaI fragment of pMPH23B with the polylinker-containing NcoI-ScaI fragment of pBADHisA. Plasmid pSKT1 can be applied to establish a papillation assay for various transposon systems in three straightforward cloning steps involving transposon ends and a cognate transposase gene.
Construction of papillation assay plasmid for IS903
The IS903 transposase gene was amplified by PCR from plasmid pNT105 using the primer pair HSP693/HSP694. The PCR fragment was trimmed with NcoI and XhoI and cloned into plasmid pSKT1 digested with the same two enzymes, yielding plasmid pSKT2. The ends of IS903 were then inserted as follows. First, oligonucleotides HSP690 and HSP691 were annealed and cloned into pSKT2 digested with SpeI and NotI, resulting in plasmid pSKT3. Second, oligonucleotides HSP690 and HSP692 were annealed and cloned into pSKT3, digested initially with XbaI and then partially with XmaI, yielding the IS903 papillation assay plasmid pSKT4.
Papillation assay
Each papillation assay plasmid was transformed into competent E. coli cells prepared and used as described previously [54]. In a standard assay, cells were plated at approximately 100 c.f.u. (colony forming units) per plate. Standard plates contained LB medium [51] supplemented with 1.5% Bacto-agar (Difco/Becton Dickinson and Company, Sparks, MD, USA), 100 μg/ml ampicillin (Ap), 20 μg/ml chloramphenicol (Cm), 0.05% lactose, 40 μg/ml X-gal, and 0.1% arabinose. For the experiments with IS903 transposition, glucose replaced arabinose for the adjustment of the transposase expression. In standard assays, the plates were incubated at 30°C for 115 h. To study the suitability of different E. coli strains, the effect of arabinose concentration, as well as growth temperature and time, were varied. The effect on papillation of the number of growing colonies per plate was studied by plating cells to produce different colony densities. For each data point, three to six representative colonies were photographed using an Olympus (Tokyo, Japan) ColorView II digital camera attached to an Olympus SZX12 stereomicroscope equipped with Zeiss (Oberkochen, Germany) KL1500 LCD cold light source. The size of the colonies was measured, and the number of papillae in each colony was enumerated manually by the use of AnalySIS software (Soft Imaging System, Olympus). The same imaging system was used for the time-lapsed video recording (Additional file 3).
Western blotting
The expression of wild-type MuA protein at different arabinose concentrations was determined by analysing whole-cell lysates using sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). For protein expression, E. coli DH5α(pLHH4) cells were grown in LB-Ap-Cm medium (5 ml) at 37°C to an OD600 of 0.5, and MuA expression was induced by the addition of arabinose. Following incubation at 28°C for 2 h, cells from 0.5 ml of the culture were pelleted and resuspended in 150 μl of 1× SB loading buffer (100 mM Tris·HCl, pH 6.8, 200 mM dithiothreitol, 4% SDS, 0.2% bromophenol blue, 20% wt/wt glycerol). The proteins (5 μl of 1:8 diluted sample in 1× SB) were separated on a 10% SDS-PAGE gel and blotted onto Immobilon™P polyvinylidene difluoride membrane (Millipore, Billerica, MD, USA) using the Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, Hercules, CA, USA), according to the manufacturer's instructions. The membrane was blocked at room temperature overnight using 5% skim milk powder dissolved in phosphate-buffered saline (PBS) buffer [51]. Following a wash with PBS, the membrane was probed with the primary rabbit anti-MuA antibody (1:1,000 dilution) and subsequently with the secondary horseradish peroxidase-conjugated anti-rabbit (from donkey; Amersham Biosciences/GE Healthcare, Buckinghamshire, United Kingdom) Ig antibody (1:10,000 dilution). The bound peroxidase was detected using the ECL Plus Western Blotting System (Amersham Biosciences/GE Healthcare,) as specified by the supplier. Immunoreactive bands were visualized using a Fujifilm LAS-3000 system (Fujifilm Corporation, Tokyo, Japan).
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MIP, TSR, LJH, AL, SH-P and SK carried out the experiments. MIP, AL, SH-P and HS conceived the experiments. MIP, TSR, SK and HS wrote the manuscript.
Supplementary Material
Supp. Table 1. E. coli strains. E. coli strains used in the work.
Supp. Fig. 1. Dependency of papillae number per colony on the number of growing colonies. Bacterial colonies were grown at different densities (upper left corner) on a standard 9-cm plate. Colony size and the corresponding number of papillae were determined. (a) Strain DH10B. (b) Strain DH5α.
Supp. Video 1. Time-lapsed video of papillation assay. Shown are bacterial colonies growing on a standard papillation medium containing 1 × 10-4 % arabinose. The standard X-gal concentration was increased to 120 μg/ml to enhance contrast. The plates were incubated initially at 25°C for 46.5 h in an unilluminated chamber. Subsequently, for recording, the plates were transferred under a microscope with an artificial LCD light source. The recording was done at ~22°C with 1-h time lapse intervals for 6 days (144 h). The colonies shown represent wild-type MuA (Wild type) and MuA63-663 (Del-N).
Supp. Fig. 2. Universal papillation assay plasmid pSKT1. This plasmid is identical to pLHH4 (see Figure 1a), except that the MuA transposase gene and Mu R-ends are replaced by polylinkers (NcoI 319-HindIII 467, SpeI 3560-NotI 3567, XmaI 7777-XbaI 7789, respectively). Unique restriction sites are shown in red font.
Supp. Fig. 3. Papillation assay plasmid pSKT4 for IS903 transposition. This plasmid contains the IS903 transposase gene and the transposon ends within the polylinkers of pSKT1 (see Additional file 4).
Supp. Table 2. Plasmids. Plasmids used in the work.
Supp. Table 3. Oligonucleotides. Oligonucleotides used in the work.
Contributor Information
Maria I Pajunen, Email: maria.pajunen@helsinki.fi.
Tiina S Rasila, Email: tiina.s.luukkainen@helsinki.fi.
Lotta J Happonen, Email: lotta.happonen@helsinki.fi.
Arja Lamberg, Email: arja.lamberg@thermofisher.com.
Saija Haapa-Paananen, Email: saija.haapa-paananen@vtt.fi.
Saija Kiljunen, Email: saija.kiljunen@utu.fi.
Harri Savilahti, Email: harri.savilahti@utu.fi.
Acknowledgements
We thank George Chaconas (University of Calgary, Calgary, AB, Canada) for the generous gift of rabbit anti-MuA polyclonal antibody. We acknowledge Keith Derbyshire (University of Albany, Albany, NY, USA) for providing the E. coli strains DH10B and HT321 as well as plasmid pNT105. We thank Sari Tynkkynen and Pirjo Rahkola for excellent technical assistance. This study was funded by the Academy of Finland, the Finnish National Technology Agency (TEKES) and the Finnish Cultural Foundation.
References
- Craig NL, Craigie R, Gellert M, Lambowitz AM. Mobile DNA II. Washington, DC: ASM Press; 2002. [Google Scholar]
- Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu Rev Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES. et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Schnable PS. et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Biemont C, Vieira C. Genetics: junk DNA as an evolutionary force. Nature. 2006;443:521–524. doi: 10.1038/443521a. [DOI] [PubMed] [Google Scholar]
- Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- Robert V, Bessereau JL. Targeted engineering of the Caenorhabditis elegans genome following Mos1-triggered chromosomal breaks. EMBO J. 2007;26:170–183. doi: 10.1038/sj.emboj.7601463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KA, Goyard S, Beverley SM. In vitro shuttle mutagenesis using engineered mariner transposons. Methods Mol Biol. 2004;270:299–318. doi: 10.1385/1-59259-793-9:299. [DOI] [PubMed] [Google Scholar]
- Kekarainen T, Savilahti H, Valkonen JP. Functional genomics on potato virus A: virus genome-wide map of sites essential for virus propagation. Genome Res. 2002;12:584–594. doi: 10.1101/gr.220702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskey C, Izsvák Z, Kawakami K, Ivics Z. DNA transposons in vertebrate functional genomics. Cell Mol Life Sci. 2005;62:629–641. doi: 10.1007/s00018-004-4232-7. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Izsvák Z. Transposons for gene therapy! Curr Gene Ther. 2006;6:593–607. doi: 10.2174/156652306778520647. [DOI] [PubMed] [Google Scholar]
- VandenDriessche T, Ivics Z, Izsvak Z, Chuah MK. Emerging potential of transposons for gene therapy and generation of induced pluripotent stem cells. Blood. 2009;114:1461–1468. doi: 10.1182/blood-2009-04-210427. [DOI] [PubMed] [Google Scholar]
- Hickman AB, Chandler M, Dyda F. Integrating prokaryotes and eukaryotes: DNA transposases in light of structure. Crit Rev Biochem Mol Biol. 2010;45:50–69. doi: 10.3109/10409230903505596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaconas G, Harshey RM. In: Mobile DNA II. Craig NL, Craigie R, Gellert M, Lambowitz AM, editor. Washington, DC: ASM Press; 2002. Transposition of phage Mu DNA; pp. 384–402. [Google Scholar]
- Haapa S, Taira S, Heikkinen E, Savilahti H. An efficient and accurate integration of mini-Mu transposons in vitro: a general methodology for functional genetic analysis and molecular biology applications. Nucleic Acids Res. 1999;27:2777–2784. doi: 10.1093/nar/27.13.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapa-Paananen S, Rita H, Savilahti H. DNA transposition of bacteriophage Mu: a quantitative analysis of target site selection in vitro. J Biol Chem. 2002;277:2843–2851. doi: 10.1074/jbc.M108044200. [DOI] [PubMed] [Google Scholar]
- Orsini L, Pajunen M, Hanski I, Savilahti H. SNP discovery by mismatch-targeting of Mu transposition. Nucleic Acids Res. 2007;35:e44. doi: 10.1093/nar/gkm070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajunen M, Turakainen H, Poussu E, Peränen J, Vihinen M, Savilahti H. High-precision mapping of protein protein interfaces: an integrated genetic strategy combining en masse mutagenesis and DNA-level parallel analysis on a yeast two-hybrid platform. Nucleic Acids Res. 2007;35:e103. doi: 10.1093/nar/gkm563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turakainen H, Saarimaki-Vire J, Sinjushina N, Partanen J, Savilahti H. Transposition-based method for the rapid generation of gene-targeting vectors to produce Cre/Flp-modifiable conditional knock-out mice. PLoS One. 2009;4:e4341. doi: 10.1371/journal.pone.0004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poussu E, Jäntti J, Savilahti H. A gene truncation strategy generating N- and C-terminal deletion variants of proteins for functional studies: mapping of the Sec1p binding domain in yeast Mso1p by a Mu in vitro transposition-based approach. Nucleic Acids Res. 2005;33:e104. doi: 10.1093/nar/gni102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poussu E, Vihinen M, Paulin L, Savilahti H. Probing the a-complementing domain of E. coli b-galactosidase with use of an insertional pentapeptide mutagenesis strategy based on Mu in vitro DNA transposition. Proteins. 2004;54:681–692. doi: 10.1002/prot.10467. [DOI] [PubMed] [Google Scholar]
- Edwards WR, Busse K, Allemann RK, Jones DD. Linking the functions of unrelated proteins using a novel directed evolution domain insertion method. Nucleic Acids Res. 2008;36:e78. doi: 10.1093/nar/gkn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilen H, Aalto J-M, Kassinen A, Paulin L, Savilahti H. A direct transposon insertion tool for modification and functional analysis of viral genomes. J Virol. 2003;77:123–134. doi: 10.1128/JVI.77.1.123-134.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberg A, Nieminen S, Qiao M, Savilahti H. Efficient insertion mutagenesis strategy for bacterial genomes involving electroporation of in vitro-assembled DNA transposition complexes of bacteriophage Mu. Appl Environ Microbiol. 2002;68:705–712. doi: 10.1128/AEM.68.2.705-712.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajunen MI, Pulliainen AT, Finne J, Savilahti H. Generation of transposon insertion mutant libraries for Gram-positive bacteria by electroporation of phage Mu DNA transposition complexes. Microbiology. 2005;151:1209–1218. doi: 10.1099/mic.0.27807-0. [DOI] [PubMed] [Google Scholar]
- Paatero AO. et al. Bacteriophage Mu integration in yeast and mammalian genomes. Nucleic Acids Res. 2008;36:e148. doi: 10.1093/nar/gkn801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. A Short Course in Bacterial Genetics: A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. [Google Scholar]
- Weinreich MD, Reznikoff WS. Fis plays a role in Tn5 and IS50 transposition. J Bacteriol. 1992;174:4530–4537. doi: 10.1128/jb.174.14.4530-4537.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TJ, Lundblad V, Hanley-Way S, Halling SM, Kleckner N. Three Tn10-associated excision events: relationship to transposition and role of direct and inverted repeats. Cell. 1981;23:215–227. doi: 10.1016/0092-8674(81)90286-5. [DOI] [PubMed] [Google Scholar]
- Makris JC, Nordmann PL, Reznikoff WS. Mutational analysis of insertion sequence 50 (IS50) and transposon 5 (Tn5) ends. Proc Natl Acad Sci USA. 1988;85:2224–2228. doi: 10.1073/pnas.85.7.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Craig NL. Isolation and characterization of Tn7 transposase gain-of-function mutants: a model for transposase activation. EMBO J. 2000;19:3446–3457. doi: 10.1093/emboj/19.13.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender J, Kleckner N. IS10 transposase mutations that specifically alter target site recognition. EMBO J. 1992;11:741–750. doi: 10.1002/j.1460-2075.1992.tb05107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiss E, Coros AM, Tavakoli NP, Derbyshire KM. Transposition is modulated by a diverse set of host factors in Escherichia coli and is stimulated by nutritional stress. Mol Microbiol. 2005;57:1593–1607. doi: 10.1111/j.1365-2958.2005.04794.x. [DOI] [PubMed] [Google Scholar]
- Lee I, Harshey RM. Importance of the conserved CA dinucleotide at Mu termini. J Mol Biol. 2001;314:433–444. doi: 10.1006/jmbi.2001.5177. [DOI] [PubMed] [Google Scholar]
- Liu J, Parkinson JS. Genetics and sequence analysis of the pcnB locus, an Escherichia coli gene involved in plasmid copy number control. J Bacteriol. 1989;171:1254–1261. doi: 10.1128/jb.171.3.1254-1261.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Luo L. Identification of residues in the Mu transposase essential for catalysis. Proc Natl Acad Sci USA. 1994;91:6654–6658. doi: 10.1073/pnas.91.14.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Harshey RM. Mutational analysis of the att DNA-binding domain of phage Mu transposase. Nucleic Acids Res. 1995;23:3937–3943. doi: 10.1093/nar/23.19.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender J, Kleckner N. Tn10 insertion specificity is strongly dependent upon sequences immediately adjacent to the target-site consensus sequence. Proc Natl Acad Sci USA. 1992;89:7996–8000. doi: 10.1073/pnas.89.17.7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumann TA, Reznikoff WS. Tn5 transposase active site mutants. J Biol Chem. 2002;277:17623–17629. doi: 10.1074/jbc.M200742200. [DOI] [PubMed] [Google Scholar]
- Derbyshire KM, Grindley ND. Cis preference of the IS903 transposase is mediated by a combination of transposase instability and inefficient translation. Mol Microbiol. 1996;21:1261–1272. doi: 10.1111/j.1365-2958.1996.tb02587.x. [DOI] [PubMed] [Google Scholar]
- Huisman O, Kleckner N. A new generalizable test for detection of mutations affecting Tn10 transposition. Genetics. 1987;116:185–189. doi: 10.1093/genetics/116.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Cotterill S, Lichtenstein CP. Genetic analysis of the terminal 8-bp inverted repeats of transposon Tn7. Gene. 1995;162:41–46. doi: 10.1016/0378-1119(95)92859-6. [DOI] [PubMed] [Google Scholar]
- Tavakoli NP, Derbyshire KM. IS903 transposase mutants that suppress defective inverted repeats. Mol Microbiol. 1999;31:1183–1195. doi: 10.1046/j.1365-2958.1999.01260.x. [DOI] [PubMed] [Google Scholar]
- Siegele DA, Hu JC. Gene expression from plasmids containing the araBAD promoter at subsaturating inducer concentrations represents mixed populations. Proc Natl Acad Sci USA. 1997;94:8168–8172. doi: 10.1073/pnas.94.15.8168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuuchi M, Mizuuchi K. Efficient Mu transposition requires interaction of transposase with a DNA sequence at the Mu operator: Implications for regulation. Cell. 1989;58:399–408. doi: 10.1016/0092-8674(89)90854-4. [DOI] [PubMed] [Google Scholar]
- Clubb RT, Mizuuchi M, Huth JR, Omichinski JG, Savilahti H, Mizuuchi K, Clore GM, Gronenborn AM. The wing of the enhancer-binding domain of Mu phage transposase is flexible and is essential for efficient transposition. Proc Natl Acad Sci USA. 1996;93:1146–1150. doi: 10.1073/pnas.93.3.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Mizuuchi M, Mizuuchi K. MuB protein allosterically activates strand transfer by the transposase of phage Mu. Cell. 1991;65:1003–1013. doi: 10.1016/0092-8674(91)90552-A. [DOI] [PubMed] [Google Scholar]
- Leung PC, Harshey RM. Two mutations of phage Mu transposase that affect strand transfer or interactions with B protein lie in distinct polypeptide domains. J Mol Biol. 1991;219:189–199. doi: 10.1016/0022-2836(91)90561-J. [DOI] [PubMed] [Google Scholar]
- Mizuuchi K. Mechanism of transposition of bacteriophage Mu: polarity of the strand transfer reaction at the initiation of transposition. Cell. 1984;39:395–404. doi: 10.1016/0092-8674(84)90018-7. [DOI] [PubMed] [Google Scholar]
- Choi W, Harshey RM. DNA repair by the cryptic endonuclease activity of Mu transposase. Proc Natl Acad Sci USA. 2010;107:10014–10019. doi: 10.1073/pnas.0912615107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Orosz A, Boros I, Venetianer P. Analysis of the complex transcription termination region of the Escherichia coli rrnB gene. Eur J Biochem. 1991;201:653–659. doi: 10.1111/j.1432-1033.1991.tb16326.x. [DOI] [PubMed] [Google Scholar]
- Rees WA, Weitzel SE, Das A, von Hippel PH. Regulation of the elongation-termination decision at intrinsic terminators by antitermination protein N of phage λ. J Mol Biol. 1997;273:797–813. doi: 10.1006/jmbi.1997.1327. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Grant SG, Jessee J, Bloom FR, Hanahan D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci USA. 1990;87:4645–4649. doi: 10.1073/pnas.87.12.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Swingle B, O'Carroll M, Haniford D, Derbyshire KM. The effect of host-encoded nucleoid proteins on transposition: H-NS influences targeting of both IS903 and Tn10. Mol Microbiol. 2004;52:1055–1067. doi: 10.1111/j.1365-2958.2004.04051.x. [DOI] [PubMed] [Google Scholar]
- Baker TA, Mizuuchi M, Savilahti H, Mizuuchi K. Division of labor among monomers within the Mu transposase tetramer. Cell. 1993;74:723–733. doi: 10.1016/0092-8674(93)90519-V. [DOI] [PubMed] [Google Scholar]
- Rasila TS, Pajunen MI, Savilahti H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal Biochem. 2009;388:71–80. doi: 10.1016/j.ab.2009.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Table 1. E. coli strains. E. coli strains used in the work.
Supp. Fig. 1. Dependency of papillae number per colony on the number of growing colonies. Bacterial colonies were grown at different densities (upper left corner) on a standard 9-cm plate. Colony size and the corresponding number of papillae were determined. (a) Strain DH10B. (b) Strain DH5α.
Supp. Video 1. Time-lapsed video of papillation assay. Shown are bacterial colonies growing on a standard papillation medium containing 1 × 10-4 % arabinose. The standard X-gal concentration was increased to 120 μg/ml to enhance contrast. The plates were incubated initially at 25°C for 46.5 h in an unilluminated chamber. Subsequently, for recording, the plates were transferred under a microscope with an artificial LCD light source. The recording was done at ~22°C with 1-h time lapse intervals for 6 days (144 h). The colonies shown represent wild-type MuA (Wild type) and MuA63-663 (Del-N).
Supp. Fig. 2. Universal papillation assay plasmid pSKT1. This plasmid is identical to pLHH4 (see Figure 1a), except that the MuA transposase gene and Mu R-ends are replaced by polylinkers (NcoI 319-HindIII 467, SpeI 3560-NotI 3567, XmaI 7777-XbaI 7789, respectively). Unique restriction sites are shown in red font.
Supp. Fig. 3. Papillation assay plasmid pSKT4 for IS903 transposition. This plasmid contains the IS903 transposase gene and the transposon ends within the polylinkers of pSKT1 (see Additional file 4).
Supp. Table 2. Plasmids. Plasmids used in the work.
Supp. Table 3. Oligonucleotides. Oligonucleotides used in the work.