

Abstract
CD4+CD25+ regulatory T cells (Treg) transform the HIV-1 infected macrophage from a neurotoxic to a neuroprotective phenotype. This was demonstrated previously in a murine model of HIV-1 encephalitis induced by intracranial injection of HIV-1/vesicular stomatitis virus-infected bone marrow macrophages. Relationships between Treg ingress of end organ tissues, notably the brain, and neuroprotection were investigated. Treg from EGFP-transgenic donor mice were expanded, labeled with indium-111, and adoptively transferred. Treg distribution was assayed by computed tomography/single photon emission computed tomography and immunohistochemistry. Treg readily migrated across the blood brain barrier and were retained within virus-induced neuroinflammatory sites. In non-inflamed peripheral tissues (liver and spleen) Treg were depleted. These observations demonstrate that Treg migrate to sites of inflammation and modulate immune responses at disease sites.
1. Introduction
Despite the widespread use of antiretroviral therapies (ART), the incidence of milder forms of HIV-associated neurocognitive disorders (HAND) continues to rise (Letendre et al., 2007). HAND is seen in up to 50% of infected people and characterized by cognitive, motor and behavioral abnormalities ranging from asymptomatic neurocognitive disorder to frank dementia (Antinori et al., 2007). In its most severe form, HIV-1 associated dementia (HAD) occurs often with an overt encephalitis with the formation of multinucleated giant cells, myelin pallor, astrogliosis, microglial nodules and neuronal drop-out. Disease is heralded through the trafficking of virus infected monocyte-derived macrophages that migrate across the blood brain barrier (BBB) (Gartner, 2000). Cell numbers that ingress the brain are commonly viewed as the principal pathological indicator for disease severity (Gendelman et al., 1994). Microglial inflammatory responses with cytokine and chemokine dysregulation are the benchmarks of disease (Buckner et al., 2006; Chadwick et al., 2008; Kolson and Pomerantz, 1996; Mollace et al., 2001; Roberts et al., 2003).
Ways to modulate microglial inflammatory responses continue to be a major thrust towards adjuvant therapies in HAND. One means to achieve such microglial immunoregulation is through CD4+CD25+ regulatory T cells (Treg). Natural Treg originate in the thymus, immigrate and exist in the periphery as a small fraction of circulating CD4+ T cells commonly identified by expression of high cell surface levels of IL-2Rα (CD25) and the transcription factor FoxP3 (Sakaguchi, 2004). In the last two decades, Treg have been observed to functionally modulate a variety of immune responses and maintain peripheral tolerance (Hara et al., 2001; Thornton and Shevach, 1998). Importantly, Treg modulate anti-inflammatory activities in experimental models of HAND. In these models, Treg readily elicit neuroprotection associated with up-regulation of brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factor (GDNF) expression and potent down-regulation of pro-inflammatory cytokines, oxidative stress and control of viral replication (Liu et al., 2009).
Treg are involved in modulating host cellular immunity in cancer (Curiel et al., 2004; Wang et al., 2004), infectious diseases (Beilharz et al., 2004; Dittmer et al., 2004), and autoimmune diseases (Boyer et al., 2004), and are being developed for therapeutics for inflammatory- and autoimmune-mediated disorders (Bluestone, 2005; Mekala and Geiger, 2005; Tarbell et al., 2004; Trenado et al., 2003). Indeed, expanded Treg maintain regulatory function and suppress proliferation and cytokine secretion of responder peripheral blood mononuclear cells in co-cultures stimulated with CD3-specific antibodies (Hoffmann et al., 2004; Ring et al., 2007; Tang and Bluestone, 2006). In healthy individuals, lymphocyte traffic into the CNS is very low and tightly controlled by the highly specialized BBB. However, several pathological conditions of the CNS, such as viral or bacterial infections or inflammation-mediated disorders, induce circulating lymphocytes to cross the BBB and gain access to inflammatory foci (Engelhardt and Ransohoff, 2005). This may be a critical step for Treg to perform their biological function in inflammatory diseases of the CNS. Alternatively, in the absence of Treg extravasation, their function could be achieved by soluble factors. The present study was aimed at resolving this important issue for Treg-CNS function. Using a murine model of human HIV-1 encephalitis (HIVE), we demonstrated that Treg migrate across the BBB reaching the areas of the brain affected by virus-induced inflammatory responses. The observations, taken together, provide evidence that Treg readily cross the BBB and accumulate in sites associated with virus-induced inflammation and can potentially be beneficial as an immunotherapeutic modality in CNS disease.
2. Materials and Methods
2.1 Mice
Male transgenic mice (C57BL/6-Tg (CAG-EGFP)1Osb/J) 6–8 weeks of age that express enhanced green fluorescent protein transgenic (EGFP-Tg) and wild type C57BL/6J male mice 4–6 weeks of age were purchased from Jackson Laboratories (Bar Harbor, ME) and housed in pathogen-free conditions. All experiments were conducted using protocols approved by University of Nebraska Medical Center Institutional Animal Care and Use Committee.
2.2. HIV-1/VSV infection of bone marrow-derived macrophages (BMM)
Bone marrow cells were obtained from femurs of 6 week old C57BL/6J mice and were cultured in Teflon flasks at 2×106 cells/ml in complete DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in the presence of human macrophage colony stimulating factor (MCSF) at 2 μg/ml (a gift from Wyeth, Cambridge, MA) (Tang and Bluestone, 2006). After 5 days, bone marrow-derived macrophages (BMM) were infected overnight with vesicular stomatitis virus (VSV)-pseudotyped HIV-1YU2 (HIV-1/VSV) at a concentration of 1 pg of HIV-1 p24 per cell (Gorantla et al., 2007). Cells were washed and cultured for 5 days post-infection in the absence of MCSF. Greater than 90% of BMM were HIV-1p24 positive as demonstrated by immunochemistry (Dako North America, Inc., Carpinteria, CA) (Gorantla et al., 2007; Gorantla et al., 2008).
2.3. Murine model of human HIVE
To induce pathological features that mimic human HIVE in mice, HIV-1/VSV-infected BMM (1 × 106 cells/5 μl/mouse) were delivered by intracerebral (i.c.) injection into the basal ganglia of 4-wk old C57BL/6J mice using stereotactic coordinates as previously described (Persidsky et al., 1996). Mice injected i.c. with PBS served as sham operated controls.
2.4. Isolation and expansion of Treg
Spleen and lymph node T cells were isolated from EGFP-Tg mice. GFP+CD4+ cells were enriched by negative selection columns (R&D System, Inc., Minneapolis, MN), stained with PE-conjugated anti-CD25 (BD Pharmingen, San Jose, CA), and sorted for CD4+CD25+ Treg using a FACSAria (BD Biosciences, San Jose, CA). The analysis and sort gates were restricted to the small lymphocyte gate as determined by their characteristic forward and side scatter properties. The purity of CD4+CD25+ T cells was >90% by flow cytometric analyses. CD4+CD25+ regulatory T cells were cultured in a 24-well plate at a concentration of 106/ml in the presence of anti-CD3/anti-CD28-conjugated magnetic beads (Dynabeads® Mouse T-Activator CD3 CD28, Cat. No. 11452D, Invitrogen, Carlsbad, CA), at a bead-to-cell ratio of 2:1 and 2000 U/ml of recombinant human IL-2 (Novartis Vaccines and Diagnostics, Inc. Emeryville, CA). Cells were split when density exceeded 2.5×106/ml. After one week, old beads were removed and fresh beads were added at 1:4 bead-cell ratios. After two weeks of culture, beads were removed by exposure to a static magnetic field for 1–2 minutes, and Treg-containing supernatant was harvested.
2.5. qRT-PCR analyses
Total RNA from Treg was extracted using TRIzol (Invitrogen) and cleaned by RNA mini columns (Qiagen, Valencia, CA). Real-time qPCR was performed by ABI PRISM 7000 sequence detector (Applied Biosystems, Life Technologies Corporation, Carlsbad, CA), using the SYBR Green detection system and murine-specific primers. RNA values were normalized to sample Gapdh expression. Primers for the experiments were purchased from Invitrogen and sequences are listed below:
Il10: 5′-CAG TTA TTG TCT TCC CGG CTG TA-3′ and
5′-CTA TGC TGC CTG CTC TTA CTG ACT-3′
Tgfb: 5′-CTG CTG CTT TCT CCC TCA AC-3′ and
5′-GAC TGG CGA GCC TTA GTT TG-3′
2.6. CT/SPECT
Treg were labeled with 111-indium (111In) oxyquinoline (Indium oxine, GE Healthcare, Arlington Heights, IL) at a dose of 100 μCi per 20×106 cells/ml RPMI 1640 supplemented with 10 mM HEPES for 45 min at 37°C. Cells were washed and resuspended in Hanks’ buffered salt solution (HBSS). Labeling efficiency was determined by γ-scintillation spectrometry (Packard Instrument Co., Meriden, CT) and was > 75% of total input isotope. Each recipient received 20 × 106 111In-labeled Treg by i.v. injection. Mice were anesthetized with 0.5–1% isoflurane delivered in a 2:1 mixture of nitrous oxide and oxygen. Image acquisition was accomplished by an x-ray computed tomography/single photon emission computed tomography system (CT/SPECT) (FLEX Triumph, Gamma Medica-Ideas, Northridge, CA) fitted with a 5-pinhole (1.0 mm/pinhole) collimator. Co-registration of anatomical CT images and functional SPECT was performed by 3D image visualization and analysis software VIVID Gamma Medica-Ideas, Northridge, CA). The specific tissue and region of interest (ROI) of each tissue were electronically defined and relative radioactivity for each area determined.
2.7. Immunocytochemical tests
Brain tissues were derived from perfused mice and processed as previously described (Gorantla et al., 2007; Liu et al., 2009). Anti-HIV-1p24 antibodies (Dako North America, Inc.) were used to identify HIV-1 infected cells. Treg were identified by dual staining with anti-CD4 (BD Pharmigen) and anti-GFP (abCAM, Inc., Cambridge, MA) antibodies. Murine microglia and astrocytes were detected with rabbit polyclonal antibodies to ionizing calcium-binding adaptor molecule 1 (Iba1) (1:500; Wako, Richmond, VA) and glial fibrillary acidic protein (GFAP) (1:1,000; Dako North America, Inc.), respectively. Antibodies to neuronal nuclei (NeuN) (1/100; Chemicon International, Millipore, Billerica, MA) and microtubule-associated protein 2 (MAP2) (1/1,000; Chemicon International) were used to identify neurons. Mouse cross-reactive chicken anti-GDNF (1:50; Promega, Madison, WI, USA) was used for growth factor expression. Primary antibodies were visualized with DyLight-649-conjugated (purple) secondary Abs (Jackson ImmunoResearch Laboratories, Inc. West Grove, PA), Alexa Fluor-488 (green)-, Alexa Fluor-594 (red)-conjugated secondary Abs (Invitrogen, Molecular Probes). Tissues were mounted using anti-fade Pro-Long Gold mounting reagent (Invitrogen) and examined by confocal microscopy (Zeiss LSM 510 META NLO microscope, Zeiss MicroImaging, Inc., Thornwood, NY).
2.8. Flow cytometry
Samples from expanded cell fractions were labeled with fluorescently labeled Abs and analyzed by flow cytometry with a FACSCalibur flow cytometer (BD Biosciences). mAbs (all from BD Biosciences unless indicated) used for staining were PE-Cy5 (CyChrome, CyC)-conjugated anti-CD4; PE-conjugated anti-CD25; and allophycocyanin (APC) anti-mouse Foxp3 (eBioscience, San Diego, CA)
2.9. Statistical analyses
Results are expressed as mean ± SEM for each group. Statistical significance between groups was analyzed by Student’s t test using Microsoft Excel. Differences were considered statistically significant at p ≤ 0.05.
3. Results
3.1. Treg immunity and neuroprotection in a murine HIVE model
We induced HIVE in C57BL/6 mice using BMM infected with HIV-1/VSV pseudotyped virus and injected i.c. into the basal ganglia of syngeneic mice; wherein, a defined viral-mediated encephalitis is induced along the injection track (Dou et al., 2005; Gendelman et al., 1994; Gorantla et al., 2007; Liu et al., 2009; Persidsky et al., 1996; Poluektova et al., 2002). To evaluate the roles of Treg in regulating neuroinflammatory responses in a model of HIVE in mice, 1×106 activated Treg were adoptively transferred to HIV-1/VSV-infected recipients 24 h after initiation of HIVE-associated pathology. By 7 days post infection, immunohistochemistry of tissues surrounding the injection tracks indicated that adoptive transfer of Treg in HIV-1/VSV-infected mice reduced GFAP and Iba1 expression compared with HIV-1/VSV infected controls (Fig. 1A). Additionally, Treg-treated animals exhibited up to 90% reduction in HIV-1 p24 expression levels compared with those in HIV-1/VSV control group. These results indicate that Treg attenuate the neuroinflammatory responses induced by HIV-1/VSV infection.
FIGURE 1. Histologic and immunocytochemical analyses of murine HIVE.
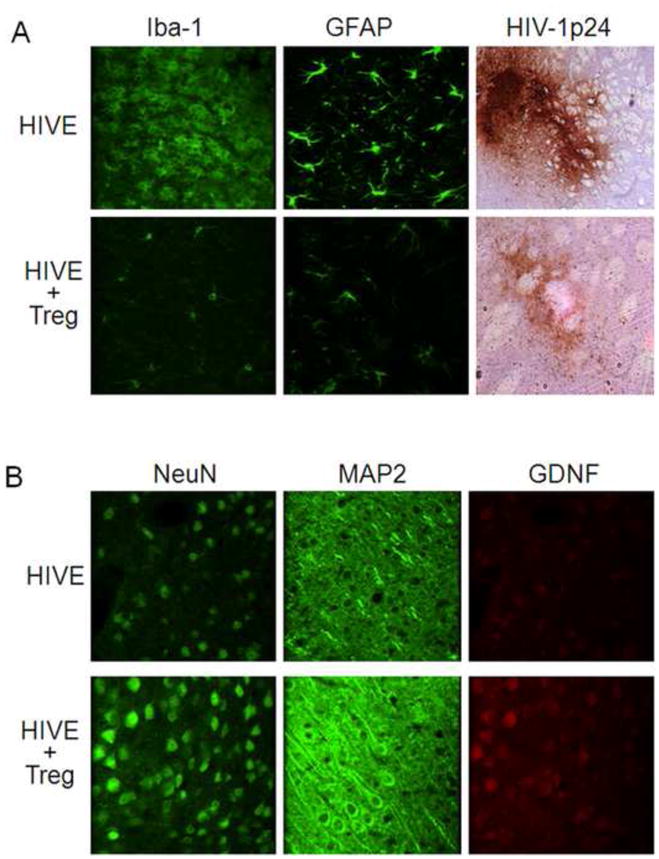
To induce HIVE, HIV-1/VSV infected BMM were stereotactically injected i.c. into the basal ganglia of syngeneic C57BL/6J mice. PBS vehicle was injected i.v. or 1×106 Treg in 200 μl PBS were adoptively transferred into HIVE mice 1 day post infection. Brains were collected 7 days later, processed, sectioned and analyzed by immunohistochemical staining. A, Brain tissues were evaluated for expression of Iba1 (green), GFAP (green), and HIV-1 p24 (DAB+ staining brown). B, Serial brain sections were stained for expression of NeuN (green), MAP2 (green), and GDNF (red). Sections were evaluated by confocal laser-scanning microscopy. Images are shown at ×400 original magnification. This figure is representative of data obtained from three independent experiments.
To evaluate the neuroprotective abilities of Treg in an HIVE model, we assessed the expression of NeuN, MAP2, and GDNF around the brain injection track area. Compared with the HIV/VSV-infected group, HIV/VSV-infected mice treated with Treg exhibited empirically higher neuronal expression of NeuN, MAP2 and GDNF (Fig. 1B). These data, taken together, indicate that Treg enhance neurotrophin secretion and protect neurons in mice with features of human HIVE.
3.2. GFP-Treg adoptive transfers
To determine the infection efficiency of BMM by HIV-1/VSV, we assessed p24 expression in non-infected and HIV-1/VSV-infected BMM after 5 days of culture post-infection. Infection efficiency was greater than 90% as determined by ratios of p24+ BMM to total BMM amongst infected BMM cultures (Fig 2A). To determine whether infected BMM retained p24 expression after i.c. infection, we examined basal ganglia tissues at the i.c. injection site 7 days post-injection. Immunofluorescence analyses of those tissues showed that p24 expression by BMM was indeed retained after 7 days post-initiation of HIVE-like pathology (Fig. 2A) at a time when microglial activation and astrogliosis in those tissues were also present (Fig 1A). To evaluate Treg expansion, lymphocyte suspensions from spleens and lymph nodes of EGFP-Tg mice were first enriched to >95% for CD4+ T cells and to 6% CD25+GFP+ Treg by negative selection (Fig 2B). CD4+CD25+GFP+ T cells were further enriched to 92% as analyzed by FACS.
FIGURE 2. Adoptive transfer of GFP-Treg to HIVE mice.
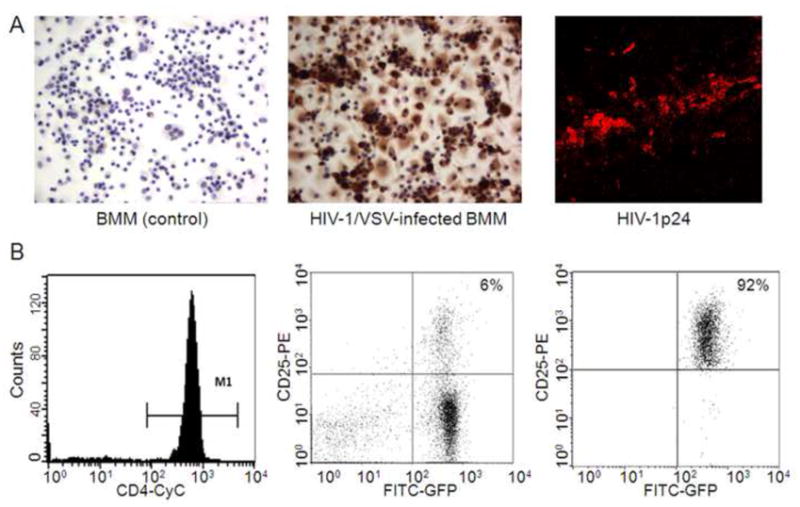
Vesicular stomatitis virus (VSV)-pseudotyped HIV-1YU2 (HIV-1/VSV) was used to infect BMM at a concentration of 1 pg of HIV-1 p24 per cell for 24 h. Infected BMM were washed and cultured in fresh media for 5 days. A, Cultured, non-infected BMM (left panel) and HIV-1/VSV-infected BMM (middle panel) were stained for expression of p24 revealing an infection efficiency of greater than 90%. Infected BMM were stereotactically injected i.c. into the basal ganglia of syngeneic C57BL/6J mice. Brain tissues around the injection line were dissected on day 7 and analyzed by immunostaining for expression of HIV-1 p24 (red). B, CD4+ cells from spleen and lymph nodes of EGFP-Tg mice were enriched by negative selection columns and analyzed by flow cytometric analysis (left panel). Enriched CD4+GFP+ cells were stained with PE-anti-CD25, analyzed (middle panel) and sorted for CD25+ T cells by FACS. Purity of CD4+CD25+GFP+ was >90% by flow cytometric analyses (right panel). This figure is representative of data obtained from three independent experiments done with four replicates.
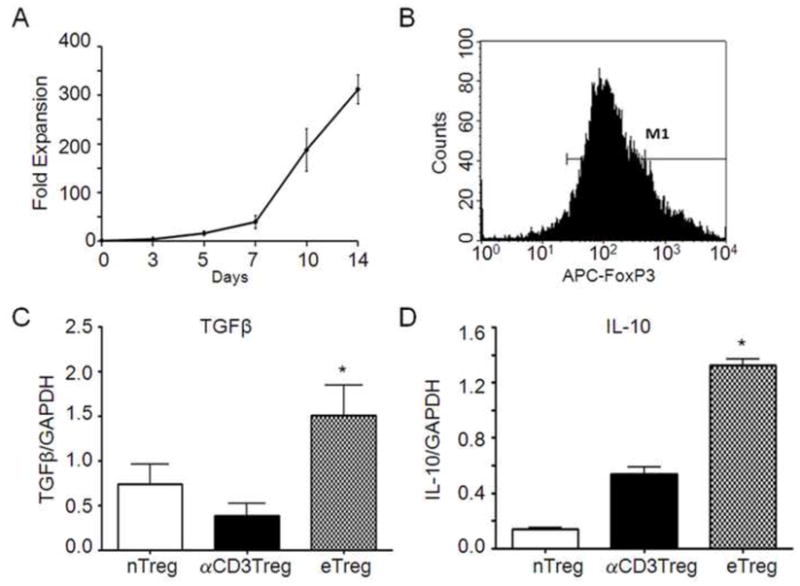
3.3. Treg expansion and characterization
Enriched CD4+CD25+GFP+ Tregs were expanded by co-culture with anti-CD3/anti-CD28 micro beads and high concentrations of rHu IL-2 (2000U/ml). Under these culture conditions, Treg were expanded over 2 weeks to yield 200–300 times the initial input number (Fig 3A). Flow cytometric analyses showed that greater than 90% of expanded CD4+CD25+GFP+ (data not shown) Treg expressed FoxP3 (Fig 3B). We compared cytokine gene expression from expanded Treg with that from fresh, sorted Treg isolates that were unstimulated or anti-CD3-stimulated. Expanded Treg significantly increased expression of TGF-β mRNA compared to unstimulated or stimulated fresh Treg isolates (Fig 3C). Most strikingly, IL-10 mRNA expression by expanded Treg significantly exceeded that of freshly isolated Treg cells (Fig 3D). These data demonstrated that enhanced cytokine gene expression levels were afforded by expansion of CD4+CD25+FoxP3+ Treg compared with fresh isolates of Treg.
FIGURE 3. Expansion and characterization of GFP-Treg.
A, Freshly sorted CD4+CD25+GFP+ Treg cells were co-cultured anti-CD3/anti-CD28 superparamagnetic beads at a bead-to-cell ratio of 2:1 and 2000 U/ml rHu IL-2. Numbers of cultured Treg over 2 weeks were determined from viable cell counts using trypan blue exclusion. B, Expanded CD4+CD25+GFP+ Treg were permeabilized, stained for FoxP3 expression and assessed by flow cytometric analysis. C and D, Total RNA was extracted from freshly isolated CD4+CD25+GFP+ Treg (nTreg), anti-CD3/anti-CD28 stimulated CD4+CD25+GFP+ Treg (αCD3Treg), or anti-CD3/anti-CD28/IL-2 expanded CD4+CD25+GFP+ Treg (eTreg). Real-time qPCR was performed to assess Tgfb (C) and Il10 (D) gene expression. RNA values were normalized to Gapdh expression. This figure is representative of data obtained from three independent experiments.
3.4. CT/SPECT analyses of 111In-labeled Treg tissue migration
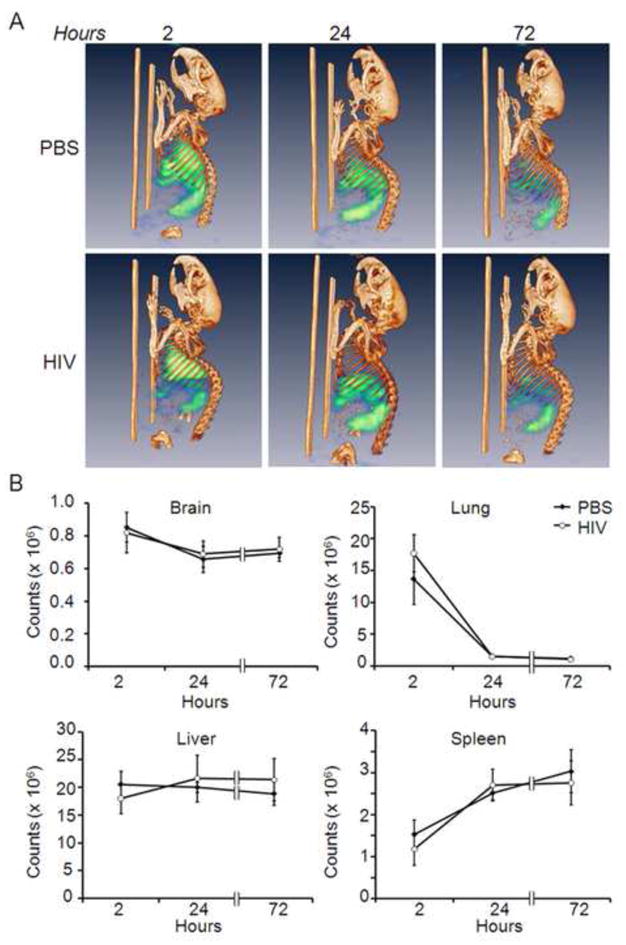
To assess real-time migration of Treg cells, 20 × 106 111In-labeled CD4+CD25+GFP+ Treg (Fig 3) were adoptively transferred to HIVE and sham mice; and mice were assessed by CT/SPECT on days 0, 1, and 3 post-transfer. On day 0, the majority of 111In-labeled Treg were detected in the lungs with lower intensity signals in spleen and liver (Fig 4A). By day 1 post-transfer and times thereafter, the predominant signals were found within spleen and liver. Progressively diminishing signal intensities exhibited over 72 h were in part due to 111In decay (t1/2=2.8 days). To quantify 111In-labeled Treg within each tissue, ROI were electronically drawn to encompass those tissues and total relative radioactivity for each tissue was measured and corrected for radioactive decay. Total counts of 111In-labeled Treg in lungs were higher 2 h after transfer then decreased rapidly thereafter (Fig 4B). Numbers of labeled Treg in liver over 72 h remained relatively unchanged; whereas, accumulation of labeled Treg in spleen was lowest at 2 h post transfer but increased at 24 h and 72 h.
FIGURE 4. CT/SPECT distribution of Treg.
CD4+CD25+GFP+ Treg were labeled with 111In oxine, and 20×106 labeled Treg were adoptively transferred to HIVE mice or sham-injected control mice. Distribution of 111In-labeled Treg was evaluated by computed tomography/single photon emission computed tomography (CT/SPECT) analyses at 2, 24 and 72 hours post transfer. A, Representative CT/SPECT images of 111In-labeled PBS sham control mice or HIVE mice acquired at 2, 24 and 72 hours. Intensities of radioactive signals are depicted as pseudo-colored blue-green signals. B, Three-dimensional electronic bit maps were drawn encompassing brain, lung, liver or spleen. Total radioactive counts of 111In-labeled Treg distributions in mice were calculated by digital image analysis using VIVID software. Counts at each time point were decay-corrected to time of injection. Relative radioactive counts as a function of pixel intensities were determined and presented as the means ± SEM for 3 mice per group. No statistical differences in 111In-labeled Treg intensities were detected between HIVE mice or PBS sham controls at any time point. In lungs of both HIVE and sham control mice, significant diminutions of 111In-labeled Treg counts were observed at 24 h and 72 h compared with 2 h post-transfer (p < 0.001). No other tissues exhibited significant increases in radioactive counts at any times compared with 2 h post-transfer, although intensities trended to increase at 24 h (p < 0.1) and 72 h (p < 0.07) in both HIVE and PBS sham controls. The data is representative of three replicate experiments.
3.5. Preferential Treg accumulation in a murine HIVE
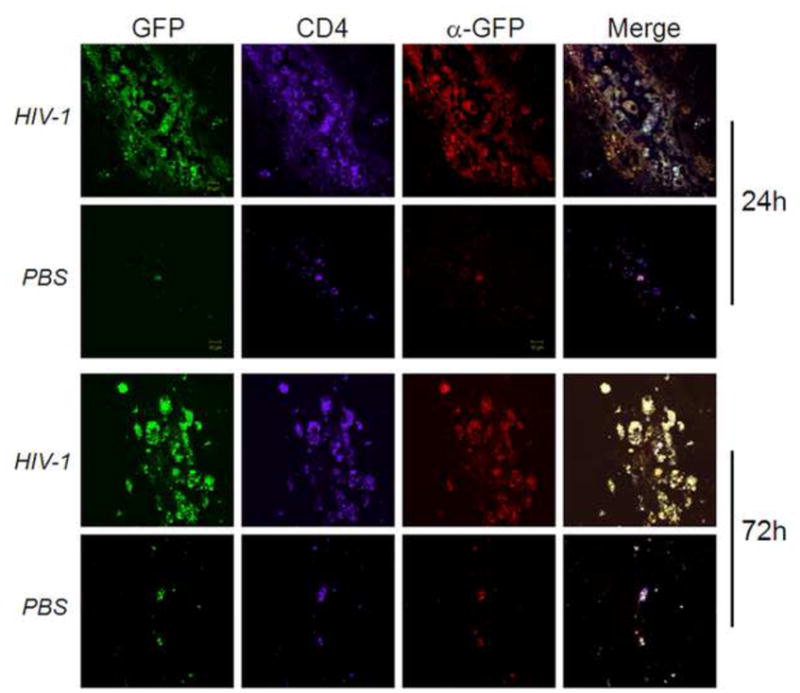
We previously demonstrated a potential role in modulating the immune response to HIV infection suggesting that CD4+CD25+ Treg exert an immunosuppressive role by a cell–cell interaction involving cell surface TGF-β1 (Nakamura et al., 2001); and predicted that for the necessitate cell-cell contact in brain, Tregs must migrate to inflammatory sites of HIVE associated pathology. To test that possibility, we adoptively transferred expanded CD4+CD25+GFP+ Treg (Fig 3) to HIVE recipients and sham control recipients after 7 days post-i.c. injection with HIV-1/VSV infected BMM or PBS for sham controls. Tissues were harvested from recipient mice at 24h and 72h post-transfer and evaluated for presence of donor-derived Treg by immunohistochemical analyses. Confocal microscopy of areas around the needle track revealed that by 24 h after adoptive transfer CD4+GFP+ donor Treg migrated to the needle tracks and sites of neuroinflammation in mice with HIVE associated pathology (Fig 5). In comparison, very few; <95% reduction in cell numbers seen in brains with HIVE linked pathology of CD4+GFP+ Treg were detected in tissues around the needle tracks from animals of the PBS sham group. By 72 h post-transfer, CD4+GFP+ donor Treg were still present within the inflammatory areas of HIVE-like pathology with remarkably few donor Treg in the PBS sham control mice. Our results indicate that Treg migrate to areas of HIV-induced inflammation and remain within inflammatory sites, thus supporting a direct involvement in modulating inflammatory processes.
FIGURE 5. Treg migrate to sites of HIV-1 induced encephalitis.
HIV-1/VSV-infected BMM were stereotactically injected i.c. into the basal ganglia of syngeneic C57BL/6J mice. Sham controls were injected i.c. with PBS. After 7 days post-infection, 20×106 expanded CD4+CD25+GFP+ Treg were adoptively transferred to each HIVE and sham control recipient. Recipients were sacrificed and perfused after 24 h and 72 h post-transfer. Brains were harvested, processed, sectioned and evaluated by immunohistochemistry for expression of GFP (green), CD4 (purple) and GFP via anti-GFP primary antibodies (α-GFP) (red). Anti-CD4 and anti-GFP were visualized by secondary antibodies conjugated to DyLight-649 (purple) or Alexa Fluor-594 (red). Tissues were analyzed by confocal laser-scanning microscopy and images are shown at ×600 magnification. The data is representative of three replicate experiments.
3.6. Depletion of Treg in peripheral noninflammatory tissues
Finally to assess tissue distribution of donor Treg in mice with HIVE, peripheral blood was assessed after 2 h and 24 h post-transfer. By 2 h after adoptive transfer, donor GFP+ Treg comprised 6% of the lymphocyte population in the peripheral circulation as determined by flow cytometric analysis; whereas, by 24 hours only 0.6% GFP+ Treg cells were detected (Fig 6A). After acquisition of CT/SPECT images 72 h post-adoptive transfer, mice were sacrificed and perfused. Brain, heart, liver, spleen, lung, kidney and lymph nodes were harvested and radioactivity of 111In-labeled Treg within tissues was measured by γ-scintillation spectrometry. Based on radioactivity levels, by 72 h most of the Treg resided in liver and spleen (up to 50% of total counts) with no detectable differences in counts between similar peripheral tissues of mice demonstrating HIVE-like pathology compared with PBS sham controls (Fig 2B). Liver tissues from perfused HIVE mice at 24 h and 72 h post-Treg transfer were assessed for donor Treg by immunohistochemistry. Densities of donor Treg in the liver at 24 h were at or above 10X greater than those at 72 h (Fig 6C) suggesting that high radioactivity in tissues at later times may be due in part to 111In released from Treg and retained in those tissues. Taken together, the data showed that the majority of in vivo expanded Tregs did not accumulate within the peripheral noninflammatory tissues such as liver and spleen. However, the small numbers of Treg that accumulated within inflammatory foci remained morphologically intact and as such are capable of modulating neuroinflammation, inhibiting viral infection, and providing neuroprotection to the host.
FIGURE 6. Treg are depleted in peripheral tissues.
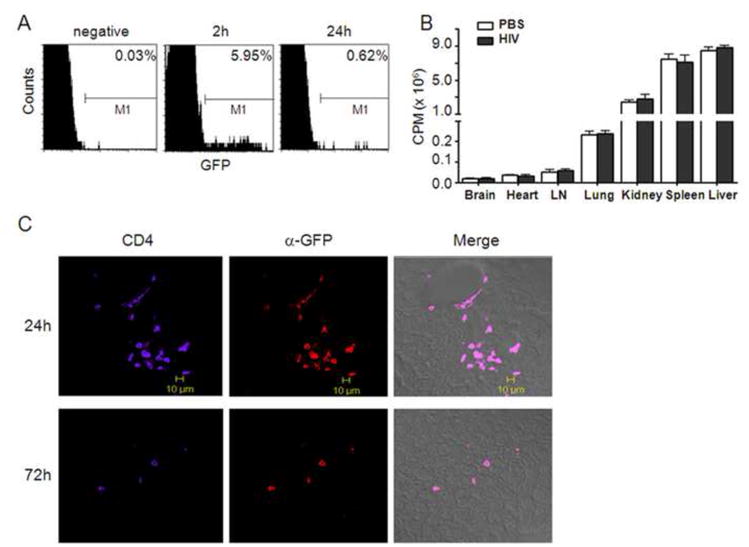
Twenty million 111In-labeled CD4+CD25+GFP+ Treg were adoptively transferred to HIVE and PBS (sham-injected) control mice. A, Peripheral blood was collected via tail vein at 2 h and 24 h post-transfer and frequencies of CD4+CD25+GFP+ donor Treg were assessed among the recipient lymphocyte population by flow cytometric analysis. B, Seventy-two hours post-transfer, mice were sacrificed, perfused and tissues from brain, heart, lymph nodes, lung kidney, spleen and liver collected. Tissue distribution of 111In-labeled Treg was assessed by γ-scintillation spectrometry. C, Liver samples from HIVE mice administered CD4+CD25+GFP+ Treg were obtained after 24 h and 72 h post-transfer and 30 μm sections were assessed by immunohistochemistry for expression of CD4 (purple) and GFP (red) by confocal laser-scanning microscopy as described in Fig 5. Merged images (right panels) are for CD4, GFP and phase contrast. Images are shown at ×400 magnification. The data is representative of three replicate experiments.
4. Discussion
Up to one third of adults and half of children with the AIDS eventually have neurologic complications, which are directly attributable to infection of the brain by HIV-1. Neurologic problems include impaired mental concentration, slowness of hand movements, and difficulty in walking, all important clinical manifestations of HAND (Antinori et al., 2007; Letendre et al., 2007). Following the wide spread use of antiretroviral drugs, the severity of disease has diminished. With major and sustained declines in opportunistic complications, HIV infection is increasingly becoming a more chronic disease (Letendre et al., 2007). Therefore, as ART are used more commonly and over longer time periods, drug toxicities become an increasingly important issue in the management of infected patients (Carr, 2003) with increasing research activities now focused on developing adjunctive treatments for disease (Perry et al., 2005; Pett and Emery, 2001). As the major factors involved in inducing neural injury in HAND are viral and cellular products secreted by immune competent mononuclear phagocytes, immunomodulatory drugs that combat those released factors are heralded for their abilities to improve neuronal function, synaptic and dendritic degeneration, and prevent neuronal apoptosis (Garden, 2002; Schifitto et al., 2006). Treatments that interfere with neuroinflammation or that protect neurons from damage can be expected to have a positive effect in the pathogenesis of HAND.
Can Treg be considered as a proposed treatment for HAND? Treg numbers are limited, being present in only 5–10% of the mature CD4+ T cell pool in mice and less than 5% in humans and therefore restrict the functional analysis and clinical application of such cells. Greater numbers of Tregs would be necessary for intervention, thus requiring in vivo or ex vivo expansion of existing Treg numbers or activity. To this end and in order to considerably increase the number of Treg, methods of polyclonal or antigen-specific in vitro expansion of both murine and human CD4+CD25+ Treg were developed and are known to exert fundamental control over a range of immune responses. Data generated in preclinical animal models indicate that adoptive transfer of Treg prevents or ameliorates several T cell-mediated disorders, including autoimmune diseases and allograft rejection, by regulating immune tolerance to self antigens (Battaglia et al., 2006; Hoffmann et al., 2002; Tarbell et al., 2007). The advantages of Treg adoptive transfer compared to other treatments are numerous and include: 1) the potential for antigen specificity with the lack of general immunosuppression; 2) the possibility of inducing natural and accurate long-lasting physiological regulation in vivo; and 3) the fact that Treg-based immunotherapy could be a custom-made product, designed for each patient, with very limited side effects (Roncarolo and Battaglia, 2007). Recent studies in others and our own laboratories have shown that Treg modulate inflammation, attenuate microglial activation, and promote neuronal survival in neurodegeneration diseases (Hara et al., 2001; Reynolds et al., 2009a; Reynolds et al., 2009b). Thus, we theorized that Treg could be used in therapy for neurodegenerative disorders by regulating neuroinflammatory responses. However, relatively few in vivo studies have assessed the ability of adoptively-transferred Treg to migrate to target sites involved in disease or biological function. Herein, we developed an HIVE animal model and used dual-labeled Treg to assess whether they reach a targeted site. As Treg are thought to function by cell-cell contact, a critical parameter of cell-based therapies would be to estimate whether there are adequate numbers of Treg available to penetrate the inflammatory target site induced by HIV-1, and also to estimate whether excess Treg that are not necessary to host protection were depleted in vivo. To determine if Treg could migrate to the inflammation area in brain, we tracked GFP+ Treg using immuofluorescence staining and confocal microscopy, and found that Treg infiltrated the CNS BBB and remained exactly at the sites of brain inflammation in HIVE, which is congruent with the notion that Treg function in a cell–cell contact dependent manner (Bluestone and Tang, 2005).
Using CT/SPECT for assessment of migration and distribution of adoptively transferred 111In-labeled cells in vivo (Bennink et al., 2004), we showed that radioactivity in lungs was greater at 2 h post-transfer compared with that in spleen and liver. By day 1 and times thereafter, radioactivity decreased in lungs and increased in spleen and liver; however, no significant differences were detected between HIVE and PBS sham-operated mice. Similarly, by γ-scintillation spectrometry and histological examination results, no significant differences were observed in the distribution of adoptively transferred Treg in HIVE and control animals. Comparable radioactivity results amongst HIVE and sham control mice demonstrate the test’s level of sensitivity to ascertain small changes in cell ingress within the CNS compared to the periphery. Importantly, despite radioactivity results that would suggest the persistence of Treg over 72 hours in the liver and spleen, the numbers of Treg in those tissues detected by immunofluorescence diminished by over an order of magnitude. This contrasts significantly to the brain; wherein, Treg infiltrated HIVE affected brain regions and were retained in sites of viral-induced inflammation. This seemingly paradoxical result between the imaging data and immunohistochemical evaluations likely reflected increased leakage of radiolabel from the adoptively-transferred Treg that have been noted in other replicate systems (Botti et al., 1997; Kuyama et al., 1997; Ritchie et al., 2007).
The experiments showed herein used an efficient method to expand polyclonal murine Treg up to 300-fold within 3 weeks. Expanded Treg maintained their phenotype and suppressive capacity but showed increased activity compared with freshly isolated cells. On a functional level, Treg expanded by anti-CD3/anti-CD28 beads differed in their capacity to produce IL-10. Anti-CD3/anti-CD28 stimulation usually generates high levels of IL-10 in murine, as well as in human, Treg (Levings et al., 2001; Tang et al., 2004). IL-10-mediated inhibition of cytokine production is believed to act by inhibition of the antigen presentation capacity of macrophages and dendritic cells, blockade of cytokine production and chemokine secretion, thus limiting the magnitude of immune responses (Fiorentino et al., 1989; Pestka et al., 2004). Kootstra showed that IL-10 inhibits HIV-1 replication specifically in macrophages, but not T cells, in a dose-dependent manner, that seemed to involve inhibition of viral protein processing (Kootstra et al., 1994). Similarly, TGF-β regulates adaptive immune components, such as T cells (Wan and Flavell, 2007), as well as innate immune components, such as natural killer (NK) cells (Ghiringhelli et al., 2006; Ralainirina et al., 2007; Wahl, 2007). Regulation of immune responses by TGF-β proceeds by inhibition of inflammatory cell function and promotion of Treg function (Wan and Flavell, 2008), the latter through induction of Foxp3 expression, which is implicated in Treg function (Komatsu et al., 2009). Taken together, these data indicate that CD4+CD25+FoxP3+ Treg retained their phenotype and function after in vitro expansions.
Substantial progress in understanding the biology of Treg and their roles in HAND has led to increasing interests in their potential utility as adjunct biological therapies in HIV disease. However, as in most cellular therapeutic strategies, risks of uncontrolled proliferation and/or development of unforeseen functional activities pose potential limitations. This is underscored by the observation that some Treg lose Foxp3 expression and convert to conventional T cells (Komatsu et al., 2009). Another concern for Treg-based therapeutics is that given the potency by which Treg function, excess Tregs could truncate immune responses necessary for surveillance of neoplasia and control of microbial infections. In the context of HIV disease, Tregs used to therapeutically control inflammation associated with neuroAIDS may inhibit the same immune responses that maintain control of viral loads and thus lead to acute viral relapses under otherwise chronic disease states (Fantini et al., 2004; Nelson, 2004). Nevertheless, Food and Drug Administration mandates for demonstrable reproducibility and control for sterility, purity, and potency of an exact defined cell therapy product present formidable barriers to clinical translation. In our study, we found that extra adoptive transferred Treg within non-inflamed tissues were depleted; whereas, Treg within inflamed sites in HIVE mice were indeed neuroprotective. Thus taken together, our results demonstrate the importance of Treg in HAND control and raise the possibility of their utility for therapeutic strategies.
Acknowledgments
This work was supported by NIH grant P20 RR15635, 1 P01 NS043985-01, 2R37 NS36126 and 5 P01 MH64570-03 (to H. E. G.).
We thank Dr. Harris Gelbard, University of Rochester Medical Center, for active discussions and critical insights into immune strategies used; and University of Nebraska Medical Center staff: Mrs. Victoria Smith and Dr. Charles Kuszynski for FACS analyses, Ms. Valerie K. Shostrom, Statistical Coordinator, College of Public Health Department of Biostatistics; and Dr. Michael Boska for important support and communication for the CT/SPECT tests, and Ms. Robin Taylor for editorial and graphic support.
Footnotes
Disclosures
The authors have no financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- Beilharz MW, Sammels LM, Paun A, Shaw K, van Eeden P, Watson MW, Ashdown ML. Timed ablation of regulatory CD4+ T cells can prevent murine AIDS progression. J Immunol. 2004;172:4917–4925. doi: 10.4049/jimmunol.172.8.4917. [DOI] [PubMed] [Google Scholar]
- Bennink RJ, van Montfrans C, de Jonge WJ, de Bruin K, van Deventer SJ, te Velde AA. Imaging of intestinal lymphocyte homing by means of pinhole SPECT in a TNBS colitis mouse model. Nucl Med Biol. 2004;31:93–101. doi: 10.1016/s0969-8051(03)00109-4. [DOI] [PubMed] [Google Scholar]
- Bluestone JA. Regulatory T-cell therapy: is it ready for the clinic? Nat Rev Immunol. 2005;5:343–349. doi: 10.1038/nri1574. [DOI] [PubMed] [Google Scholar]
- Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol. 2005;17:638–642. doi: 10.1016/j.coi.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Botti C, Negri DR, Seregni E, Ramakrishna V, Arienti F, Maffioli L, Lombardo C, Bogni A, Pascali C, Crippa F, Massaron S, Remonti F, Nerini-Molteni S, Canevari S, Bombardieri E. Comparison of three different methods for radiolabelling human activated T lymphocytes. Eur J Nucl Med. 1997;24:497–504. doi: 10.1007/BF01267680. [DOI] [PubMed] [Google Scholar]
- Boyer O, Saadoun D, Abriol J, Dodille M, Piette JC, Cacoub P, Klatzmann D. CD4+CD25+ regulatory T-cell deficiency in patients with hepatitis C-mixed cryoglobulinemia vasculitis. Blood. 2004;103:3428–3430. doi: 10.1182/blood-2003-07-2598. [DOI] [PubMed] [Google Scholar]
- Buckner CM, Luers AJ, Calderon TM, Eugenin EA, Berman JW. Neuroimmunity and the blood-brain barrier: molecular regulation of leukocyte transmigration and viral entry into the nervous system with a focus on neuroAIDS. J Neuroimmune Pharmacol. 2006;1:160–181. doi: 10.1007/s11481-006-9017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr A. Toxicity of antiretroviral therapy and implications for drug development. Nat Rev Drug Discov. 2003;2:624–634. doi: 10.1038/nrd1151. [DOI] [PubMed] [Google Scholar]
- Chadwick W, Magnus T, Martin B, Keselman A, Mattson MP, Maudsley S. Targeting TNF-alpha receptors for neurotherapeutics. Trends Neurosci. 2008;31:504–511. doi: 10.1016/j.tins.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Dittmer U, He H, Messer RJ, Schimmer S, Olbrich AR, Ohlen C, Greenberg PD, Stromnes IM, Iwashiro M, Sakaguchi S, Evans LH, Peterson KE, Yang G, Hasenkrug KJ. Functional impairment of CD8(+) T cells by regulatory T cells during persistent retroviral infection. Immunity. 2004;20:293–303. doi: 10.1016/s1074-7613(04)00054-8. [DOI] [PubMed] [Google Scholar]
- Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 2005;26:485–495. doi: 10.1016/j.it.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Fantini MC, Becker C, Monteleone G, Pallone F, Galle PR, Neurath MF. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25- T cells through Foxp3 induction and down-regulation of Smad7. J Immunol. 2004;172:5149–5153. doi: 10.4049/jimmunol.172.9.5149. [DOI] [PubMed] [Google Scholar]
- Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med. 1989;170:2081–2095. doi: 10.1084/jem.170.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia. 2002;40:240–251. doi: 10.1002/glia.10155. [DOI] [PubMed] [Google Scholar]
- Gartner S. HIV infection and dementia. Science. 2000;287:602–604. doi: 10.1126/science.287.5453.602. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Lipton SA, Tardieu M, Bukrinsky MI, Nottet HS. The neuropathogenesis of HIV-1 infection. J Leukoc Biol. 1994;56:389–398. doi: 10.1002/jlb.56.3.389. [DOI] [PubMed] [Google Scholar]
- Ghiringhelli F, Menard C, Martin F, Zitvogel L. The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression. Immunol Rev. 2006;214:229–238. doi: 10.1111/j.1600-065X.2006.00445.x. [DOI] [PubMed] [Google Scholar]
- Gorantla S, Liu J, Sneller H, Dou H, Holguin A, Smith L, Ikezu T, Volsky DJ, Poluektova L, Gendelman HE. Copolymer-1 induces adaptive immune anti-inflammatory glial and neuroprotective responses in a murine model of HIV-1 encephalitis. J Immunol. 2007;179:4345–4356. doi: 10.4049/jimmunol.179.7.4345. [DOI] [PubMed] [Google Scholar]
- Gorantla S, Liu J, Wang T, Holguin A, Sneller HM, Dou H, Kipnis J, Poluektova L, Gendelman HE. Modulation of innate immunity by copolymer-1 leads to neuroprotection in murine HIV-1 encephalitis. Glia. 2008;56:223–232. doi: 10.1002/glia.20607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Kingsley CI, Niimi M, Read S, Turvey SE, Bushell AR, Morris PJ, Powrie F, Wood KJ. IL-10 is required for regulatory T cells to mediate tolerance to alloantigens in vivo. J Immunol. 2001;166:3789–3796. doi: 10.4049/jimmunol.166.6.3789. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Eder R, Kunz-Schughart LA, Andreesen R, Edinger M. Large-scale in vitro expansion of polyclonal human CD4(+)CD25high regulatory T cells. Blood. 2004;104:895–903. doi: 10.1182/blood-2004-01-0086. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med. 2002;196:389–399. doi: 10.1084/jem.20020399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolson DL, Pomerantz RJ. AIDS Dementia and HIV-1-Induced Neurotoxicity: Possible Pathogenic Associations and Mechanisms. J Biomed Sci. 1996;3:389–414. doi: 10.1007/BF02258044. [DOI] [PubMed] [Google Scholar]
- Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci U S A. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kootstra NA, van ‘t Wout A, Huisman HG, Miedema F, Schuitemaker H. Interference of interleukin-10 with human immunodeficiency virus type 1 replication in primary monocyte-derived macrophages. J Virol. 1994;68:6967–6975. doi: 10.1128/jvi.68.11.6967-6975.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyama J, McCormack A, George AJ, Heelan BT, Osman S, Batchelor JR, Peters AM. Indium-111 labelled lymphocytes: isotope distribution and cell division. Eur J Nucl Med. 1997;24:488–496. doi: 10.1007/BF01267679. [DOI] [PubMed] [Google Scholar]
- Letendre S, Ances B, Gibson S, Ellis RJ. Neurologic complications of HIV disease and their treatment. Top HIV Med. 2007;15:32–39. [PubMed] [Google Scholar]
- Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gong N, Huang X, Reynolds AD, Mosley RL, Gendelman HE. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J Immunol. 2009;182:3855–3865. doi: 10.4049/jimmunol.0803330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekala DJ, Geiger TL. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Blood. 2005;105:2090–2092. doi: 10.1182/blood-2004-09-3579. [DOI] [PubMed] [Google Scholar]
- Mollace V, Nottet HS, Clayette P, Turco MC, Muscoli C, Salvemini D, Perno CF. Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends Neurosci. 2001;24:411–416. doi: 10.1016/s0166-2236(00)01819-1. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol. 2004;172:3983–3988. doi: 10.4049/jimmunol.172.7.3983. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Gelbard HA. Adjunctive therapies for HIV-1 associated neurologic disease. Neurotox Res. 2005;8:161–166. doi: 10.1007/BF03033827. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, Patil A, Nottet HS, Epstein L, Gelbard H, Flanagan E, Reinhard J, Pirruccello SJ, Gendelman HE. Human immunodeficiency virus encephalitis in SCID mice. Am J Pathol. 1996;149:1027–1053. [PMC free article] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- Pett SL, Emery S. Immunomodulators as adjunctive therapy for HIV-1 infection. J Clin Virol. 2001;22:289–295. doi: 10.1016/s1386-6532(01)00201-3. [DOI] [PubMed] [Google Scholar]
- Poluektova LY, Munn DH, Persidsky Y, Gendelman HE. Generation of cytotoxic T cells against virus-infected human brain macrophages in a murine model of HIV-1 encephalitis. J Immunol. 2002;168:3941–3949. doi: 10.4049/jimmunol.168.8.3941. [DOI] [PubMed] [Google Scholar]
- Ralainirina N, Poli A, Michel T, Poos L, Andres E, Hentges F, Zimmer J. Control of NK cell functions by CD4+CD25+ regulatory T cells. J Leukoc Biol. 2007;81:144–153. doi: 10.1189/jlb.0606409. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J Immunol. 2009a;182:4137–4149. doi: 10.4049/jimmunol.0803982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Proteomic studies of nitrated alpha-synuclein microglia regulation by CD4+CD25+ T cells. J Proteome Res. 2009b;8:3497–3511. doi: 10.1021/pr9001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S, Thome M, Pretsch L, Enk AH, Mahnke K. Expanded murine regulatory T cells: analysis of phenotype and function in contact hypersensitivity reactions. J Immunol Methods. 2007;326:10–21. doi: 10.1016/j.jim.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Ritchie D, Mileshkin L, Wall D, Bartholeyns J, Thompson M, Coverdale J, Lau E, Wong J, Eu P, Hicks RJ, Prince HM. In vivo tracking of macrophage activated killer cells to sites of metastatic ovarian carcinoma. Cancer Immunol Immunother. 2007;56:155–163. doi: 10.1007/s00262-006-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ES, Zandonatti MA, Watry DD, Madden LJ, Henriksen SJ, Taffe MA, Fox HS. Induction of pathogenic sets of genes in macrophages and neurons in NeuroAIDS. Am J Pathol. 2003;162:2041–2057. doi: 10.1016/S0002-9440(10)64336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol. 2007;7:585–598. doi: 10.1038/nri2138. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- Schifitto G, Peterson DR, Zhong J, Ni H, Cruttenden K, Gaugh M, Gendelman HE, Boska M, Gelbard H. Valproic acid adjunctive therapy for HIV-associated cognitive impairment: a first report. Neurology. 2006;66:919–921. doi: 10.1212/01.wnl.0000204294.28189.03. [DOI] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA. Regulatory T-cell physiology and application to treat autoimmunity. Immunol Rev. 2006;212:217–237. doi: 10.1111/j.0105-2896.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, Yang H, Suthanthiran M, Mojsov S, Steinman RM. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med. 2007;204:191–201. doi: 10.1084/jem.20061631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. 2004;199:1467–1477. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, Cohen JL. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest. 2003;112:1688–1696. doi: 10.1172/JCI17702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM. Transforming growth factor-beta: innately bipolar. Curr Opin Immunol. 2007;19:55–62. doi: 10.1016/j.coi.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol Rev. 2007;220:199–213. doi: 10.1111/j.1600-065X.2007.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. TGF-beta and regulatory T cell in immunity and autoimmunity. J Clin Immunol. 2008;28:647–659. doi: 10.1007/s10875-008-9251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DA, Peng G, Guo Z, Li Y, Kiniwa Y, Shevach EM, Wang RF. Tumor-specific human CD4+ regulatory T cells and their ligands: implications for immunotherapy. Immunity. 2004;20:107–118. doi: 10.1016/s1074-7613(03)00359-5. [DOI] [PubMed] [Google Scholar]