

Abstract
Mutations or deletions in the cyclin-dependent kinase inhibitor p16INK4A are associated with multiple cancer types, but more commonly found in melanoma tumors and associated with familial melanoma predisposition. Although p16 is thought to function as a tumor suppressor by negatively regulating the cell cycle, it remains unclear why genetic compromise of p16 predisposes to melanoma over other cancers. Here we describe a novel role for p16 in regulating oxidative stress in several cell types, including melanocytes. Expression of p16 was rapidly upregulated following UV-irradiation, and in response to H2O2-induced oxidative stress in a p38 stress-activated protein kinase-dependent manner. Knockdown of p16 using siRNA increased intracellular reactive oxygen species (ROS) and oxidative (8-oxoguanine) DNA damage which was further enhanced by H2O2 treatment. Elevated ROS were also observed in p16-depleted human keratinocytes, and in whole skin and dermal fibroblasts from Cdkn2a-deficient mice. Aberrant ROS and p38 signaling in Cdkn2a-deficient fibroblasts were normalized by expression of exogenous p16. The effect of p16 depletion on ROS was not recapitulated by knockdown of retinoblastoma protein (Rb) and did not require Rb. Finally, p16-mediated suppression of ROS could not be attributed to potential effects of p16 on cell cycle phase. These findings suggest a potential alternate Rb-independent tumor-suppressor function of p16 as an endogenous regulator of carcinogenic intracellular oxidative stress. Compared to keratinocytes and fibroblasts, we also found increased susceptibility of melanocytes to oxidative stress in the context of p16 depletion, which may explain why compromise of p16 predisposes to melanoma over other cancers.
Keywords: p16, oxidative stress, melanocyte, 8-oxoguanine
Introduction
Inactivation of the p16INK4A (hereafter referred to as p16) gene commonly occurs in many tumors (Sharpless et al., 1999), although germ-line mutations in p16 are more commonly associated with hereditary melanoma predisposition (Goldstein et al., 2006). Somatic mutations of p16 (Flores et al., 1996) or allelic deletions of the p16-containing CDKN2a locus at 9p21 (Curtin et al., 2005) have also been described in a large percentage of sporadic melanomas. The p16 tumor suppressor protein normally inhibits the kinase activity of cyclin-dependent kinases (CDK) 4 and 6, thereby inhibiting the hyperphosphorylation of retinoblastoma (Rb)-related pocket proteins required for cell-cycle progression (Lukas et al., 1995). Native p16 thus functionally serves to prevent inappropriate division of stressed or damaged cells by holding them in the late G1-S transition, and may promote irreversible exit from the cell cycle into a senescent state (Alcorta et al., 1996).
Acute or chronic exposure to ultraviolet (UV) radiation produces reactive oxygen species (ROS) in the skin (Herrling et al., 2006), which may contribute to the development of skin cancers including melanoma. There is a good deal of correlative evidence to suggest that the link between UV radiation and melanoma may lie in the generation of oxidative damage (Meyskens et al., 2001). Interestingly, melanocytes isolated from melanoma patients display increased sensitivity to peroxidizing agents that correlates with endogenous antioxidant imbalance (Grammatico et al., 1998), and elevated ROS have been found in melanocytes from dysplastic nevi relative to normal skin of the same individuals (Pavel et al., 2004). In addition, mutation or loss of the enzyme hOGG-1, which repairs mutagenic oxidative DNA lesions (namely 8-oxoguanine, 8-OG), has been associated with melanoma progression (Pashaei et al., 2008; Zyrek-Betts et al., 2008). Recently, we demonstrated a role for UV-induced oxidative stress and damage in an animal model of UV-induced melanoma (Cotter et al., 2007).
Given the role of p16 as a melanoma tumor suppressor, and recent implication of oxidative stress in UV-induced melanoma noted above, we investigated a potential link between p16 and regulation of intracellular ROS. We found that multiple cell types exhibit increased levels of intracellular ROS in the context of deficiency or loss of p16, which can be restored upon re-expression of p16. Regulation of ROS by p16 occurred independent of Rb and potential effects on cell cycle. Thus the tumor suppressor function of p16 may extend beyond cell-cycle control and include a novel role in regulating oxidative stress in skin cells. Oxidative dysregulation in the context of loss of p16 function may lead to accumulation of mutations which could predispose to tumor development. The increased susceptibility of melanocytes (compared to keratinocytes and fibroblasts) to oxidative stress in the context of p16 depletion may explain why compromise of p16 predisposes to melanoma over other cancers.
Results
Oxidative stress upregulates p16 in melanocytes, which are more susceptible to oxidative stress than other cell types
The p16 tumor suppressor is known to function by inducing cell-cycle arrest or senescence when cells encounter potentially oncogenic DNA damage (Shapiro et al., 1998). In response to UV exposure, melanocytes acutely upregulate p16 at both protein and mRNA levels (Piepkorn, 2000). We confirmed that p16 protein levels are elevated in normal human melanocytes following UV exposure, detected as early as 1 h and peaking at 5 h (Figure 1a, top). Direct induction of oxidative stress had comparable effects on p16, as melanocytes treated with H2O2 demonstrated similar upregulation of p16 protein levels which was blocked by pre-addition of the antioxidant N-acetylcysteine (NAC) (Figure 1a, bottom). The induction of ROS under these conditions, and the capacity of NAC to reduce ROS levels when added prior to H2O2 treatment, was confirmed by addition of 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) and subsequent fluorimetric analysis (Supplementary Figure S1a). In addition, p16 upregulation following H2O2 treatment was associated with dose-dependent increased positive staining for 8-oxoguanine (8-OG), which was reduced by pre-addition of NAC (Supplementary Figure S1b). Addition of NAC to untreated cells reduced endogenous oxidative stress and p16 levels, and pre-addition of NAC significantly reduced UV-induced oxidative stress (Figure 1b, top) and attenuated UV-induced upregulation of p16 (Figure 1b, bottom).
Figure 1.

Exogenous oxidative stress acutely upregulates p16 in human skin cells, and melanocytes are more susceptible than other cell types to oxidative stress and damage. (a) Melanocytes were untreated (0 h) or UV-irradiated (480 J/m2, lanes 2-5), and cell lysates were prepared over a 24-h period and blotted with antibodies against p16 and Actin (upper panel). Melanocytes were treated with H2O2 at the concentrations indicated, in the absence (lanes 1-3) or presence (lanes 4-6) of 5 mM NAC, and 5 h later cell lysates were blotted for p16 and Actin (lower panel). (b) Melanocytes were untreated (0 h) or UV-irradiated (480 J/m2) in the absence or presence of 5 mM NAC, and 5 h later ROS levels were measured by DCFDA assay (upper panel, values normalized to mean of control conditions which were set at 1) and cell lysates were blotted for p16 and Actin (lower panel). Error bars indicate SEM from three independent experiments. *P<.001 (one-sample t test), **P<.001 (two-sample t test). (c) Keratinocytes (KC), fibroblasts (FB), and melanocytes (MC) isolated from each of six donors were untreated or treated with 0.05 mM H2O2 for 1.5, 3, or 5 h. RNA was isolated and expression of p16 and GAPDH was quantitated by qRT-PCR, with p16 levels normalized to GAPDH at each time point (and then normalized to control conditions which were set at 1). Error bars indicate SEM from six independent determinations. *P<.05 (one-sample t tests). (d) Melanocytes (MC), keratinocytes (KC), and fibroblasts (FB) isolated from each of seven donors were untreated or treated with 0.05 mM H2O2 for 5 h, and ROS levels were measured by DCFDA assay (left panel). Error bars indicate SEM from seven independent determinations. *P<.001 (repeated measures ANOVA analysis, P values adjusted for multiple comparisons). Cells isolated from each of three donors were untreated or treated with 0.5 mM H2O2 for 48 h, then fixed and immobilized for 8-OG staining. Error bars indicate SEM of percent 8-OG positive cells assessed under each condition from three independent determinations. *P=.06, **P<.001 (adjusted for multiple comparisons).
Significant upregulation of p16 was also observed at the RNA level in H2O2-treated melanocytes, as well as in similarly treated human keratinocytes and fibroblasts (Figure 1c). This response of p16 to oxidative stress may be particularly important in melanocytes, however, which demonstrated significantly higher levels of basal and H2O2-induced oxidative stress (Figure 1d, left) and 8-OG (Figure 1d, right) than these other cell types.
ROS-mediated p16 upregulation occurs through the p38 SAPK
Previous studies in hematopoietic stem cells found that p38 stress-activated protein kinase (SAPK) phosphorylation is associated with upregulation of p16 (Ito et al., 2006). To investigate this pathway in melanocytes, cells were tested for upregulation of p16 by exogenous oxidative stress in the presence of a p38 phosphorylation inhibitor. As shown in Figure 2a, H2O2 treatment resulted in p38 phosphorylation and addition of the inhibitor attenuated H2O2-induced upregulation of p16. Addition of NAC inhibited both upregulation of p16 and phosphorylation of p38 in H2O2-treated cells (Figure 2a). Thus ROS-induced p38 phosphorylation is required for the upregulation of p16, demonstrating a functional ROS-dependent p38 SAPK-p16 signaling pathway in melanocytes.
Figure 2.
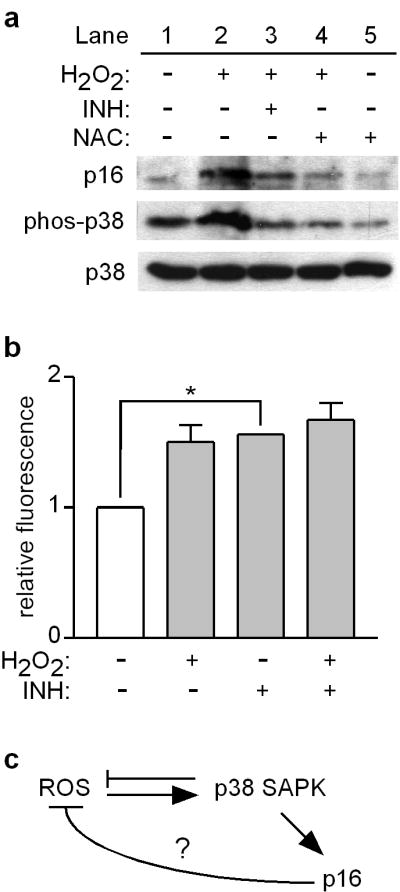
ROS upregulate p16 through the p38 SAPK in human melanocytes. (a) Melanocytes were cultured for 5 h alone (lane 1) or in the presence of 0.05 mM H2O2 (lanes 2-4), phospho-p38 inhibitor (INH), and with or without pre-addition of 5 mM NAC (lanes 4 and 5). Cell lysates were blotted for p16, phospho-p38 and p38, with p38 serving as a loading control. (b) Melanocytes were cultured for 5 h alone or in the presence of 0.05 mM H2O2 and/or phospho-p38 inhibitor (INH). ROS levels were measured by DCFDA assay, and values normalized to those of control conditions which were set at 1. Error bars indicate SEM from three independent experiments. *P<.001 (one-sample t test). (c) Schematic depicting the ROS-dependent p38-p16 signaling pathway, and the possibility that p16 suppresses endogenous ROS.
We further investigated this pathway by examining the effect of p38 phosphorylation on intracellular ROS levels. Interestingly, in the absence of an exogenous oxidative insult, inhibition of p38 phosphorylation significantly increased intracellular ROS to levels comparable to that observed in H2O2-treated cells (Figure 2b). This finding is consistent with a recent report in which inhibition or genetic deficiency of p38 resulted in increased intracellular ROS (Naidu et al., 2009), suggesting that activated p38 may also act to suppress ROS levels (Figure 2c). However, the induction of ROS upon inhibition of p38 phosphorylation also suggests the possible presence of a negative feedback loop in the ROS-dependent p38-p16 pathway in which p16 may act to suppress endogenous ROS (Figure 2c).
Intracellular ROS and oxidative DNA damage is increased in p16-deficient cells
We next asked whether p16 is required for normal regulation of intracellular ROS levels. Depletion of p16 in melanocytes by RNAi (Figure 3a) was associated with an increase in intracellular ROS both in the presence and absence of exogenous oxidative (H2O2) stress (Figure 3b). ROS in p16-depleted cells were normalized by pre-addition of NAC (Figure 3b). We next assessed the extent of oxidative DNA damage (ie. 8-OG) in p16-depleted cells. As shown in Figure 3c, the fraction of untreated melanocytes positive for 8-OG was significantly higher in cells transfected with p16-specific compared to control siRNA. While 8-OG positivity increased in both groups following treatment with H2O2, an increased fraction of 8-OG positive cells was observed in the p16-depleted group (Figure 3c). Thus p16 functions as a negative regulator of oxidative stress in melanocytes, lowering intracellular ROS and reducing oxidative DNA damage both in untreated cells and under conditions of exogenous oxidative stress.
Figure 3.

Intracellular ROS and oxidative DNA damage is increased in p16-depleted cells. (a) Melanocytes were transfected with control scrambled (Scr) or p16-specific siRNA, and 48 h later cell lysates were blotted for p16 and Actin. (b) Melanocytes were transfected with siRNA, and 48 h later either untreated or treated with 5 mM NAC and/or 0.05 mM H2O2. After 5 h, ROS levels were measured by DCFDA assay, and values normalized to those of control conditions which were set at 1. Error bars indicate SEM from three independent experiments. *P=.005 (one-sample t test). (c) Melanocytes were transfected with siRNA, and 48 h later either untreated or treated with 0.5 mM H2O2. After an additional 48 h, cells were fixed and immobilized for 8-OG staining. Error bars indicate SEM of percent 8-OG positive cells assessed under each condition in three independent experiments. *P=.05 (two-sample t test). (d) Melanocytes (MC), keratinocytes (KC), and fibroblasts (FB) isolated from four individual donors were transfected with p16-specific siRNA, and then 48 h later cultured in the absence or presence of 0.05 mM H2O2. After 5 h, intracellular ROS levels were measured by DCFDA assay. Error bars indicate SEM from four independent experiments with different donor cells. *P<.001, **P=.002 (repeated measures analysis of variance, ANOVA).
To extend these findings to other cell types, melanocytes, fibroblasts, and keratinocytes were propagated from four individual donors, and depleted of p16 using siRNA (Supplementary Figure S2). When intracellular ROS levels were compared in different cell types from the same donor, p16-depleted melanocytes exhibited significantly higher levels both basally and following H2O2 treatment (Figure 3d). Thus p16 appears to regulate oxidative stress in multiple cell types, and p16 depletion results in significantly greater levels of intracellular ROS in melanocytes compared to other skin cell types.
The Cdkn2a locus regulates oxidative stress in vivo
The p16-encoding Cdkn2a locus is mutated in approximately half of familial melanoma cases (Curtin et al., 2005). We next investigated Cdkn2a-dependent regulation of oxidative stress in vivo by measuring ROS levels in the skin of Cdkn2a-deficient mice. As shown in Figure 4a, we observed a significant increase in basal ROS in skin isolated from these mice compared to wild-type animals.
Figure 4.

Suppression of ROS by Cdkn2a in vivo, and normalization of ROS in Cdkn2a-deficient cells upon restoration of p16 expression. (a) Dorsal skin was removed from wild-type and Cdkn2a-deficient mice, and ROS levels were measured in tissue lysates by DCFDA assay. Mean values normalized to those of wild-type mice which were set at 1. Error bars indicate SEM from independent measurements in wild-type (n=10) and Cdkn2a-deficient (n=11) mice. *P=.03 (two-sample t test). (b) Two lines of fibroblasts were derived from both wild-type and Cdkn2a-deficient mice, and ROS levels determined by DCFDA assay. Error bars indicate SEM from five independent experiments. *P<.001 (repeated measures analysis of variance, ANOVA). (c) Wild-type (WT) and Cdkn2a-deficient fibroblasts were infected with either GFP control lentivirus, or lentivirus expressing p16/GFP as indicated. Cell lysates were prepared 48 h later and subjected to DCFDA assay for ROS (left panel) and Western blotting for p16, phospho-p38, p38, or Actin (right panel). Error bars indicate SEM from three independent experiments. *P=.01 (two-sample t test).
Oxidative dysregulation in Cdkn2a-deficient cells and restoration by exogenous p16
Two fibroblast lines were derived both from wild-type and CDKN2a-null mice. Fibroblast lines from knockout mice demonstrated significantly elevated ROS levels compared to lines derived from wild-type mice (Figure 4b), recapitulating our findings in whole mouse skin and p16-depleted human cells. Having demonstrated that p16 is required for normal regulation of oxidative stress, we next asked whether restoration of p16 expression in p16-deficient cells would be sufficient to normalize intracellular ROS levels. It is important to note that the Cdkn2a-null mice and derived fibroblasts used in these experiments are also deficient in p14ARF, although the siRNA we employed (Figure 3) is specific for p16 (and not cross-reactive with ARF). Fibroblasts isolated from wild-type and Cdkn2a-null mice were infected with control lentivirus expressing GFP, and Cdkn2a-null fibroblasts were seperately infected with lentivirus expressing p16/GFP which conferred a level of p16 expression comparable to that of wild-type cells (Figure 4c, right). We found that restoration of p16 expression in Cdkn2a-null fibroblasts was sufficient to neutralize elevated oxidative stress, as ROS levels significantly decreased (Figure 4c, left) and p38 phosphorylation was attenuated (Figure 4c, right). Thus p16 is both necessary and sufficient for proper maintenance of endogenous cellular ROS levels.
p16 regulates oxidative stress independently of Rb pathway
Given the prominent upstream role of p16 in negative regulation of the Rb pathway (Lukas et al., 1995), we examined the phosphorylation status of Rb in H2O2-treated human melanocytes and its requirement for p16-mediated regulation of oxidative stress. Following exposure to H2O2, there was loss of Rb phosphorylation (as observed previously in similarly-treated endothelial cells (Cicchillitti et al., 2003)) coincident with upregulation of p16 at 5 h, with recovery of Rb phosphorylation and normalization of p16 expression by 24 h (Figure 5a, left). Cell cycle analysis revealed modest but statistically significant increase in the G1 fraction and decrease in the G2M fraction in H2O2-treated melanocytes at 5 h (Figure 5a, right). Next, p16 and Rb were either separately or simultaneously depleted in melanocytes by two-step RNAi (Figure 5b, lower left). While p16 knockdown was associated with elevated ROS, there was no effect of Rb knockdown alone and furthermore, combined Rb and p16 knockdown increased oxidative stress comparable to that seen with p16 knockdown alone (Figure 5b, upper left). Thus the dysregulation of ROS by depletion of p16 cannot be recapitulated by depletion of Rb, and Rb is not required for p16 regulation of ROS levels.
Figure 5.

p16 regulation of ROS occurs independently of Rb. (a) Melanocytes were untreated (0 h), or treated with 50 μM H2O2 for 5h or 24 h. Cells were lysed for Western blotting (left panel), or analyzed by flow cytometry to determine cell cycle phase (right panel). Error bars represent SEM of triplicate determinations, *P<.001 (two-sample t test). Representative of two experiments performed. (b) Melanocytes were transfected with scrambled (Scr) or indicated specific siRNA, and then 48 h later ROS levels were measured by DCFDA assay, cell lysates were prepared for Western blotting, and cycle analysis was performed with percentages of cells in each phase (G1, S, G2M) indicated. Error bars indicate SEM of duplicate or triplicate determinations. *P=.01, **P<.001; ns, not significant. Representative of two experiments performed.
Oxidative dysregulation in p16-depleted cells not due to effects on cell-cycle
Finally, we considered the possibility that the observed suppressive effect of p16 on ROS levels could be related to its role in cell cycle regulation. It is established that p16 is a negative regulator of the cell cycle, and cells with p16 mutations exhibit faster proliferation rates and decreased fraction of cells in the G1 phase compared to cells with wild-type p16 (Serrano et al., 1996). Consistent with these findings, we observed an increased G2M fraction in fibroblasts isolated from Cdkn2a-null mice compared to cells from wild-type mice, as well as increased BrdU staining in skin epidermis of Cdkn2a-null compared to wild-type mice (not shown). It is also known that increased proliferation is associated with increased mitochondrial respiration and increased ROS leakage from the mitochondrial chain into the cytoplasm (Chung et al., 2009). Thus dysregulation of intracellular ROS in p16-deficient cells could be secondary to increased proliferation resulting from loss of p16-mediated control of the cell cycle.
Indeed, we observed that p16 depletion was associated with a modest but significant shift in cell cycle phase distribution reflected by decreased (81 to 65%) G1 and increased (15 to 26%) G2M fractions (Figure 5b, right). However, a significant but less pronounced effect was also seen on G1 and G2M fractions with Rb knockdown (Figure 5b, right) which was not associated with any increase in ROS levels (Figure 5b, upper left). Thus small increases in the cycling fraction are not necessarily associated with increased ROS levels. We noted that the degree of p16 knockdown using two-step RNAi (Figure 5b, lower left) was more complete than in our earlier experiments depleting p16 by single-step RNAi in melanocytes (Figure 3a, Supplementary Figure 2a), in which a lower concentration of transfection reagent was used. We repeated single-step p16 RNAi as in earlier experiments using cells isolated from four different donors, this time assessing whether alterations in oxidative stress and damage were associated with changes in cell cycle phase. Under such conditions of partial depletion of p16 (Supplementary Figure S3a), cell cycle distribution was not significantly altered compared to control RNAi-transfected cells (Supplementary Figure S3b). In these cells matched for cell cycle phase, we confirmed increased ROS levels (Supplementary Figure S3c) and increased proportion of 8-OG positive cells (Supplementary Figure S3d) in p16-RNAi-transfected compared to control RNAi-transfected cells. Taken together, these data suggest that dysregulated oxidative stress and resulting oxidative damage observed in p16-depleted cells occurs independently of potential effects of p16 on cell-cycle control.
Discussion
It is widely accepted that p16 acts as a tumor suppressor by inhibiting Rb phosphorylation and inducing cell cycle arrest in the G1 phase in response to potentially genotoxic stimuli (Alcorta et al., 1996). This p16-mediated cell cycle arrest allows time for repair of DNA damage prior to replication, thereby reducing the chance of propagating mutations (Figure 6). However, it is unknown whether other mechanisms exist by which p16 may protect against tumor formation. Here we show that in addition to UV irradiation, exogenous oxidative stress rapidly upregulates expression of p16. We present evidence that p16 deficiency leads to dysregulation of intracellular ROS in multiple cell types and accumulation of oxidative DNA damage, and that p16 is both necessary and sufficient for normal regulation of p38 SAPK signaling and intracellular oxidative status. Our findings further suggest that compromise of p16 may allow cells to progress to S phase bearing oxidative DNA lesions that may result in carcinogenic mutations (Figure 6). Our results indicate that p16-mediated regulation of intracellular oxidative stress appears to be independent of the Rb pathway, and not secondary to potential cell cycle effects, thus confirming a novel function for p16.
Figure 6.

Potential tumor-suppressive functions of p16. In the canonical pathway, p16 mediates cell cycle arrest as part of the DNA damage response pathway, allowing time for DNA repair enzymes to correct potentially oncogenic mutations or avoiding transformation by inducing senescence. Loss of p16 leads to increased ROS levels (direct or indirect effect, indicated by dashed/solid line) and oxidative DNA damage, which in p16-deficient cells with impaired capacity for cell cycle arrest and senescence, may result in increased accumulation of mutations promoting oncogenesis. Melanocytes are particularly susceptible to oxidative stress (Figure 1d), perhaps explaining why loss of p16 predisposes to melanoma rather than other cancers.
It is unknown why compromise of p16 is more commonly associated with melanoma over other cancers. While we demonstrate here that p16 is necessary for proper oxidative regulation in multiple cell types, our data also indicate that melanocytes maintain higher levels of intracellular ROS and incur greater oxidative DNA damage in response to exogenous oxidative stress than other cell types (Figure 1d). This finding also holds true in the context of p16 depletion when different skin cell types were matched by donor and analyzed for intracellular ROS (Figure 3d). This inherent predisposition to oxidative damage may underlie an increased susceptibility of melanocytes over other cell types to transformation in the setting of p16 deficiency. The basis for higher basal levels of oxidative stress in melanocytes may relate to their synthesis of melanin pigment, which is distributed to adjacent keratinocytes for the purpose of absorbing photons and scavenging free radicals (Riley, 1997). While UV exposure stimulates melanocytes to proliferate and produce melanin that can absorb UV-generated ROS (Gilchrest et al., 1996), higher UV doses can oxidize melanin and increase ROS production (Wood et al., 2006), which may further increase oxidative stress in melanocytes (Urabe et al., 1994). Moreover, under conditions of increased oxidative stress, there is less efficient repair of 8-OG lesions (Eiberger et al., 2008). Consistent with this notion, one recent study has shown that melanocytes are deficient (compared to fibroblasts) in repair of oxidative DNA damage (Wang et al.).
It has been suggested that loss of p16 in melanocytes may lead to failure of senescence which underlies malignant transformation of melanocytic nevi (Figure 6) (Mooi et al., 2006). Oncogene activation in melanocytes also generates oxidative stress, and increased ROS and p16 expression have been implicated in oncogene-induced melanocyte senescence (Leikam et al., 2008). Consistent with this notion, it has been shown that several melanoma-associated p16 mutants lack the capacity to induce senescence (Haferkamp et al., 2008), although p16 is not consistently expressed in melanocytes of senescent nevi in vivo (Gray-Schopfer et al., 2006) and more recent studies indicate that oncogene-induced senescence does not require p16 or p14ARF (Dhomen et al., 2009; Haferkamp et al., 2009).
Our data showing negative control of ROS by p16 may seem at odds with these (Leikam et al., 2008) and other reports in the literature, suggesting that the relationship between p16 levels and oxidative stress may be highly dependent on the circumstances and cell system utilized. For example, it was reported that over-expression of p16 increases cellular ROS levels in a human diploid fibroblast line (TIG-3) and p16 knockdown decreases ROS in a conditionally immortalized human fibroblast line (SVts8) (Takahashi et al., 2006). Another study using EJ human carcinoma cells found that while tet-regulated over-expression of p21 elevated cellular ROS, induction of p16 over-expression had no discernable effect on ROS (Macip et al., 2002). By contrast, our studies utilized siRNAi to knockdown p16 in primary normal human cells expressing wild-type p16, or lentivirus to express p16 in freshly-isolated mouse fibroblasts that were genetically deficient in p16. These various findings suggest that regulation of ROS by p16 in melanocytes and melanoma cells may be context-specific, and could reflect differences between cell types, or the immortalized or senescent state of the cells. Nevertheless, it seems plausible from our findings that p16 may act to suppress tumorigenesis through two inter-related pathways (Figure 6): first mediating cell cycle arrest to facilitate repair of DNA damage, and second, to control accumulation of ROS that may cause oxidative DNA damage. Moreover, melanocytes may be more perturbed by oxidative dysregulation induced by compromise of p16 than other cell types, leading to increased susceptibility to melanoma over other cancers.
The precise mechanism by which p16 deficiency increases intracellular ROS in our system remains to be elucidated. Several recent reports also describe possible novel tumor-suppressive roles of p16 that are independent of its role in cell cycle control. For example, several p16 mutants responsible for inherited melanoma susceptibility in humans retain robust CDK4-binding capacity (Becker et al., 2001). It has been reported that p16 interacts with brahma-related gene 1 (BRG1), a chromatin remodeling factor (Bochar et al., 2000) whose expression is frequently lost in primary and metastatic melanomas (Becker et al., 2009). Given the myriad of possible transcriptional targets of the BRG1-p16 complex, it is possible that p16 may regulate cellular oxidative stress through this newly discovered interaction. It has also been reported that p16 can bind to c-Jun N-terminal kinase (JNK) 3, thereby blocking JNK-mediated phosphorylation of c-Jun following UV exposure and activation of the Ras-JNK-Jun-AP-1 signaling cascade (Choi et al., 2005). Therefore it is possible that loss of p16 removes an important inhibitor of Ras-JNK-Jun-AP-1 signaling (and cellular transformation), resulting in an upregulation of genes responsible for increased intracellular ROS. Interestingly, the residue Arg24 of p16 is thought to play a crucial role in the stabilization of the p16-JNK3 complex, and an Arg24Pro mutation in p16 co-segregates in nine melanoma-prone families (Becker et al., 2001).
Taken together, our work presented here suggests that p16 may exert tumor-suppressive effects that extend beyond its known function as a cell cycle regulator. Further study of the mechanistic basis for oxidative regulation by p16 may shed further light on why loss of p16 commonly occurs in tumors, and why inherited p16 mutations predispose to melanoma susceptibility.
Materials and Methods
Skin cells
Normal human melanocytes and keratinocytes were prepared from neonatal foreskins as described previously (Bowen et al., 2003). Human fibroblasts were also prepared from foreskins following overnight treatment with dispase and removal of the epidermis (Bowen et al., 2003). Briefly, tissue fragments were incubated sequentially with collagenase (260 U per mL, Sigma Chemical Co, St. Louis, MO) for 1.5 h and then with trypsin/EDTA (0.05%, Invitrogen, Carlsbad, CA) for 15 min at 37 °C. Cultures were expanded in high-glucose DMEM supplemented with 10% FBS (complete medium). Mouse fibroblasts were isolated from neonatal wild-type (C57/BL6, National Cancer Institute, Rockville, MD) and background-matched CDKN2a homozygous-null mice (B6.129-Cdkn2atm1Rdp, MMHCC, National Cancer Institute) in sterile fashion as previously described (Bockholt et al., 1995). Briefly, the skin was dissected away and the ribcage/torso region was removed, and then mechanically disrupted using a razor blade in complete medium. Cultures were expanded in complete medium. All cell cultures were maintained at 37 °C in a humidified incubator with 5% CO2. Mouse fibroblasts were stored at -80 °C, thawed, maintained in exponential growth phase, and used for experiments within two to three weeks.
Generation and measurement of oxidative stress
Cells were UV-irradiated as previously described (Cotter et al., 2007), or H2O2 (Sigma) was added to cultures. Endogenous ROS of protein equivalents were quantified by DCFDA assay as previously described (Cotter et al., 2007). Although DCFDA is somewhat non-specific, fluorescence levels likely reflect H2O2 levels (Cathcart et al., 1983). For measurements in whole skin, dorsal skin was excised from newborn pups and incubated in high glucose DMEM supplemented with 10% FBS and 20 μM DCFDA for 30 min at 37 °C. Skins were then rinsed in PBS, manually homogenized in a disposable eppendorf tube with plastic pestle (Fisher Scientific, Pittsburgh, PA) containing 150 μL lysis buffer (2% SDS, 50 mM Tris and 10% glycerol), then 30 μg lysate was subjected to fluorimetric analysis. NAC solution (American Regent, Shirley, NY) diluted from newly-opened vials, was added at a final concentration of 5 mM to cells 30 min prior to other treatments. The phospho-p38 specific inhibitor (SB203580, EMD Chemicals, Gibbstown, NJ) was added directly to cultures at a final concentration of and 20 μM.
Western Blotting
Specific proteins were detected in cell lysates as previously described (Raj et al., 2008). Primary antibodies were used against p16 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA), β-Actin (1:10,000, A-3853, Sigma, St. Louis, MO), p38 SAPK, (1:1000, ab31828, Abcam, Cambridge, MA), phospho-p38 (1:1000, ab32557, Abcam), HO-1 (1:1000, OSA 110, Stressgen, Ann Arbor, MI), Rb (1:1000, sc-50, Santa Cruz Biotechnology), and Rb phosphorylated at residues 780 and 795 (1:1000, 9307 and 9301, respectively, Cell Signaling Technology, Danvers, MA).
Oxidative damage in melanocytes
The presence of 8-OG was detected in cells immobilized in agarose by immunohistochemistry as previously described (Cotter et al., 2007). Approximately 300 cells were counted per experimental condition for each experiment.
RNAi
Cells were transfected at 70% confluency in 6-well plates with oligonucleotides targeting p16 (sc-36143 targeting exon 1β and non-overlapping with p14ARF, Santa Cruz Biotechnology), Rb (Raj et al., 2008), or a control non-specific siRNA (Raj et al., 2008). For p16 and the control non-specific siRNA, 6 μL RNAi duplexes (10 μM stocks) were mixed with 100 μL transfection medium (sc-36868, Santa Cruz Biotechnology). For Rb knockdown two duplexes were used in combination at a final concentration of 50 nM each in 100 μL transfection medium. These mixtures were added to 6 μL transfection reagent (sc-29528, Santa Cruz Biotechnology) and 100 μL transfection medium for each well, and after 30 min added drop-wise to wells containing 800 μL transfection media. For (two-step) knockdown of p16 and Rb, twice the amount of transfection reagent was used. After 6 h, 1 mL of normal media for the given cell type with 20% FBS was added to the cells. After an additional 24 h, normal media was exchanged. Conditions were optimized for p16 and Rb knockdown at 48 h.
qRT-PCR
Cellular RNA (0.5 μg) prepared using the RNeasy kit (Qiagen, Valencia, CA) was reverse-transcribed using SuperScript II Reverse Transcriptase (Invitrogen), and equal volumes of cDNA (1 μl of each 20 μl reaction) were subjected to PCR using primers specific for p16 (5′-CCCAACGCACCGAATAGTTAC-3′ and 5′-ACCACCAGCGTGTCCAGGAA-3′) or GAPDH (5′-CCCTCAACGACCACTTTGTC-3′ and 5′-GGGTCTACATGGCAACTGTG-3′) for up to 40 cycles (95 °C for 10 s, 59 °C for 10 s and 72 °C for 20 s). The SyBR Advantage qPCR Premix Kit (Clontech, Mountain View, CA) was used according to the manufacturer's instructions, and samples were run using a CFD-3240 Chromo4 detector (MJ Research, Waltham, MA) equipped with Opticon Monitor software (Promega Corporation, Madison, WI) for data acquisition, monitoring, and analysis. Expression levels for each gene were normalized to GAPDH. GAPDH expression was not affected by H2O2 treatment or p16 knockdown.
p16-expressing lentivirus
The p16 gene was PCR-amplified from human melanocyte cDNA using primers (5′-GGACTGCAGCATGGAGCCGGCGG-3′ and 5′-TTTCTCGAGCCTCTCTGGTTCTTTCA-3′) and PfuUltra II Fusion HS polymerase (Stratagene, La Jolla, CA). Underlined nucleotides indicate PstI and XhoI sites introduced by PCR for further subcloning. The PCR product was cloned into pSC-B-Amp/Kan (Stratagene), confirmed by sequencing, then subcloned into the SbfI/XhoI sites of the modified pEI2 lentiviral expression vector (Welm et al., 2008) obtained from Bryan Welm (Huntsman Cancer Institute). The lentiviral construct was validated for p16 expression by transient transfection into HeLa cells followed by Western blotting. For viral production, HEK 293T/17 cells (ATCC, Manassas, VA) grown in DMEM with 10% FBS were co-transfected with 5 μg lentiviral vector and helper plasmids (Lenti-X HT packaging mix, Clontech) and 30 μg of polyethylenimine (pH 7.0, Sigma) in 1 mL of OptiMEM (Invitrogen) as described (Welm et al., 2008). Viral particles were collected 48 h and 72 h after transfection, purified by centrifugation (×1500 g) and filtration (0.45 μm), followed by ultracentrifugation (×100,000 g) to achieve 100-fold concentration. Viral titers were determined by limiting dilution and visualization of GFP-positive cells. For cellular infection, 8 μg/mL polybrene (Sigma) was added as previously described (Raj et al., 2008).
Cell cycle analysis
Human melanocytes at 60-70% confluency were cultured in 6-well plates and subjected to RNAi. After 48 h, cells harvested by trypsinization were washed with cold PBS, resuspended in 500 μl of 70% ethanol, and then stored overnight at 4 °C. After washing in PBS, cells were resuspended in PBS containing 50 μg/ml propidium iodide (Sigma) and then 10,000 cells were analyzed on a FACSort using ModFit LT version 3.1 software (Verity Software House, Topsham, ME) as described previously (Raj et al., 2008).
Statistics
Statistical analysis was performed using Prism 3.0 (GraphPad Software, La Jolla, CA) and R 2.80 (R Foundation for Statistical Computing, Vienna, Austria) and supervised by a biostatistician (K.M.B.). Data from experimental groups were subjected to standard one- and two-sample t tests. For experiments using cells from multiple donors, a repeated measures ANOVA analysis was performed with P values adjusted for multiple comparisons using Tukey's Honest Significant Difference. For experiments in which individual cells were counted (ie. determination of 8-OG), data were analyzed using logistic regression. The pairing of donors was taken into account as a factor in the analysis, and a Bonferonni correction was applied for two comparisons. P values ≤ .05 were considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported in part by NIH grants AR050102 and CA125761, the Department of Dermatology, and the Huntsman Cancer Foundation. We acknowledge use of the DNA/peptide and Flow Cytometry core facilities and a pilot project grant supported by P30 CA042014 awarded to Huntsman Cancer Institute. We thank Bryan Welm for providing the pEI2 plasmid and technical support for our lentiviral production, and the MMHCC at the National Cancer Institute for providing B6.129-Cdkn2atm1Rdp mice.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker TM, Haferkamp S, Dijkstra MK, Scurr LL, Frausto M, Diefenbach E, et al. The chromatin remodelling factor BRG1 is a novel binding partner of the tumor suppressor p16INK4a. Mol Cancer. 2009;8:4. doi: 10.1186/1476-4598-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker TM, Rizos H, Kefford RF, Mann GJ. Functional impairment of melanoma-associated p16(INK4a) mutants in melanoma cells despite retention of cyclin-dependent kinase 4 binding. Clin Cancer Res. 2001;7:3282–3288. [PubMed] [Google Scholar]
- Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102:257–265. doi: 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- Bockholt SM, Burridge K. An examination of focal adhesion formation and tyrosine phosphorylation in fibroblasts isolated from src-, fyn-, and yes- mice. Cell Adhes Commun. 1995;3:91–100. doi: 10.3109/15419069509081279. [DOI] [PubMed] [Google Scholar]
- Bowen AR, Hanks AN, Allen SM, Alexander A, Diedrich MJ, Grossman D. Apoptosis regulators and responses in human melanocytic and keratinocytic cells. J Invest Dermatol. 2003;120:48–55. doi: 10.1046/j.1523-1747.2003.12010.x. [DOI] [PubMed] [Google Scholar]
- Cathcart R, Schwiers E, Ames BN. Detection of picomole levels of hydroperoxides using a fluorescent dichlorofluorescein assay. Anal Biochem. 1983;134:111–116. doi: 10.1016/0003-2697(83)90270-1. [DOI] [PubMed] [Google Scholar]
- Choi BY, Choi HS, Ko K, Cho YY, Zhu F, Kang BS, et al. The tumor suppressor p16(INK4a) prevents cell transformation through inhibition of c-Jun phosphorylation and AP-1 activity. Nat Struct Mol Biol. 2005;12:699–707. doi: 10.1038/nsmb960. [DOI] [PubMed] [Google Scholar]
- Chung JS, Lee SB, Park SH, Kang ST, Na AR, Chang TS, et al. Mitochondrial reactive oxygen species originating from Romo1 exert an important role in normal cell cycle progression by regulating p27(Kip1) expression. Free Radic Res. 2009;43:729–737. doi: 10.1080/10715760903038432. [DOI] [PubMed] [Google Scholar]
- Cicchillitti L, Fasanaro P, Biglioli P, Capogrossi MC, Martelli F. Oxidative stress induces protein phosphatase 2A-dependent dephosphorylation of the pocket proteins pRb, p107, and p130. J Biol Chem. 2003;278:19509–19517. doi: 10.1074/jbc.M300511200. [DOI] [PubMed] [Google Scholar]
- Cotter MA, Thomas J, Cassidy P, Robinette K, Jenkins N, Florell SR, et al. N-acetylcysteine protects melanocytes against oxidative stress/damage and delays onset of ultraviolet-induced melanoma in mice. Clin Cancer Res. 2007;13:5952–5958. doi: 10.1158/1078-0432.CCR-07-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Eiberger W, Volkmer B, Amouroux R, Dherin C, Radicella JP, Epe B. Oxidative stress impairs the repair of oxidative DNA base modifications in human skin fibroblasts and melanoma cells. DNA Repair (Amst) 2008;7:912–921. doi: 10.1016/j.dnarep.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Flores JF, Walker GJ, Glendening JM, Haluska FG, Castresana JS, Rubio MP, et al. Loss of the p16INK4a and p15INK4b genes, as well as neighboring 9p21 markers, in sporadic melanoma. Cancer Res. 1996;56:5023–5032. [PubMed] [Google Scholar]
- Gilchrest BA, Park HY, Eller MS, Yaar M. Mechanisms of ultraviolet light-induced pigmentation. Photochem Photobiol. 1996;63:1–10. doi: 10.1111/j.1751-1097.1996.tb02988.x. [DOI] [PubMed] [Google Scholar]
- Goldstein AM, Chan M, Harland M, Gillanders EM, Hayward NK, Avril MF, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66:9818–9828. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- Grammatico P, Maresca V, Roccella F, Roccella M, Biondo L, Catricala C, et al. Increased sensitivity to peroxidizing agents is correlated with an imbalance of antioxidants in normal melanocytes from melanoma patients. Exp Dermatol. 1998;7:205–212. doi: 10.1111/j.1600-0625.1998.tb00325.x. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer VC, Cheong SC, Chong H, Chow J, Moss T, Abdel-Malek ZA, et al. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br J Cancer. 2006;95:496–505. doi: 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferkamp S, Becker TM, Scurr LL, Kefford RF, Rizos H. p16INK4a-induced senescence is disabled by melanoma-associated mutations. Aging Cell. 2008;7:733–745. doi: 10.1111/j.1474-9726.2008.00422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferkamp S, Scurr LL, Becker TM, Frausto M, Kefford RF, Rizos H. Oncogene-Induced Senescence Does Not Require the p16(INK4a) or p14ARF Melanoma Tumor Suppressors. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.5. in press. [DOI] [PubMed] [Google Scholar]
- Herrling T, Jung K, Fuchs J. Measurements of UV-generated free radicals/reactive oxygen species (ROS) in skin. Spectrochim Acta A Mol Biomol Spectrosc. 2006;63:840–845. doi: 10.1016/j.saa.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- Leikam C, Hufnagel A, Schartl M, Meierjohann S. Oncogene activation in melanocytes links reactive oxygen to multinucleated phenotype and senescence. Oncogene. 2008;27:7070–7082. doi: 10.1038/onc.2008.323. [DOI] [PubMed] [Google Scholar]
- Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, et al. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. 1995;375:503–506. doi: 10.1038/375503a0. [DOI] [PubMed] [Google Scholar]
- Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. Embo J. 2002;21:2180–2188. doi: 10.1093/emboj/21.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyskens FL, Jr, Farmer P, Fruehauf JP. Redox regulation in human melanocytes and melanoma. Pigment Cell Res. 2001;14:148–154. doi: 10.1034/j.1600-0749.2001.140303.x. [DOI] [PubMed] [Google Scholar]
- Mooi WJ, Peeper DS. Oncogene-induced cell senescence--halting on the road to cancer. N Engl J Med. 2006;355:1037–1046. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- Naidu S, Vijayan V, Santoso S, Kietzmann T, Immenschuh S. Inhibition and genetic deficiency of p38 MAPK up-regulates heme oxygenase-1 gene expression via Nrf2. J Immunol. 2009;182:7048–7057. doi: 10.4049/jimmunol.0900006. [DOI] [PubMed] [Google Scholar]
- Pashaei S, Li L, Zhang H, Spencer HJ, Schichman SA, Fan CY, et al. Concordant loss of heterozygosity of DNA repair gene, hOGG1, in melanoma in situ and atypical melanocytic hyperplasia. J Cutan Pathol. 2008;35:525–531. doi: 10.1111/j.1600-0560.2007.00865.x. [DOI] [PubMed] [Google Scholar]
- Pavel S, van Nieuwpoort F, van der Meulen H, Out C, Pizinger K, Cetkovska P, et al. Disturbed melanin synthesis and chronic oxidative stress in dysplastic naevi. Eur J Cancer. 2004;40:1423–1430. doi: 10.1016/j.ejca.2003.11.035. [DOI] [PubMed] [Google Scholar]
- Piepkorn M. The expression of p16(INK4a), the product of a tumor suppressor gene for melanoma, is upregulated in human melanocytes by UVB irradiation. J Am Acad Dermatol. 2000;42:741–745. doi: 10.1067/mjd.2000.103988. [DOI] [PubMed] [Google Scholar]
- Raj D, Liu T, Samadashwily G, Li F, Grossman D. Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis. 2008;29:194–201. doi: 10.1093/carcin/bgm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley PA. Melanin. Int J Biochem Cell Biol. 1997;29:1235–1239. doi: 10.1016/s1357-2725(97)00013-7. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Shapiro GI, Edwards CD, Ewen ME, Rollins BJ. p16INK4A participates in a G1 arrest checkpoint in response to DNA damage. Mol Cell Biol. 1998;18:378–387. doi: 10.1128/mcb.18.1.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9:22–30. doi: 10.1016/s0959-437x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, et al. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006;8:1291–1297. doi: 10.1038/ncb1491. [DOI] [PubMed] [Google Scholar]
- Urabe K, Aroca P, Tsukamoto K, Mascagna D, Palumbo A, Prota G, et al. The inherent cytotoxicity of melanin precursors: a revision. Biochim Biophys Acta. 1994;1221:272–278. doi: 10.1016/0167-4889(94)90250-x. [DOI] [PubMed] [Google Scholar]
- Wang HT, Choi B, Tang MS. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1005244107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welm BE, Dijkgraaf GJ, Bledau AS, Welm AL, Werb Z. Lentiviral transduction of mammary stem cells for analysis of gene function during development and cancer. Cell Stem Cell. 2008;2:90–102. doi: 10.1016/j.stem.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SR, Berwick M, Ley RD, Walter RB, Setlow RB, Timmins GS. UV causation of melanoma in Xiphophorus is dominated by melanin photosensitized oxidant production. Proc Natl Acad Sci U S A. 2006;103:4111–4115. doi: 10.1073/pnas.0511248103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyrek-Betts J, Micale M, Lineen A, Chaudhuri PK, Keil S, Xue J, et al. Malignant blue nevus with lymph node metastases. J Cutan Pathol. 2008;35:651–657. doi: 10.1111/j.1600-0560.2007.00878.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.